

Abstract
BMS-433771 was found to be a potent inhibitor of respiratory syncytial virus (RSV) replication in vitro. It exhibited excellent potency against multiple laboratory and clinical isolates of both group A and B viruses, with an average 50% effective concentration of 20 nM. Mechanism-of-action studies demonstrated that BMS-433771 inhibits the fusion of lipid membranes during both the early virus entry stage and late-stage syncytium formation. After isolation of resistant viruses, resistance was mapped to a series of single amino acid mutations in the F1 subunit of the fusion protein. Upon oral administration, BMS-433771 was able to reduce viral titers in the lungs of mice infected with RSV. This new class of orally active RSV fusion inhibitors offers potential for clinical development.
Respiratory syncytial virus (RSV) belongs to the Pneumovirus genus of the Paramyxovirus virus family. RSV is the leading cause of virus-induced lower respiratory tract disease among infants and children (6, 21, 26) and is the most common pathogen found in children under 5 years of age admitted to the hospital. Essentially every child develops an RSV infection during the first 2 years of life, and recurrent infections are common (20, 24, 42, 44). RSV is especially serious in premature infants and children with bronchopulmonary dysplasia or congenital heart disease. Additionally, in recent studies, RSV was the most common virus identified in the middle-ear fluid of children suffering from acute otitis media (25, 43). RSV infection has also been implicated in the development of childhood asthma and other long-term conditions involving pulmonary dysfunction (54-56).
RSV is also a significant etiologic agent in the elderly (17, 18, 42, 65). In this population, RSV infection manifests as a flu-like illness that can be misdiagnosed as influenza (39, 46). A recent epidemiology study illustrates the impact of RSV on mortality. In that study it was estimated that in the United States >17,000 deaths/year are attributable to RSV infection, with >78% of these deaths occurring in persons over 65 years of age (62). Patients in nursing homes are at greatest risk, with outbreak rates measured as high as 40% (39, 58). For adults, RSV infection is most dangerous in immunosuppressed patients, as it is life threatening. For instance, in bone marrow transplant patients, there can be progression to severe lower respiratory pneumonia, leading to high mortality rates (16, 20, 38). Recent data also suggest that RSV is a significant pathogen in healthy adults, with one study showing that 43% of the adults with confirmed RSV infections missed work for periods of up to 2 weeks (12, 19, 44).
At present, the only clinically approved therapeutic agent against RSV is aerosolized ribavirin (Virazole). However, this drug is of limited use due to its mode of administration, among other issues (22, 52). In addition, a humanized monoclonal antibody, Synagis, has been approved for prophylactic use (52). However, its use is restricted to high-risk children up to 2 years of age.
Recently, a number of small-molecule inhibitors of RSV infection in cell culture have been described (27, 33, 41, 60; K. Andries et al., Abstr. 40th Intersci. Conf. Antimicrob. Agents Chemother., abstr. 130, 2000). Interestingly, although these small molecules show little structural homology with each other, three of these compounds have been reported to inhibit RSV in a similar fashion, by interfering with virus-cell membrane fusion (27, 41; Andries et al., 40th ICAAC). However, none of these compounds exhibited pharmacokinetic properties that would allow for oral dosing. We have identified a series of small-molecule RSV inhibitors from the proprietary compound library at Bristol-Myers Squibb. Following a synthetic chemistry effort, BMS-433771 was generated as a potential drug candidate. Unlike other small-molecule RSV inhibitors, BMS-433771 has pharmacokinetic properties that allow for oral efficacy in an in vivo animal model of infection.
MATERIALS AND METHODS
Compounds.
BMS-233675 (47, 48) was the original lead compound identified from the screen of the Bristol-Myers Squibb proprietary chemical deck. BMS-433771 and BMS-243458 were prepared by the Medicinal Chemistry group at Bristol-Myers Squibb. Bis(5-amidino-2-benzimidazolyl)methane (BABIM), a known inhibitor of RSV, was synthesized by a previously published procedure (64) (Fig. 1). For in vitro experiments, all compounds were dissolved in dimethyl sulfoxide to a concentration of 20 mM.
FIG. 1.

Structures of key fusion inhibitors used in this study.
RSV growth.
All tissue culture reagents were obtained from GIBCO/BRL (Grand Island, N.Y.). HEp-2 cells and the Long, A2, and B Wash/18537/62 (BWash) RSV strains were obtained from the American Type Culture Collection (Manassas, Va.). HEp-2 cells were propagated at 37°C in Dulbecco's modified Eagle's medium (DMEM) supplemented with heat-inactivated 10% fetal bovine serum (FBS), 200 U of penicillin G per ml and 200 μg of streptomycin per ml (PEN-STR), and l-glutamine (Glu). Human RSV laboratory stains and clinical isolates were propagated on 85% confluent HEp-2 monolayers in DMEM supplemented with 2% FBS, PEN-STR, and Glu. Cells were infected at a multiplicity of infection (MOI) of 0.002 to 0.01 PFU/cell. Infectivity was monitored by examining the cell monolayers for cytopathic effect (CPE) and the appearance of syncytia. RSV was harvested when sufficient cytopathology was observed, usually at 4 to 6 days postinfection. Clinical isolates of RSV were obtained from Ann Falsey (University of Rochester) or Peter Wright (Vanderbilt University). After amplification the viral titers ranged from 1.0 × 106 to 2.5 × 107 PFU/ml, as determined by plaque assays with methylcellulose overlays.
Plaque titration of RSV.
Virus titrations were performed on ∼85% confluent HEp-2 monolayers in 35-mm2 tissue culture dishes. Monolayers were rinsed with phosphate-buffered saline (PBS) and infected with 100 μl of diluted virus stock. Following 1 h of adsorption, the dishes were carefully overlaid with 2 ml of minimal essential medium (MEM) supplemented with 2% FBS, PEN-STR, and Glu containing 0.75% methylcellulose (4,000 cP) and an additional 0.1% NaHCO3. At 6 days post infection, the overlay was removed and the infected monolayers were stained with 0.2% crystal violet in 25% methanol for 2 h, followed by gentle rinsing. During examination of compound efficacy, the compound was present in both the inoculum and the methylcellulose overlay.
Assay for cell protection against RSV.
An assay for cell protection against RSV was used to screen for compounds that inhibit RSV-induced CPE in tissue culture. The RSV Long strain was used to infect HEp-2 cells that were seeded in flat-bottom 96-well plates at 1.5 × 104 cells/well, and cell viability and cytotoxicity were examined by determination of the ability of cell mitochondria to metabolize 3-(4,5-dimethylthiazole-2-yl)-2,5-diphenyltetrazolium bromide (MTT), as described previously (49). All assays were run in quadruplicate, and a series of uninfected HEp-2 cells containing test compounds were run in parallel to assess the cytoxicities of the test compounds. MTT was added to the cells following a 6-day incubation, at which time complete cell death was observed for the cells in control wells with virus-infected cells.
The cell protection assays with other viruses used a similar protocol, with specific modifications based on virus type. For influenza virus, Madin-Darby bovine kidney (MDBK) or Madin-Darby canine kidney (MDCK) cells were used. Influenza virus A/WSN/33 was added to wells in 100 μl of medium at an MOI of 0.002, and MTT incorporation assays were conducted after a 3-day incubation at 37°C (36). Rhinovirus (human rhinovirus type 16), vesicular stomatitis virus (VSV; Indiana strain), and poliovirus were assayed with H1-HeLa cells plated into black, clear-bottom 96-well plates (nontreated) at MOIs of 0.1, 0.01, and 0.1, respectively. Cell viabilities in assays with these three viruses were measured by Alamar Blue (Biosource International, Camarillo, Calif.) reduction. Following appropriate incubation periods, each well of the plates was incubated with 20 μl of Alamar Blue for 5 h at 34°C, and the results were read on an LJL microplate reader equipped with a dichroic 561-rhodamine filter (Molecular Devices, Sunnyvale, Calif.). The plates were processed at 3 days postinfection for rhinovirus and at 2 days postinfection for VSV and poliovirus. For human immunodeficiency virus (HIV), MT-2 cells were used along with the T-cell-tropic LAI strain of HIV at an MOI of 0.001 in DMEM supplemented with 10% FBS, PEN-STR, and Glu. Following a 5-day incubation at 37°C, antiviral activity was measured by harvesting cell media and quantitating HIV by determination of reverse transcriptase activity (51). Cytotoxicity was assessed by the standard MTT assay (49).
Viral protein expression assay.
The abilities of the test compounds to inhibit RSV replication was measured by the expression of virus-specific proteins in infected cells by a [35S]methionine incorporation assay. RSV was used to infect 80% confluent HEp-2 cells in 35-mm2 dishes in the presence of the test compounds. For single-cycle replication studies, cells were infected at MOIs of 2 to 10 in the presence or absence of test compound in DMEM supplemented with 2% FBS, PEN-STR, and Glu. At 16 h postinfection, the medium was removed and the infected cells were starved in 1 ml of DMEM without methionine for 30 min. The medium was removed and replaced with 200 μl of DMEM that lacked methionine and that was supplemented with 20 μCi of [35S]-methionine ([35S]Protein Labeling Mix; NEN Life Science, Boston, Mass.). After 90 min at 37°C, the labeling medium was removed and the monolayers were carefully washed with 2 ml of PBS. The cells were lysed in 200 μl of radioimmunoprecipitation assay buffer (50 mM Tris [pH 8.0], 150 mM NaCl, 1% Nonidet P-40, 0.5% sodium dexoycholate, 0.1% sodium dodecyl sulfate), and RSV proteins were precipitated with 5 μg of goat anti-RSV polyclonal immunoglobulin G (Fitzgerald Industries, Concord, Mass.) and a 25-μl suspension of protein G-Sepharose (Amersham Pharmacia Biotech, Piscataway, N.J.) (1). Samples were analyzed on 12% acrylamide Tris-Glycine Ready Gels (Bio-Rad, Hercules, Calif.), and the gels were fixed and treated with En3Hance (NEN Life Science) according to the instructions of the manufacturer. The radiolabeled proteins in dried gels were quantitated by scanning the autoradiographs with a Molecular Dynamics SI Personal Densitometer with ImageQuaNT software (Molecular Devices, Sunnyvale, Calif.). The intensity of the RSV-specific matrix band was used for EC50pro determinations. EC50pro represents the concentration of inhibitor yielding 50% of the level of protein synthesis obtained for the untreated virus-infected control. For multiple-cycle protein expression studies, infections were carried out at MOIs of 0.08 to 0.4, and the cells were incubated for 64 h before radiolabeling, as described above.
Virus-specific protein expression studies were also conducted for parainfluenza virus type 3 and Sendai virus type 52 on ∼85% confluent MDBK cell monolayers in 35-mm2 dishes. Cells infected with Sendai virus were labeled with [35S]methionine at 16 h postinfection, viral protein bands were identified directly by sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and dried gels were used for quantitation, as described above. Parainfluenza virus type 3-infected cells were radiolabeled with [35S]methionine at 32 h postinfection, and proteins were identified after immunoprecipitation with goat anti-parainfluenza virus type 1 polyclonal immunoglobulin G (which is cross-reactive with parainfluenza virus type 3; Biodesign International, Saco, Maine) and protein G-Sepharose.
Syncytium formation assay.
HEp-2 cells in 35-mm2 dishes were infected with RSV at MOIs of 5 to 10 PFU/cell in 1 ml of DMEM supplemented with 2% FBS, PEN-STR, and Glu. At 16 h postinfection, 25 or 250 nM compound was added to the medium. Monolayers were fixed 12 h later in 100% methanol for 5 min, stained with Accustain (Sigma Diagnostics, St. Louis, Mo.), washed with water, and air dried. Syncytia were photographed with a Polaroid camera mounted on an Olympus inverted microscope.
Generation of resistant viruses.
Resistant RSVs were generated through serial passages (MOIs, 0.01 to 0.1) of the wild-type Long strain in liquid medium successively containing 1, 2, 5, 10, 20, and 40 μg of test compound per ml. Resistant viruses were plaque purified on HEp-2 monolayers overlaid with 0.75% agarose containing 5 μg of compound per ml. Individual plugs containing single plaques were removed with a pipette tip and amplified in T-150 flasks with or without 1 to 2 μg of test compound per ml. Viruses resistant to BMS-233675, BMS-243458, or BABIM were generated (Fig. 1).
Genotypic characterization of resistant viruses.
Genomic RNAs from wild-type and resistant viruses were obtained from infected HEp-2 monolayers through cell lysis, phenol extraction, and ethanol precipitation (1). Viral RNAs were reverse transcribed and amplified by PCR with primers specific for the fusion (F) protein, short hydrophobic (SH) protein, and the glycoprotein (G protein). Multiple individual clones of each gene were isolated and sequenced. In addition, the reverse transcription-PCR gene product was sequenced directly to obtain a consensus sequence.
Reverse genetics.
The infectious cDNA clone derived from the A2 strain (D53) was obtained from Peter Collins (National Institutes of Health). The avian poxvirus MVA, which expresses the bacteriophage T7 polymerase, was obtained from Bernard Moss (National Institutes of Health). Isolation of a rescued virus by use of transfected full-length RSV DNA, various T7 polymerase-based RSV protein expression plasmids, and the MVA vaccinia virus was performed exactly as described previously (8). The infectious clone was modified to contain the single lysine-to-arginine change at amino acid 394 of the F protein that was found in a resistant virus. In order to do this, a unique BssHII sequence was inserted at nucleotide 7317 of the infectious clone. This was accomplished by altering the nucleotides at positions 7317 (C to G), 7318 (A to C), and 7320 (A to C) to create the BssHII site without changing the amino acid coding sequence. The identical BssHII site was also inserted into the cloned 458R7 F-protein gene, which contains a lysine-to-arginine mutation at amino acid 394. A 1,706-nucleotide fragment created through a unique StuI restriction site (present at nucleotide 5611 of the full-length clone) and the BssHII site was recovered from the modified 458R7 F-protein gene and used to replace the homologous BssHII-StuI fragment in the modified D53 full-length clone. This created an infectious full-length clone with only one amino acid difference (K394R) from the sequence of the wild-type BMS-433771-sensitive virus, designated the RSV A2 K394R transfectant.
Mouse model of RSV infection.
The inbred BALB/c mouse host model of RSV infection (2, 61) was used to examine BMS-433771 for in vivo efficacy. The compound was tested by oral administration to female BALB/c mice (age, 6 to 10 weeks; weight, between 18 and 22 g). BMS-433771 was dissolved in a solution of 50% polyethylene glycol 400 (Sigma, St. Louis, Mo.) in water. The mice were dosed by oral gavage with 0.2 ml of solution in water 1 h before virus inoculation. To initiate RSV infection, the mice were anesthetized by intraperitoneal injection of ketamine (70 mg/kg of body weight) and xylazine (20 mg/kg) and were inoculated via the intranasal route with 105 50% tissue culture infectious doses (TCID50s) of the Long strain of RSV in 50 μl of cell culture medium.
At 4 days after RSV infection, the mice were euthanized by CO2 asphyxiation. Excised lungs were prepared as homogenates (10%; wt/vol) in Hanks balanced salt solution containing 0.21 M sucrose, 25 mM HEPES, and 5 mM sodium l-glutamate supplemented with 20 U of penicillin G per ml, 20 μg of streptomycin per ml, and 0.05 μg of amphotericin B per ml (GIBCO/BRL). Lung homogenates were frozen on dry ice and thawed to release cell-associated virus and were then held on ice until clarification by centrifugation at 300 × g for 10 min at 4°C. For TCID50 determination, lung homogenate samples were titrated for RSV infectivity in HEp-2 cells. Each test sample was assayed in quadruplicate sets of serial threefold dilutions in serum-free MEM supplemented with 10 mM HEPES buffer, 2 mM sodium l-glutamine, 10 U of penicillin G per ml, 10 μg of streptomycin per ml, and 0.025 μg of amphotericin B per ml. Each 100-μl sample dilution was plated onto HEp-2 cells in flat-bottom 96-well polystyrene plates (Corning, Acton, Mass.). An additional 100 μl of supplemented MEM containing 5% (vol/vol) fetal calf serum was added to each well. After 6 days of incubation, 65 μl of supernatant from each well was transferred, in a replica-plate format, to fresh 96-well microculture plates of HEp-2 cell monolayers containing 50 μl of serum-free supplemented MEM. Following 90 min of incubation at 37°C, 100 μl of supplemented MEM containing 5% (vol/vol) fetal calf serum was added to each well and the cultures were incubated for an additional 6 days. This transfer provided an additional amplification of virus and allowed for massive syncytium formation after 4 days of incubation, with cell death evident by day 6. The replica plates were stained and fixed with a solution of crystal violet dye and formalin (0.05% crystal violet, 7.4% formalin, 20% ethanol). The dye-fixative mixture was left on the cells for at least 20 min and was removed by gentle rinsing with water. The dye remaining in the wells was dissolved in 200 μl of a 33% acetic acid solution and quantitated by measuring the absorbance at 590 nm with an automated spectrophotometer (SpectraMax 250). From the comparison of the absorbance readings of these wells with those of control wells, the end-point dilution of each test sample titration that achieved the TCID50 was determined by using the SOFTmax PRO Macintosh Microplate Analysis Software applications system (Molecular Devices). The final RSV titers in the lung were calculated as the log10 TCID50 per gram of lung. The lower limit of detection for this assay was ∼2.5 log10 TCID50 per g of lung.
Nucleotide sequence accession numbers.
The RSV Long strain fusion protein nucleotide sequences for BMS-433771-resistant and wild-type viruses were submitted to GenBank and have been given accession numbers AY330611, AY330612, AY330613, A330614, AY330615, and AY330616.
RESULTS
Identification and in vitro activities of RSV inhibitor compounds.
The ability to protect HEp-2 cells from a virus-induced CPE was used as a means to screen the Bristol-Myers Squibb proprietary compound deck for inhibitors of RSV. Agents that inhibit the growth of RSV are able to protect HEp-2 cells from the CPE generated by replicating RSV in this 6-day assay. From this screen, BMS-233675 (Fig. 1) was identified as an inhibitor of RSV-induced cytopathology. The effective concentration of compound yielding 50% protection (EC50) in the assay was calculated to be 0.34 μM, whereas the 50% cytotoxic concentration (CC50) was 84 μM (Fig. 2). Ribavirin, the only agent approved for use for the treatment of RSV infection, had an EC50 and a CC50 in this assay of 2.7 and 34 μM, respectively (data not shown).
FIG. 2.

Levels of cell protection from RSV by BMS-233675 (EC50s; filled circles) and cytotoxicity of BMS-233675 (CC50s; open circles).
By using BMS-233675 as a starting point, a chemistry initiative was undertaken in an attempt to increase the potency of this series against RSV and to optimize various parameters required for development of the compound as an antiviral agent for use by humans. The initial structure-activity relationship of the benzotriazole series exemplified by BMS-233675 has been reported previously (67), and additional reports are in preparation. These studies resulted in the identification of BMS-433771 (Fig. 1) as a potential clinical candidate. In dose titration experiments, BMS-433771 was able to protect HEp-2 cell cultures from RSV Long strain-induced CPE with an EC50 of 12 nM (Fig. 3A). The relative CC50 required for reduction of cellular MTT metabolism in the absence of virus was >218 μM, the highest concentration tested (data not shown).
FIG. 3.

BMS-433771 inhibition of multiple-cycle RSV replication. (A) Measurement of BMS-433771-induced protection of HEp-2 cells from RSV-induced CPE. Data are plotted as the percent protection compared to the CPE for untreated, infected controls. (B) BMS-433771 inhibition of RSV multiple-cycle viral replication as measured by viral protein expression assay. Data are plotted as the percent replication compared to that of an untreated, infected control by quantitating the virus-expressed matrix protein.
The cell protection assay for RSV is an indirect assay, since it examines the health of the infected cell. In order to directly examine viral replication, a virus-specific protein-radiolabeling assay was used. In this assay, the extent of viral protein expression was measured after single or multiple cycles of replication through metabolic labeling, radioimmunoprecipitation, and quantitation of the RSV matrix protein. This assay was found to be highly reproducible, and the results correlated well with those of the cell protection assay. By this assay, BMS-433771 inhibited RSV Long strain protein expression in infected cells in a multiple-cycle assay, with an EC50pro of 13 nM (Fig. 3B).
BMS-433771 was also examined for its activity against RSV in plaque reduction assays. Compounds were added to cells infected with RSV (∼50 PFU) and overlaid with methylcellulose for 5 days before crystal violet staining and analysis. The EC50s of BMS-433771 for the Long strain RSV in HEp-2 cells were found to range from 2 to 40 nM (data not shown). Therefore, BMS-433771 inhibited multiple-cycle RSV replication in tissue culture, as measured by cell protection, viral protein expression, and plaque reduction assays.
Activities of BMS-433771 against laboratory and clinical isolates.
All of the experiments described above were performed with the Long strain (subgroup A). In order to examine the spectrum of activity of BMS-433771 against multiple RSVs, representatives of both the A and the B subgroups were obtained and analyzed in the viral protein expression assay. BMS-433771 was efficacious against laboratory strains A2 and B Washington, with EC50pros of 10 and 18 nM, respectively (Table 1). In addition, BMS-433771 showed appreciable activities against eight clinical isolates (six subgroup A isolates and two subgroup B isolates), with EC50pros ranging from 9 to 50 nM (Table 1). The average EC50pro for the 11 viral strains was 20.4 nM.
TABLE 1.
Inhibition of RSV strains by BMS-433771
| Virus | Group | EC50pro (nM)a |
|---|---|---|
| Long | A | 13 ± 1.6 |
| A2 | A | 10 ± 2.1 |
| B Washington | B | 18 ± 4.0 |
| V8612-22b | A | 14 ± 2.7 |
| V911-73b | A | 26 ± 4.6 |
| HOU-0915b | A | 22 ± 1.8 |
| RUG-0420b | A | 24 ± 2.2 |
| JEN-1133b | A | 50 ± 15.6 |
| LEO-0713b | A | 23 ± 3.2 |
| MUL-0721b | B | 16 ± 1.8 |
| BEN-0819b | B | 9 ± 2.5 |
The results are averages of two experiments.
Clinical isolate.
Selectivity of BMS-433771 against various viruses.
In order to examine the specificity of BMS-4337771, efficacy experiments were performed with a number of related and unrelated viruses. These included parainfluenza virus type 3, Sendai virus, VSV, influenza A virus (WSN strain), human rhinovirus, HIV, and poliovirus. When the activity of BMS-433771 was examined in either cell protection or viral protein expression assays with these viruses, BMS-433771 did not exhibit any inhibitory activity at the highest concentrations examined (Table 2). The highest concentrations used range from 2,000- to 16,000-fold above the EC50 of BMS-433771 for the Long strain of RSV. The results demonstrate that BMS-433771 is a specific inhibitor of RSV. Additionally, since tests with these viruses were conducted in four separate cell lines, cytotoxicity values for BMS-433771 in these cells were also measured. BMS-433771 did not exhibit significant cytotoxicity in any of the cell lines in which it was evaluated, with the CC50s being greater than the maximum concentrations examined (Table 2).
TABLE 2.
Activity of BMS-433771 against other RNA viruses
| Virus | Cell line | EC50 (μM) | CC50 (μM) |
|---|---|---|---|
| Influenza virus | MDCK/MDBK | >200 | >200 |
| Human rhinovirus | H1 HeLa | >200 | >200 |
| Poliovirus | H1 HeLa | >200 | >200 |
| VSV | H1 HeLa | >200 | >200 |
| HIV | MT-2 | >200 | >200 |
| Parainfluenza virus type 3 | MDBK | >25 | >25 |
| Sendai virus | MDBK | >25 | >25 |
BMS-433771 is a reversible inhibitor of viral fusion.
Experiments were carried out with BMS-433771 to determine the mechanism of action of the compound in cell culture. Time-of-addition experiments with high viral MOIs (2 to 10 PFU/cell) illustrated that BMS-433771 is an inhibitor of an early step of viral infection (Fig. 4). The readout in these experiments was the expression of virus-specific proteins at 16 h postinfection. The EC50pro obtained when BMS-433771 was added at the onset of Long virus infection in single-cycle replication studies was ∼20 nM (data not shown). However, at a concentration of 5 μM (250 times the EC50pro), BMS-433771 was efficient at inhibiting viral protein synthesis only when it was added at the time of infection, reducing the level of viral protein expression by >95% (Fig. 4A, lane B). When 5 μM compound was added at 2 or 4 h postinfection at 37°C, poor inhibition of viral protein expression was observed (Fig. 4A, lanes C and D, respectively). Ribavirin addition resulted in equivalent inhibition whether the compound was added at the time of infection or at 2 or 4 h postinfection at 37°C (Fig. 4A, lanes E to G). This outcome would be expected for ribavirin, which ultimately exerts its inhibitory effects at the stage of viral transcription, a postentry event (10).
FIG. 4.


BMS-433771 mechanism-of-action studies. All samples were processed for evaluation as described in the text for the viral protection assay. (A) Effect of time of addition of BMS-433771 and ribavirin on RSV replication at 37°C. Samples were kept at 37°C, 5 μM compound was added postinfection at the times indicated below each lane, and incubation was continued for another 16 h at 37°C. (B) Reversible inhibition of RSV by BMS-433771. HEp-2 cells were infected with RSV at 37°C in the presence of 5 μM BMS-433771 (771) for 3 h and then placed at 4°C and treated as described below each lane before the samples were shifted to 37°C for 16 h. Five micrograms of anti-RSV antibody (α-RSV) was added to the specified samples. (C) Effect of time of addition of BMS-433771 on RSV replication at 4°C. Five micromolar BMS-433771 was added at the times postinfection indicated below each lane at 4°C before the samples were shifted to 37°C for 16 h.
When an enveloped virus infects cells at 4°C, the virus can still bind to cellular receptors, but the low temperature prevents viral fusion (28, 45). This allows discrimination of the mechanism of action of BMS-433771 between a mechanism of virus adsorption and one of membrane fusion. Reversibility experiments were performed by adding BMS-433771 along with RSV to HEp-2 cells and incubation for 3 h at 37°C. The cells were then placed at 4°C and washed five times with 2 ml of cold PBS to remove the BMS-433771. Finally, the dishes were incubated with 2 ml of either fresh medium or medium with anti-RSV antibody (20 μg) for 30 min at 4°C before the dishes were returned to 37°C. The cells were radiolabeled with [35S]methionine 16 h later to determine the extent of viral infection. As expected, treatment with BMS-433771 without washing or with anti-RSV antibody was able to significantly inhibit viral protein expression (Fig. 4B, lanes B and C, respectively). However, viral replication is essentially reversible when BMS-433771 is washed out after 3 h (Fig. 4B, lane E). Also, if BMS-433771 was washed out after 3 h and anti-RSV antibody was added at that time, viral protein expression was inhibited to an extent similar to that detected in control samples (Fig. 4B, lane G). This strongly suggests that BMS-433771 does not inhibit virus adsorption, because if it had, the input RSV would have been removed during the washing step, making the reversal of viral inhibition impossible. On the other hand, a fusion inhibitor would permit viral binding, and if the binding were reversible, the compound would be washed away, thereby allowing the infection to proceed (Fig. 4B, lane E). In addition, since RSV infection can still be blocked by the addition of anti-RSV antibody once BMS-433771 is removed (Fig. 4B, lane G), the compound is inhibiting the virus at a stage at which it is still susceptible to antibody neutralization. This result indicates that the inhibited virus has not yet entered the cells, in concordance with the hypothesis that BMS-433771 inhibits fusion of the virion envelope with the cellular surface plasma membrane.
Other experiments were performed to further elucidate the mechanism of inhibition of BMS-433771. HEp-2 cells were infected with virus and incubated at 4°C for specified times. BMS-433771 was added at various times postinfection, and cells were shifted to 37°C for 16 h before being processed. BMS-433771 can inhibit RSV replication during single-cycle infection if it is added at 0, 2, or 4 h postinfection at 4°C (Fig. 4C, lanes B, C, and D, respectively). This is in contrast to the results of the experiment performed at 37°C (Fig. 4A), in which inhibition was observed only at the zero time point. This result indicates that BMS-433771 can still inhibit fusion after virions have bound to cellular receptors. Ribavirin, on the other hand, exhibits inhibition when it is added at the time of infection or at 2 or 4 h infection at either 4°C (Fig. 4C, lanes E to G) or 37°C (Fig. 4A, lanes E to G).
BMS-433771 inhibits RSV-induced syncytium formation.
A characteristic of RSV infection in vitro is that infected cells fuse with adjacent infected or uninfected cells to form giant syncytia (Fig. 5B). If BMS-433771 is an inhibitor of virus-cell fusion during early infection, it should also be an inhibitor of late-stage cellular fusion, which results in syncytium formation. In order to properly examine the effects of BMS-433771 on syncytium formation, BMS-433771 was added at 16 h postinfection to ensure that it had no effects on viral entry or other early steps in replication. When 25 or 250 nM BMS-433771 was added to infected cells at 16 h, complete inhibition of RSV-induced syncytium formation was observed (Fig. 5C and D, respectively), showing that it inhibits F-induced membrane fusion.
FIG. 5.

Effect of BMS-433771 on RSV-induced syncytium formation. HEp-2 cells were infected with RSV, and BMS-433771 was added at 16 h postinfection. The samples were evaluated for syncytium formation after an additional 12 h of incubation. (A) Uninfected HEp-2 cells; (B) infected cells with no drug treatment; large multinucleated cells (syncytia) are clearly seen and are the result of RSV-induced cell fusion; (C) infected HEp-2 cells to which 25 nM BMS-433771 was added; (D) infected HEp-2 cells to which 250 nM BMS-433771 was added.
Isolation and genotype mapping of BMS-433771-resistant virus.
RSV expresses three identified envelope proteins (the F protein, the G [attachment] protein, and the SH protein) that may be involved in viral entry. Resistant viruses were generated in an effort to determine the molecular target of our chemotype. Viruses were isolated through multiple passages in HEp-2 cells in the presence of increasing concentrations of compound (BMS-233675, BMS-243458, or BABIM) (Fig. 1). BMS-243458 is an early synthetic analog within the series. BABIM is a previously reported fusion inhibitor that shares several common structural elements with this particular series (13, 14, 64). Five viruses in which resistance was generated with these three inhibitors were examined for cross-resistance to BMS-433771. All viruses exhibited significant resistance to BMS-433771 (Table 3).
TABLE 3.
Activity of BMS-43771 against resistant virusa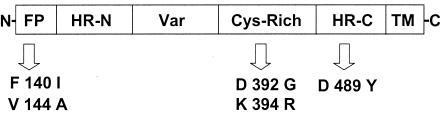
| Compound used to select for resistance peptide | F1 mutation | Resistance (fold)b |
|---|---|---|
| BMS-233675 | V144A | 150 |
| BMS-233675 | D392G | 35 |
| BMS-243458 | K394R | >1,250 |
| BMS-243458 | D489Y | >1,250 |
| BABIM | F140I | >1,250 |
| K394 transfectant | K394R | >400 |
The schematic above the table shows the F1 polypeptide containing the amino acid changes found in BMS-433771-resistant viruses. Various domains of the F1 polypeptide are indicated: N, amino terminus; FP, fusion peptide; HR-N, N-terminal heptad repeat; Cys-rich, cysteine-rich region; HR-C, C-terminal heptad repeat; TM, transmembrane domain; C, carboxy terminus.
The fold decrease in activity (resistance level) compared to that of the wild-type Long strain.
In order to further characterize these resistant viruses, the genes for the F, G, and SH proteins from independent drug-resistant viruses were amplified by a reverse transcription-PCR. Both amplified cDNA and independent clones from each resistant virus were sequenced. No amino acid changes were detected in any of the genes for the G protein or the SH protein of the resistant virus sequenced compared to the sequence of the wild-type Long strain. Sequencing of the F-protein genes from the resistant and wild-type Long viruses revealed that all resistant viruses contained single amino acid changes in the F1 subunit of the F protein compared to the sequence of our wild-type Long strain (Table 3). It should be noted that our wild-type Long F-protein gene contained two amino acid changes compared with the sequence of the Long F-protein gene entered in GenBank. One change was from Pro to Ser at amino acid 101, and the other was from Ala to Val at amino acid 442.
Among the resistant viruses, the Lys-to-Arg change at amino acid position 394 (K394R) was found within the cysteine-rich domain of the F1 subunit. The amino acid changes found in the F-protein genes of other resistant viruses included a F140I or V144A changes, which were located in the fusogenic peptide of the F1 protein, or a D489Y change, which was located in the C-terminal heptad repeat (Table 3). Although these resistant viruses were initially selected by using either the early inhibitor compounds BMS-233675 and BMS-243458 or the structurally related compound BABIM, they were all shown to possess resistance to BMS-433771, with increases in EC50s ranging from 35-fold to >1,250-fold compared to that for the wild-type Long strain (Table 3).
In additional studies conducted to prove that a single amino acid change in the F1 gene by itself can induce resistance, the K394R mutation was inserted into an infectious clone of the A2 strain of RSV (8). This was accomplished through mutagenesis of the infectious A2 clone generated by Collins et al. (8). The A2 K394R transfectant was rescued and analyzed for susceptibility to BMS-433771. The A2 K394R transfectant virus exhibits significant resistance to BMS-433771 (Table 3). Thus, these experiments provide strong evidence that a single amino acid change in the F1 subunit can impart resistance to BMS-433771, thereby confirming the importance of the F protein in the mechanism of action of the inhibitor.
Efficacy in mouse model of RSV infection.
The inbred BALB/c mouse host model of RSV infection was used to examine BMS-433771 for its antiviral efficacy in vivo. Cohorts of eight animals were used, and BMS-433771 was administered at a 50 mg/kg/dose twice a day (b.i.d.; total dose, 100 mg/kg) beginning at 1 h prior to virus infection. Infected animals were killed 4 days later, lung homogenates were prepared, and TCID50 titers were obtained. The infected control cohort, which was treated only with the dosing vehicle, exhibited titers ranging from ∼3.5 to 4.9 TCID50s (log10), with a median TCID50 of 3.99. In the BMS-433771-treated cohort, viral titers were below the detection limit in five of eight animals. In two other animals, TCID50s were just above the detection limit of 2.5, with a single animal exhibiting a TCID50 titer of 2.85 (Fig. 6). Thus, oral administration of BMS-433771 in this experiment was able to significantly reduce viral titers in the lungs of RSV-infected mice.
FIG. 6.

Oral activity of BMS-433771 in a mouse model of RSV infection. A 50-mg/kg/day BMS-433771 b.i.d. dose was administered to mice by oral gavage beginning 1 h prior to intranasal inoculation with 105 TCID50s of RSV. After 4 days, the mice were killed and TCID50 titers were determined by using lung homogenates. Each datum point represents the RSV titer for each animal in the respective treatment cohort. The horizontal line drawn for each cohort marks the geometric mean RSV titer for the group, and the viral titers are provided in parentheses. The horizontal dotted line represents the RSV titer at the limit of detection of the assay.
DISCUSSION
An orally bioavailable antiviral agent with activity against RSV could be useful for the treatment of RSV infections in a number of different human subpopulations. Herein we report on the in vitro properties of a new RSV fusion inhibitor, BMS-433771, and the initial finding of the in vivo activity of the compound in the mouse model of RSV infection following oral administration.
BMS-433771 is a highly selective and potent inhibitor of RSV replication in vitro and possesses activities against a broad range of laboratory and human clinical isolates of both the A and the B subgroups of RSV, with an average EC50 of 20.4 nM. BMS-433771 demonstrated good selectivity, showing no activity against a panel of other viruses when concentrations >1,000 times the EC50 for RSV were used. Mechanism-of-action studies demonstrated that BMS-433771 inhibits an early event in the life cycle of RSV. Inhibition is reversible and at a stage at which it remains susceptible to antibody neutralization, consistent with inhibition of RSV fusion. Furthermore, BMS-433771 can inhibit syncytium formation, another process mediated by the F protein. All of these results are consistent with the mechanism of action for BMS-433771 being the inhibition of fusion.
To aid in target identification, several analogs of BMS-433771 were used to generate viruses resistant to this class of fusion inhibitor (Fig. 1). Mutant viruses selected with these early analogs and BABIM were cross-resistant to the compounds (data not shown) as well as to BMS-433771 (Table 3). This suggests that these compounds share a common or overlapping site of action. Analyses of resistant viruses indicate that the molecular target of BMS-433771 is the F1 subunit of the RSV F protein. A rescued transfectant virus containing a single Lys-to-Arg change at amino acid 394 of the F protein exhibited resistance, proving that a single-residue substitution in the F1 subunit alone can impart resistance to BMS-433771 and further confirming the importance of the F protein in the mechanism of action.
Recent reports have described additional RSV fusion inhibitors (27, 41, 60; Andries et al., 40th ICAAC). However, BMS-433771 is the first compound shown to possess activity when it is administered orally in an animal model of RSV infection. BMS-433771 and R-170591 (Andries et al., 40th ICAAC) share some structural homology, while the other two compounds, VP-14637 (41) and RFI-647 (27), have no obvious structural similarity to any of the other compounds. Interestingly, all four RSV fusion inhibitors were identified by using cell culture screens. Cell culture screens for antiviral activity theoretically target multiple pathways throughout the virus replicative cycle. The fact that RSV fusion seems to be specifically targeted in this assay may suggest that fusion inhibitors are significantly easier to detect in this type of screen than inhibitors of other stages of the virus life cycle. One explanation may be that the F protein is more promiscuous than other viral proteins in its ability to bind to small molecules and that the fusion process, in which numerous interactions and conformational changes occur, provides multiple target sites that molecules can functionally inhibit. The variation in the structures of the multiple RSV fusion inhibitors tends to support this view. In addition, with entry inhibitors, compounds need not pass through cell membranes in order to demonstrate antiviral activity in a cell culture screen. It is interesting that reports of cell culture-based screens for influenza virus inhibitors also identified three distinct inhibitors of the hemagglutinin-catalyzed membrane fusion reaction (7, 37, 50, 59).
Single amino acid changes were found in the F-protein genes of all the resistant viruses. These viruses were generated after several passages in the presence of increasing concentrations of compound. All mutations were the result of a single nucleotide change, so it is possible that they were generated as early as the first passage. Interestingly, all of the amino acid changes in the resistant viruses occurred within the F1 subunit. The RSV F protein is translated as a single polypeptide that is enzymatically cleaved into two subunits (subunits F1 and F2) that remain connected via a disulfide bridge (9). By analogy with the influenza virus HA2 and HIV gp41 proteins, the RSV F1 subunit is responsible for the mediation of membrane fusion (3, 15, 30, 68). The F1 subunit contains the hydrophobic fusion peptide, which is buried within the molecule in its native state (57). After F is bound to a receptor, a conformational rearrangement in the F protein exposes the fusion peptide, which inserts into the opposing cell membrane and, through a series of steps that remain undefined, promotes fusion of the viral and host cell membranes (4, 68). The finding that all resistant viruses possess amino acid changes in the F1 polypeptide complements the mechanism-of-action studies that demonstrate that BMS-433771 inhibits RSV-induced membrane fusion. Additionally, since all of the amino acid changes in the resistant viruses mapped to the F1 subunit, it strongly suggests that the F1 polypeptide is the specific molecular target of BMS-433771.
The F1 subunit contains the amino-terminal fusion peptide that is believed to insert in the host cell membrane during fusion. The F1 subunit also contains the two heptad repeat regions, located at the N and C termini, that are hypothesized to associate in an antiparallel manner, bringing about apposition of viral and cellular membranes during fusion (35, 40, 68). A potential binding pocket for small inhibitor molecules has been described within a structure of the RSV fusion core obtained through cocrystallization of peptides representing the N-terminal and C-terminal heptad repeats (68). This pocket is present in the trimer of N-terminal repeats and is occupied by two phenylalanine residues (F483 and F488) from the C-terminal heptad repeat peptides. One of the resistant viruses selected in our study had an amino acid change (D489Y) near this region in the C-terminal heptad repeat. The RSV fusion inhibitors R-170591 and VP-14637 also produced resistant viruses with mutations in the same region (D486N and F488Y for R-170591 and VP-14637, respectively) of the C-terminal heptad repeat region of the F1 subunit (11; Andries et al., 40th ICAAC). Other mutations that resulted in resistance to BMS-433771 map to alternate areas of the F1 subunit, suggesting that changes throughout the F1 subunit can abrogate the inhibition by BMS-433771. Two distinct mutations (F140I and V144A) were found in the hydrophobic fusion peptide, which is the series of amino acids within the N terminus of the F1 subunit involved in host membrane insertion. Also, two additional BMS-433771-resistant viruses have amino acid changes that occur in the cysteine-rich domain (D392G or K394R) found between the heptad repeat regions. The precise function of this region is unknown at present (5). However, other fusion inhibitors induce mutations that map to this area. Resistance to R-170591 can result from an S398L change (Andries et al., 40th ICAAC), while VP-14637 can generate resistant virus with a T400A substitution (11). The similar substitution patterns in resistant viruses may suggest that these three inhibitors, even though they are quite different in structure, may share similar modes of binding to the F1 subunit. It would be of interest to examine these resistant viruses for cross-resistance to the different RSV fusion inhibitors.
The availability of the mouse model of RSV infection enables examination of potential inhibitors of RSV in vivo. However, the value of animal models of RSV infection as harbingers of human RSV infection remains questionable (18). Nevertheless, for a new chemical entity with inhibitory activity against RSV in vitro, the mouse model of infection can provide proof of principle for efficacy in vivo and spur interest in further clinical development. In mice, BMS-433771 administered orally at 50 mg/kg b.i.d. beginning 1 h prior to infection significantly reduced the titers of RSV in the lungs of mice. This is the first report of a small-molecule RSV fusion inhibitor with activity in an animal model following oral administration. BMS-433771 has also been shown to have inhibitory activity in the cotton rat model of RSV infection following oral administration (C. Cianci et al., submitted). As for the other RSV inhibitors, BABIM was reported to have antiviral activity in cotton rats after intraperitoneal administration (63), and R-170591 demonstrated efficacy in vivo after aerosol delivery in rodent models of RSV infection (Andries et al., 40th ICAAC). The efficacy of RFI-641 was shown in three models of RSV infection, but only when it was administered via the intranasal or aerosol route (27, 66).
A clear demonstration of the clinical significance of inhibition of the RSV F-protein function has been established by using Synagis, the humanized monoclonal antibody directed against the F protein (23, 29, 53). BMS-433771 targets the same RSV protein and viral function. Biochemical studies demonstrated that BMS-433771 is a specific inhibitor of membrane fusion induced by the viral F protein. Inhibition of membrane fusion is a mechanism of antiviral activity that is being explored for a number of different viruses, including HIV, with the first fusion inhibitor of HIV recently being approved by the Food and Drug Administration (31, 32, 34). Accordingly, BMS-433771 is a novel small-molecule antiviral agent that is suitable for clinical evaluation due to its mechanism of action, in vitro potency, selectivity, and, most significantly, oral efficacy in vivo.
REFERENCES
- 1.Ausubel, F. M., R. Brent, R. E. Kingston, D. D. Moore, J. G. Seidman, and J. A. Smith. 1989. Current protocols in molecular biology. John Wiley & Sons, Inc., New York, N.Y.
- 2.Byrd, L. G., and G. A. Prince. 1997. Animal models of respiratory syncytial virus infection. Clin. Infect. Dis. 25:1363-1368. [DOI] [PubMed] [Google Scholar]
- 3.Calder, L. J., L. Gonzalez-Reyes, B. Garcia-Barreno, S. A. Wharton, J. J. Skehel, D. C. Wiley, and J. A. Melero. 2000. Electron microscopy of the human respiratory syncytial virus fusion protein and complexes that it forms with monoclonal antibodies. Virology 271:122-131. [DOI] [PubMed] [Google Scholar]
- 4.Carr, C. M., and P. S. Kim. 1993. A spring-loaded mechanism for the conformational change of influenza hemagglutinin. Cell 73:823-832. [DOI] [PubMed] [Google Scholar]
- 5.Chambers, P., C. R. Pringle, and A. J. Easton. 1992. Sequence analysis of the gene encoding the fusion glycoprotein of pneumonia virus of mice suggests possible conserved secondary structure elements in paramyxovirus fusion glycoproteins. J. Gen. Virol. 73:1717-1724. [DOI] [PubMed] [Google Scholar]
- 6.Chanock, R. M., and R. H. Parrott. 1965. Acute respiratory disease in infancy and childhood: present understanding and prospects for prevention. Pediatrics 36:21-39. [PubMed] [Google Scholar]
- 7.Cianci, C., K. L. Yu, D. D. Dischino, W. Harte, M. Deshpande, G. Luo, R. J. Colonno, N. A. Meanwell, and M. Krystal. 1999. pH-dependent changes in photoaffinity labeling patterns of the H1 influenza virus hemagglutinin by using an inhibitor of viral fusion. J. Virol. 73:1785-1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Collins, P. L., M. G. Hill, E. Camargo, H. Grosfeld, R. M. Chanock, and B. R. Murphy. 1995. Production of infectious human respiratory syncytial virus from cloned cDNA confirms an essential role for the transcription elongation factor from the 5′ proximal open reading frame of the M2 mRNA in gene expression and provides a capability for vaccine development. Proc. Natl. Acad. Sci. USA 92:11563-11567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Collins, P. L., K. McIntosh, and R. M. Chanock. 2001. Respiratory syncytial virus, p. 1443-1486. In D. M. Knipe and P. M. Howley (ed.), Virology, 3rd ed. Raven Press, New York, N.Y.
- 10.De Clercq, E. 2001. Antiviral drugs: current state of the art. J. Clin. Virol. 22:73-89. [DOI] [PubMed] [Google Scholar]
- 11.Douglas, J. L., M. L. Panis, E. Ho, K. Y. Lin, S. H. Krawczyk, D. M. Grant, R. Cai, S. Swaminathan, and T. Cihlar. 2003. Inhibition of respiratory syncytial virus fusion by the small molecule VP-14637 via specific interactions with F protein. J. Virol. 77:5054-5064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dowell, S. F., L. J. Anderson, H. E. J. Gary, D. D. Erdman, J. F. Plouffe, T. M. J. File, B. J. Marston, and R. F. Breiman. 1996. Respiratory syncytial virus is an important cause of community-acquired lower respiratory infection among hospitalized adults. J. Infect. Dis. 174:456-462. [DOI] [PubMed] [Google Scholar]
- 13.DuBovi, E. J., J. D. Geratz, S. R. Shaver, and R. R. Tidwell. 1981. Inhibition of respiratory syncytial virus-host cell interactions by mono- and diamidines. Antimicrob. Agents Chemother. 19:649-656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.DuBovi, E. J., J. D. Geratz, and R. R. Tidwell. 1980. Inhibition of respiratory syncytial virus by bis(5-amidino-2-benzimidazolyl)methane. Virology 103:502-504. [DOI] [PubMed] [Google Scholar]
- 15.Dutch, R. E., T. S. Jardetzky, and R. A. Lamb. 2000. Virus membrane fusion proteins: biological machines that undergo a metamorphosis. Biosci. Rep. 20:597-612. [DOI] [PubMed] [Google Scholar]
- 16.Englund, J. A., C. J. Sullivan, M. C. Jordan, L. P. Dehner, G. M. Vercellotti, and H. H. Balfour. 1988. Respiratory syncytial virus infection in immunocompromised adults. Ann. Intern. Med. 109:203-208. [DOI] [PubMed] [Google Scholar]
- 17.Falsey, A. R., C. K. Cunningham, W. H. Barker, R. W. Kouides, J. B. Yuen, M. Menegus, L. B. Weiner, C. A. Bonville, and R. F. Betts. 1995. Respiratory syncytial virus and influenza A infections in the hospitalized elderly. J. Infect. Dis. 172:389-394. [DOI] [PubMed] [Google Scholar]
- 18.Falsey, A. R., J. J. Treanor, R. F. Betts, and E. E. Walsh. 1992. Viral respiratory infections in the institutionalized elderly: clinical and epidemiologic findings. J. Am. Geriatr. Soc. 40:115-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Falsey, A. R., and E. E. Walsh. 2000. Respiratory syncytial virus infection in adults. Clin. Microbiol. Rev. 13:371-384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Garcia, R., I. Raad, D. Abi-Said, G. Bodey, R. Champlin, J. Tarrand, L. A. Hill, J. Umphrey, J. Neumann, J. Englund, and E. Whimbey. 1997. Nosocomial respiratory syncytial virus infections: prevention and control in bone marrow transplant patients. Infect. Control Hosp. Epidemiol. 18:412-416. [DOI] [PubMed] [Google Scholar]
- 21.Glezen, W. P., L. H. Taber, A. L. Frank, and J. A. Kasel. 1986. Risk of primary infection and reinfection with respiratory syncytial virus. Am. J. Dis. Child. 140:543-546. [DOI] [PubMed] [Google Scholar]
- 22.Greenough, A. 2001. Recent advances in the management and prophylaxis of respiratory syncytial virus infection. Acta Paediatr. Suppl. 90:11-14. [DOI] [PubMed] [Google Scholar]
- 23.Greenough, A., and M. Thomas. 2000. Respiratory syncytial virus prevention: past and present strategies. Expert Opin. Pharmacother. 1:1195-1201. [DOI] [PubMed] [Google Scholar]
- 24.Hacking, D., and J. Hull. 2002. Respiratory syncytial virus-viral biology and the host response. J. Infect. 45:18-24. [DOI] [PubMed] [Google Scholar]
- 25.Heikkinen, T., M. Thint, and T. Chonmaitree. 1999. Prevalence of various respiratory viruses in the middle ear during acute otitis media. N. Engl. J. Med. 340:260-264. [DOI] [PubMed] [Google Scholar]
- 26.Holberg, C. J., A. L. Wright, F. D. Martinez, C. G. Ray, L. M. Taussig, and M. D. Lebowitz. 1991. Risk factors for respiratory syncytial virus-associated lower respiratory illnesses in the first year of life. Am. J. Epidemiol. 133:1135-1151. [DOI] [PubMed] [Google Scholar]
- 27.Huntley, C. C., W. J. Weiss, A. Gazumyan, A. Buklan, B. Feld, W. Hu, T. R. Jones, T. Murphy, A. A. Nikitenko, B. O'Hara, G. Prince, S. Quartuccio, Y. E. Raifeld, P. Wyde, and J. F. O'Connell. 2002. RFI-641, a potent respiratory syncytial virus inhibitor. Antimicrob. Agents Chemother. 46:841-847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ikeda, S., J. Neyts, S. Verma, A. Wickramasinghe, P. Mohan, and E. De Clercq. 1994. In vitro and in vivo inhibition of ortho- and paramyxovirus infections by a new class of sulfonic acid polymers interacting with virus-cell binding and/or fusion. Antimicrob. Agents Chemother. 38:256-259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Johnson, S., C. Oliver, G. A. Prince, V. G. Hemming, D. S. Pfarr, S.-C. Wang, M. Dormitzer, J. O'Grady, S. Koenig, J. K. Tamura, R. Woods, G. Bansal, D. Couchenour, E. Tsao, W. C. Hall, and J. F. Young. 1997. Development of a humanized monoclonal antibody (MEDI-493) with potent in vitro and in vivo activity against respiratory syncytial virus. J. Infect. Dis. 176:1215-1224. [DOI] [PubMed] [Google Scholar]
- 30.Kahn, J. S., M. J. Schnell, L. Buonocore, and J. K. Rose. 1999. Recombinant vesicular stomatitis virus expressing respiratory syncytial virus (RSV) glycoproteins: RSV fusion protein can mediate infection and cell fusion. Virology 254:81-91. [DOI] [PubMed] [Google Scholar]
- 31.Kilby, J. M., S. Hopkins, T. M. Venetta, B. DiMassimo, G. A. Cloud, J. Y. Lee, L. Alldredge, E. Hunter, D. Lambert, D. Bolognesi, T. Matthews, M. R. Johnson, M. A. Nowak, G. M. Shaw, and M. S. Saag. 1998. Potent suppression of HIV-1 replication in humans by T-20, a peptide inhibitor of gp41-mediated virus entry. Nat. Med. 4:1302-1307. [DOI] [PubMed] [Google Scholar]
- 32.Kilby, J. M., J. P. Lalezari, J. J. Eron, M. Carlson, C. Cohen, R. C. Arduino, J. C. Goodgame, J. E. Gallant, P. Volberding, R. L. Murphy, F. Valentine, M. S. Saag, E. L. Nelson, P. R. Sista, and A. Dusek. 2003. The safety, plasma pharmacokinetics, and antiviral activity of subcutaneous enfuvirtide (T-20), a peptide inhibitor of gp41-mediated virus fusion, in HIV-infected adults. AIDS Res. Hum. Retrovir. 19:685-693. [DOI] [PubMed] [Google Scholar]
- 33.Kimura, K., S. Mori, K. Tomita, K. Ohno, K. Takahashi, S. Shigeta, and M. Terada. 2000. Antiviral activity of NMSO3 against respiratory syncytial virus infection in vitro and in vivo. Antivir. Res. 47:41-51. [DOI] [PubMed] [Google Scholar]
- 34.Lalezari, J. P., K. Henry, M. O'Hearn, J. S. Montaner, P. J. Piliero, B. Trottier, S. Walmsley, C. Cohen, D. R. Kuritzkes, J. J. Eron, Jr., J. Chung, R. DeMasi, L. Donatacci, C. Drobnes, J. Delehanty, and M. Salgo. 2003. Enfuvirtide, an HIV-1 fusion inhibitor, for drug-tesistant HIV infection in North and South America. N. Engl. J. Med. 348:2175-2185. [DOI] [PubMed] [Google Scholar]
- 35.Lawless-Delmedico, M. K., P. Sista, R. Sen, N. C. Moore, J. B. Antczak, J. M. White, R. J. Greene, K. C. Leanza, T. J. Matthews, and D. M. Lambert. 2000. Heptad-repeat regions of respiratory syncytial virus F1 protein form a six-membered coiled-coil complex. Biochemistry 39:11684-11695. [DOI] [PubMed] [Google Scholar]
- 36.Luo, G., R. Colonno, and M. Krystal. 1996. Characterization of a hemagglutinin-specific inhibitor of influenza A virus. Virology 226:66-76. [DOI] [PubMed] [Google Scholar]
- 37.Luo, G. X., A. Torri, W. E. Harte, S. Danetz, C. Cianci, L. Tiley, S. Day, D. Mullaney, K. L. Yu, C. Ouellet, P. Dextraze, N. Meanwell, R. Colonno, and M. Krystal. 1997. Molecular mechanism underlying the action of a novel fusion inhibitor of influenza A virus. J. Virol. 71:4062-4070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Martin, M. A., M. J. Bock, M. A. Pfaller, and R. P. Wenzel. 1988. Respiratory syncytial virus infections in adult bone marrow transplant recipients. Lancet i:1396-1397. [DOI] [PubMed] [Google Scholar]
- 39.Mathur, U., D. W. Bentley, and C. B. Hall. 1980. Concurrent respiratory syncytial virus and influenza A infections in the institutionalized elderly and chronically ill. Ann. Intern. Med. 93:49-52. [DOI] [PubMed] [Google Scholar]
- 40.Matthews, J. M., T. F. Young, S. P. Tucker, and J. P. Mackay. 2000. The core of the respiratory syncytial virus fusion protein is a trimeric coiled coil. J. Virol. 74:5911-5920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McKimm-Breschkin, J. 2000. VP-14637 ViroPharma. Curr. Opin. Investig. Drugs 1:425-427. [PubMed] [Google Scholar]
- 42.Mills, J. 1996. Management of respiratory syncytial virus infections. Plenum Press, New York, N.Y. [DOI] [PubMed]
- 43.Moyse, E., M. Lyon, G. Cordier, J. F. Mornex, L. Collet, and P. Froehlich. 2000. Viral RNA in middle ear mucosa and exudates in patients with chronic otitis media with effusion. Arch. Otolaryngol. Head Neck Surg. 126:1105-1110. [DOI] [PubMed] [Google Scholar]
- 44.Murry, A. R., and S. F. Dowell. 1997. Respiratory syncytial virus: not just for kids. Hosp. Pract. 15:87-88. [DOI] [PubMed] [Google Scholar]
- 45.Osiowy, C., and R. Anderson. 1995. Neutralization of respiratory syncytial virus after cell attachment. J. Virol. 69:1271-1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Osterweil, D., and D. Norman. 1990. An outbreak of an influenza-like illness in a nursing home. J. Am. Geriatr. Soc. 38:659-662. [DOI] [PubMed] [Google Scholar]
- 47.Pagani, F., and F. Sparatore. 1965. Benzotriazolyl-alkyl-benzimidazoles and their dialkyl-aminoalkyl derivatives. Boll. Chim. Farm. 104:427-431. [PubMed] [Google Scholar]
- 48.Paglietti, G., V. Boido, and F. Sparatore. 1975. Dialkylaminoalkylbenzimidazoles of pharmacological importance. IV. Il Farmaco 30:505-511. [PubMed] [Google Scholar]
- 49.Pauwels, R., J. Balzarini, M. Baba, R. Snoeck, D. Schols, P. Herdewijn, J. Desmyter, and E. De Clercq. 1988. Rapid and automated tetrazolium-based colorimetric assay for the detection of anti-HIV compounds. J. Virol. Methods 20:309-321. [DOI] [PubMed] [Google Scholar]
- 50.Plotch, S. J., B. O'Hara, J. Morin, O. Palant, J. LaRocque, J. D. Bloom, S. A. Lang, Jr., M. J. DiGrandi, M. Bradley, R. Nilakantan, and Y. Gluzman. 1999. Inhibition of influenza A virus replication by compounds interfering with the fusogenic function of the viral hemagglutinin. J. Virol. 73:140-151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Popovic, M., M. G. Sarngadharan, E. Read, and R. C. Gallo. 1984. Detection, isolation, and continuous production of cytopathic retroviruses (HTLV-III) from patients with AIDS and pre-AIDS. Science 224:497-500. [DOI] [PubMed] [Google Scholar]
- 52.Prince, G. A. 2001. An update on respiratory syncytial virus antiviral agents. Expert Opin. Investig. Drugs 10:297-308. [DOI] [PubMed] [Google Scholar]
- 53.Schmidt, A. C., R. B. Couch, G. J. Galasso, F. G. Hayden, J. Mills, B. R. Murphy, and R. M. Chanock. 2001. Current research on respiratory viral infections: Third International Symposium. Antivir. Res. 50:157-196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sigurs, N. 2001. Epidemiologic and clinical evidence of a respiratory syncytial virus-reactive airway disease link. Am. J. Respir. Crit. Care Med. 163:S2-S6. [DOI] [PubMed] [Google Scholar]
- 55.Sigurs, N., R. Bjarnason, F. Sigurbergsson, B. Kjellman, and B. Bjorksten. 1995. Asthma and immunoglobulin E antibodies after respiratory syncytial virus bronchiolitis: a prospective cohort study with matched controls. Pediatrics 95:500-505. [PubMed] [Google Scholar]
- 56.Simoes, E. A. 2001. Treatment and prevention of respiratory syncytial virus lower respiratory tract infection. Long-term effects on respiratory outcomes. Am. J. Respir. Crit. Care Med. 163:S14-S17. [DOI] [PubMed] [Google Scholar]
- 57.Smith, B. J., M. C. Lawrence, and P. M. Colman. 2002. Modeling the structure of the fusion protein from human respiratory syncytial virus. Protein Eng. 15:365-371. [DOI] [PubMed] [Google Scholar]
- 58.Sorvillo, F. J., S. F. Huie, M. A. Strassburg, A. Butsumyo, W. X. Shandera, and S. L. Fannin. 1984. An outbreak of respiratory syncytial virus pneumonia in a nursing home for the elderly. J. Infect. 9:252-256. [DOI] [PubMed] [Google Scholar]
- 59.Staschke, K. A., S. D. Hatch, J. C. Tang, W. J. Hornback, J. E. Munroe, J. M. Colacino, and M. A. Muesing. 1998. Inhibition of influenza virus hemagglutinin-mediated membrane fusion by a compound related to podocarpic acid. Virology 248:264-274. [DOI] [PubMed] [Google Scholar]
- 60.Sudo, K., K. Konno, W. Watanabe, S. Shigeta, and T. Yokota. 2001. Mechanism of selective inhibition of respiratory syncytial virus by a benzodithiin compound (RD3-0028). Microbiol. Immunol. 45:531-537. [DOI] [PubMed] [Google Scholar]
- 61.Taylor, G., E. J. Stott, M. Hughes, and A. P. Collins. 1984. Respiratory syncytial virus infection in mice. Infect. Immun. 43:649-655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Thompson, W. W., D. K. Shay, E. Weintraub, L. Brammer, N. Cox, L. J. Anderson, and K. Fukuda. 2003. Mortality associated with influenza and respiratory syncytial virus in the United States. JAMA 289:179-186. [DOI] [PubMed] [Google Scholar]
- 63.Tidwell, R. R., J. D. Geratz, W. A. Clyde, K. U. Rosenthal, and E. J. Dubovi. 1984. Suppression of respiratory syncytial virus infection in cotton rats by bis(5-amidino-2-benzimidazolyl)methane. Antimicrob. Agents Chemother. 26:591-593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tidwell, R. R., J. D. Geratz, and E. J. Dubovi. 1983. Aromatic amidines: comparison of their ability to block respiratory syncytial virus induced cell fusion and to inhibit plasmin, urokinase, thrombin, and trypsin. J. Med. Chem. 26:294-298. [DOI] [PubMed] [Google Scholar]
- 65.Treanor, J., and A. Falsey. 1999. Respiratory viral infections in the elderly. Antivir. Res. 44:79-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Weiss, W. J., T. Murphy, M. E. Lynch, J. Frye, A. Buklan, B. Gray, E. Lenoy, S. Mitelman, J. O'Connell, S. Quartuccio, and C. Huntley. 2003. Inhalation efficacy of RFI-641 in an African green monkey model of RSV infection. J. Med. Primatol. 32:82-88. [DOI] [PubMed] [Google Scholar]
- 67.Yu, K. L., Y. Zhang, R. L. Civiello, K. Kadow, C. Cianci, M. Krystal, and N. A. Meanwell. 2003. Fundamental structure-activity relationships associated with a new structural class of respiratory syncytial virus inhibitor. Bioorg. Med. Chem. Lett. 13:2141-2144. [DOI] [PubMed] [Google Scholar]
- 68.Zhao, X., M. Singh, V. N. Malashkevich, and P. S. Kim. 2000. Structural characterization of the human respiratory syncytial virus fusion protein core. Proc. Natl. Acad. Sci. USA 97:14172-14177. [DOI] [PMC free article] [PubMed] [Google Scholar]