

Abstract
TRF1 is a key component of the telomere-capping complex and binds double-strand telomeric DNA as homodimers. So far, it is not clear whether TRF1 dimerization coincides with its telomere binding or is actively controlled before it binds the telomere, and in the latter case, how this event might affect its telomere association. We previously found that TRF1 dimerization and its telomere binding can be increased by GNL3L, which is the vertebrate paralogue of nucleostemin (NS). Here, we show that NS and GNL3L bind TRF1 directly but competitively through two separate domains of TRF1. In contrast to GNL3L, NS prevents TRF1 dimerization through a mechanism not determined by its ability to displace TRF1-bound GNL3L. Furthermore, NS is capable of shortening the dynamic association of TRF1 with the telomere in normal and TRF2ΔBΔM-induced telomere-damaged cells without affecting the amount of telomere-bound TRF1 proteins in vivo. Importantly, NS displays a protective function against the formation of telomere-dysfunction-induced foci. This work demonstrates that TRF1 dimerization is actively and oppositely regulated by NS and GNL3L extrachromosomally. Changing the relative amount of TRF1 monomers versus dimers in the nucleoplasm might affect the dynamic association of TRF1 with the telomere and the repair of damaged telomeres.
Key words: Dimerization, FLIP, GNL3L, Nucleostemin, Telomere damage, TRF1
Introduction
Telomeric repeat-binding factor 1 (TRF1) is a component of the telomere-capping complex, also known as shelterin or telosome, which consists of six core proteins: TRF1, TRF2, TIN2 (TRF1-interacting nuclear protein 2), POT1 (protection of telomeres protein 1), TPP1 (also known as adrenocortical dysplasia homolog, ACD) and RAP1 (de Lange, 2005; Songyang and Liu, 2006). The telomere-capping complex organizes chromosomal ends into a high-order structure and has a crucial role in maintaining their stability. TRF1 serves the important functions of protecting telomeres (Martinez et al., 2009), preventing telomere elongation (van Steensel and de Lange, 1997), controlling the mitotic transit (Zhou et al., 2003) and regulating ALT (alternative lengthening of telomere)-associated PML body (APB) formation in telomerase-inactive cells (Jiang et al., 2007; Potts and Yu, 2005). TRF1 protein is post-translationally modified by phosphorylation, ADP ribosylation, ubiquitylation, and homodimerization (Chang et al., 2003; Chong et al., 1995; Kim et al., 2008; Lee et al., 2006; Smith et al., 1998; Wu et al., 2008). Dimerization allows TRF1 to form proper configuration and bind double-strand telomeric DNA repeats through its Myb domain (Bianchi et al., 1997; Broccoli et al., 1997). Even though TRF1 dimerization was observed both in a complex with DNA and by a yeast two-hybrid assay (Bianchi et al., 1997), it is not clear whether this event is actively regulated while TRF1 is in the nucleoplasm or occurs concomitantly with the formation of the telomere-capping complex.
We first discovered that nucleostemin (NS) is a TRF1-binding protein that accelerates the degradation of TRF1 (Zhu et al., 2006). NS belongs to a novel family of nucleolar GTP-binding proteins (Tsai and Meng, 2009), and is found preferentially expressed by multiple stem cell types, human cancers and adult regenerating tissues (Baddoo et al., 2003; Maki et al., 2007; Ohmura et al., 2008; Siddiqi et al., 2008; Tsai and McKay, 2002). It is required for early embryogenesis and the maintenance of continuous cell proliferation (Tsai and Meng, 2009; Zhu et al., 2006). In later studies, we found that its vertebrate paralogue, GNL3L (guanine nucleotide binding protein-like 3-like), can also bind TRF1 and promote its homodimerization and telomere association without being physically bound to the telomere (Zhu et al., 2009). In contrast to the NS effect on TRF1, GNL3L shows the ability to promote TRF1 stability by preventing its ubiquitylation and degradation. Those previous findings indicate that the TRF1 protein complex might be modulated actively and dynamically before it binds the telomere (Tsai, 2009).
To further our understanding of this new ‘pre-telomeric’ regulation of TRF1, we set out to investigate how NS controls the biochemical interaction and telomeric association of TRF1. Our results showed that NS and GNL3L both bind TRF1 directly but competitively. Whereas GNL3L promotes the dimerization and telomeric association of TRF1 (Zhu et al., 2009), NS diminishes TRF1 dimerization independently of its ability to dissociate GNL3L from TRF1. Functional studies demonstrated that NS is capable of shortening the dynamic association of TRF1 with the telomere in normal and TRF2ΔBΔM-induced telomere-damaged cells without affecting the amount of telomere-bound TRF1 proteins in vivo. TRF2ΔBΔM is a TRF2 mutant deleted of its N-terminal basic domain and C-terminal Myb domain. TRF2ΔBΔM lacks telomere-binding activity, thus acting as a dominant-negative allele of TRF2, and has been used to destabilize the telomere-capping complex and trigger telomere damage (van Steensel et al., 1998). Importantly, we found that NS has a protective role against TRF2ΔBΔM-induced telomere damage. This work reveals a novel TRF1 regulatory mechanism that operates bidirectionally and extrachromosomally.
Results
NS and GNL3L bind directly to separate domains of TRF1
We have previously shown that TRF1 coexists in the same protein complex with NS or with GNL3L (Zhu et al., 2009; Zhu et al., 2006). To demonstrate that NS or GNL3L directly binds TRF1, we isolated bacterially expressed NS and GNL3L proteins and examined their binding with purified GST–TRF1 proteins by affinity pull-down assays. Immunoblotting of the input, unbound supernatant (Sup) and retained fractions (Ret) confirmed that both NS and GNL3L can specifically and directly bind TRF1 (Fig. 1A). The amounts of NS in the unbound (Sup) and bound (Ret) fractions relative to the input are less than that of GNL3L, which might be caused by the rapid degradation of purified NS proteins. The TRF1-interactive domain of NS was mapped by deletions made on the basic (B), coiled-coil (C), GTP-binding (G), intermediate (I) or A-domain of NS (Fig. 1B). Unlike the TRF1-interacting G-domain of GNL3L (Zhu et al., 2009), binding between TRF1 and NS requires the I-domain of NS. Conversely, the NS-interactive domain of TRF1 was determined by testing the NS binding of TRF1 mutants deleted of the A-domain, HBD (homodimerization domain), UD (undefined) or Myb domain (Fig. 1C). Whereas GNL3L interacts with the HBD domain of TRF1 (Zhu et al., 2009), binding between TRF1 and NS requires the UD domain of TRF1. The lack of binding capability of NSΔI or TRF1ΔUD should not be caused by protein misfolding because these proteins are capable of binding MDM2 and GNL3L, respectively (Meng et al., 2008; Zhu et al., 2009). To exclude another possibility that the differential NS-binding activities of TRF1 mutants might result from their distinct subcellular localization, confocal studies were conducted in HeLa cells and showed that both the NS binding (TRF1ΔMyb) and non-binding (TRF1ΔUD) mutants are located outside the nucleolus (Fig. 1D). Similar findings were observed in U2OS cells (data not shown). These results demonstrate that NS and GNL3L bind directly to separate domains of TRF1.
Fig. 1.

NS and GNL3L bind directly to two separate domains of TRF1. (A) Purified NS and GNL3L proteins (40 μg) were incubated with purified Sepharose-bound GST–TRF1 protein (4 μg). Fractions of the input (1/100th), unbound supernatant (Sup, 1/100th), and retained lysates (Ret, 1/10th–1/20th) were immunoblotted (IB) with anti-NS and anti-GNL3L (G3L) antibodies. The pull-down results confirmed that NS and GNL3L bind TRF1 directly. (B) GST pull-down assays showing that the I-domain of NS is required for its TRF1 interaction. (C) Conversely, the UD domain of TRF1 is required for its interaction with NS. Bottom panels in B and C depict the deletion mutants of NS and TRF1. Bent lines and numbers denote the deleted regions and amino acid positions, respectively. B, basic; C, coiled-coil; G, GTP-binding; I, intermediate; A, acidic; HBD, homodimerization; UD, undefined domain. (D) Confocal images of GFP-fused wild-type and mutant TRF1 proteins in HeLa cells. Nucleoli are labeled by anti-B23 staining. Scale bar: 20 μm.
NS negatively regulates the GNL3L binding and homodimerization of TRF1
To interrogate the TRF1-binding relationship between NS and GNL3L, we asked whether NS and GNL3L form hetero-trimeric complexes with TRF1 or bind TRF1 as separate hetero-dimeric complexes. To address this issue, we first examined the coimmunoprecipitation of endogenous NS, TRF1 and GNL3L, and found only TRF1 in the NS protein complex (Fig. 2A). Next, we tested whether these three proteins can form hetero-trimeric complexes. NS (Myc), GNL3L (HA) and TRF1 (FLAG) were coexpressed in HEK293 cells, and the TRF1 and GNL3L co-complex was purified by sequential anti-FLAG and anti-HA immunoprecipitation. Anti-Myc western blots showed that the hetero-trimeric complex does exist, but accounts for only a small fraction of the hetero-dimeric complex of GNL3L and TRF1 even upon overexpression (Fig. 2B). These results indicate that the majority of NS and GNL3L bind TRF1 separately, but a small portion might form a heterotrimeric complex with TRF1. To examine the possibility that NS and GNL3L compete against each other for TRF1 binding, GST-fused TRF1 proteins were purified to pull down lysates containing a fixed amount of Myc-tagged GNL3L and increasing amounts of HA-tagged NS (Fig. 2C, left). The total protein concentrations were adjusted to the same levels in all samples. The results showed that NS is capable of blocking GNL3L binding to TRF1 in a dose-dependent manner. Reciprocally, GNL3L can also compete against NS for TRF1 binding (Fig. 2C, right). One major TRF1-binding protein in the telomere-capping complex is TIN2. GST-binding competition experiments showed that NS and TIN2 do not interfere with each other's binding to TRF1 (Fig. 2D). TRF1 is also known to form homodimers. To investigate the NS role in regulating TRF1 dimerization, GST–TRF1 was used to pull down lysates with the same amount of Myc-tagged TRF1 and increasing amounts of NS. In contrast to the effect of GNL3L on TRF1 dimerization (Zhu et al., 2009), addition of NS reduced the association between the Myc-tagged TRF1 and GST–TRF1 in a dose-dependent manner (Fig. 2E). Together, these results demonstrate that NS prevents TRF1 from binding to GNL3L and to itself, but does not interfere with the interaction of TRF1 with its partner at the telomere, TIN2.
Fig. 2.

NS inhibits GNL3L binding and homodimerization of TRF1. (A) Immunoprecipitation (IP) of endogenous NS complex copurified TRF1 but not GNL3L. (B) Two-step coimmunoprecipitation (coIP) was used to determine whether NS (Myc-tagged), GNL3L (HA-tagged) and TRF1 (FLAG-tagged) are capable of forming hetero-trimeric complexes. The TRF1 complex was precipitated using anti-FLAG antibody and eluted using FLAG peptide. Then the TRF1–GNL3L co-complex was purified by anti-HA immunoprecipitation. The amount of the hetero-trimeric complex (detected by immunoblotting the TRF1–GNL3L co-complex with anti-Myc antibody) represents a small fraction of the TRF1–GNL3L co-complex even upon overexpression. (C) Agarose-bound GST–TRF1 was incubated with lysates containing a fixed amount of GNL3L (Myc) and increasing amounts of NS (HA) or vice versa. Increasing the amount of NS significantly reduces the signal of TRF1-bound GNL3L (left). Reciprocally, increasing the amount of GNL3L also decreases NS binding to GST–TRF1 (right). (D) GST pull-down competition experiments show that NS (HA) does not affect the binding between TIN2 (Myc) and GST–TRF1 (left); nor does TIN2 (Myc) change the binding between NS (HA) and GST–TRF1 (right). (E) Increasing the amount of NS significantly decreases the binding efficiency between TRF1 (Myc) and GST–TRF1 in a dose-dependent manner.
NS structures required for its inhibition of GNL3L binding and dimerization of TRF1
NS inhibition of TRF1 homodimerization might be mediated by its role in competing against GNL3L for TRF1 binding, because GNL3L is known to promote TRF1 homodimerization (Zhu et al., 2009). If true, its abilities to block TRF1 dimerization and GNL3L binding should correlate. To test this idea, we examined the NS protein structures required to inhibit the GNL3L binding or dimerization of TRF1. Our results showed that the ability of NS to displace TRF1-bound GNL3L was mostly abolished by deleting its TRF1-interactive I-domain (Fig. 3A), but not affected by the deletion of the B-, G- or A-domain of NS, or by the G256V mutation that abolishes its GTP-binding capability (Fig. 3B–E). Reciprocally, the ability of GNL3L to displace TRF1-bound NS only required its TRF1-interactive G-domain (Fig. 3G), and was not decreased by the deletion of its BC- or I-domain (Fig. 3H,I) or the G253V mutation that corresponds to the G256V mutation of NS (Fig. 3J).
Fig. 3.

Identification of NS domain required for its ability to inhibit GNL3L binding to TRF1 and vice versa. Affinity binding between GST–TRF1 and GNL3L (Myc) was conducted in the presence of different NS deletion mutants. The NS ability to block GNL3L binding to TRF1 requires its TRF1-interacting I-domain (A), but is unaffected by any other deletions (B–D) or the G256V mutation (E). (F) GNL3L mutants used to map the domain(s) required for its competition with NS for TRF1 binding. The ability of GNL3L to block NS binding to TRF1 requires only its TRF1-interacting G-domain (G) and does not depend on its BC-domain (H), I-domain (I) or GTP binding (J).
With respect to the NS effect on blocking TRF1 homodimerization, our data showed that this NS activity was completely abolished by deleting its I-domain (NSΔI, Fig. 4A), B-domain (NSΔB, Fig. 4B) or G-domain (NSΔG, Fig. 4C), and partially blocked by the non-GTP-binding mutation, G256V (Fig. 4D). This anti-TRF1 dimerization effect of NS was unaffected by the A-domain deletion (NSΔA, Fig. 4E). Given that the NSΔB and NSΔG mutants can both displace TRF1-bound GNL3L, but cannot block TRF1 homodimerization, we conclude that the anti-TRF1 dimerization effect of NS is not determined by its ability to compete against GNL3L for TRF1 binding.
Fig. 4.

The anti-TRF1 dimerization effect of NS requires its basic and GTP-binding domains, in addition to its TRF1-binding I-domain. Agarose-bound GST–TRF1 was incubated with lysates containing a fixed amount of TRF1 (Myc) and increasing amounts of NS deletion mutants (HA). The NS ability to block TRF1 dimerization is completely abolished by deletions of its I-domain (A), B-domain (B) or G-domain (C), partially blocked by the GTP-binding mutant (G256V, D), but is completely unaffected by the A-domain deletion of NS (E). Agarose-bound (R) and supernatant fractions (S) are indicated.
NS decreases TRF1 binding to telomeric DNA in vitro
Because TRF1 binds telomeric DNA as homodimers, we speculate that NS might regulate the telomeric association of TRF1. To test whether NS has such a function, we first used the electrophoretic mobility shift assay (EMSA) to determine the direct effect of NS on TRF1 binding to double-stranded (TTAGGG)6 probes. Compared with the probe alone (Fig. 5A1, lane 1) and vector-transfected sample (Fig. 5A1, lane 2), the Myc-tagged TRF1-transfected sample (Fig. 5A1, lane 3) yielded a specific TRF1–DNA complex (black arrow) that could be competed by excess unlabeled probes (Fig. 5A1, lane 4) and supershifted by anti-Myc antibody (Fig. 5A1, grey arrow in lane 5). NS by itself did not bind the (TTAGGG)6 probe (Fig. 5A1, lanes 7 and 8). A 40% reduction in the TRF1–DNA complex intensity was noted in the NS overexpression samples (Fig. 5A1, lanes 9 and 10). Next, we performed chromatin immunoprecipitation (ChIP) to confirm that NS has an effect on the telomere binding of TRF1 in vivo. The ChIP results showed that NS does not bind the telomere by itself or through TRF1 or TRF2 (Fig. 5B, left). In addition, the ChIP experiments showed that NS does not change the amount of telomere-bound TRF1 (Fig. 5B, right). These results suggest that NS has little or no effect on regulating the amount of telomere-bound TRF1 proteins in vivo.
Fig. 5.
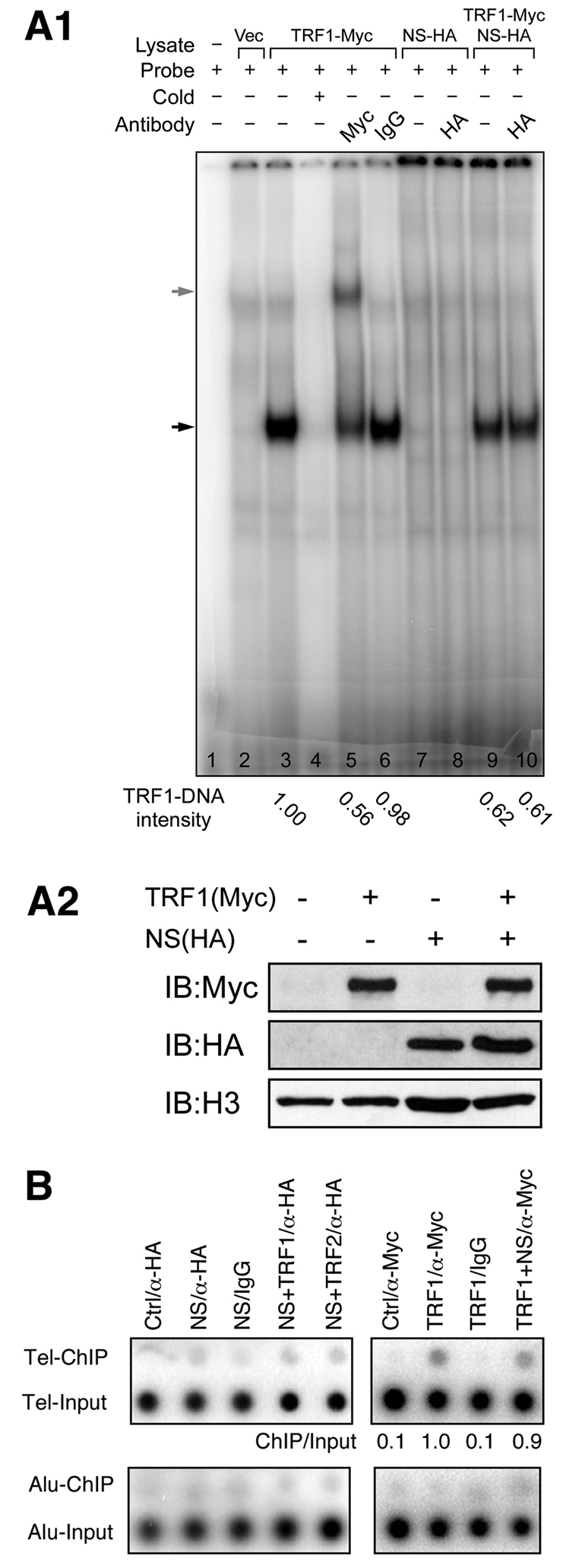
NS reduces the amount of telomere-bound TRF1 in the electrophoretic mobility shift assay but not in the chromatin immunoprecipitation experiment. (A1) EMSA experiments identified a specific TRF1-probe complex (lane 3, indicated by black arrow) that can be competed by unlabeled probes (lane 4) and supershifted by anti-Myc antibody (lane 5, indicated by grey arrow). Coexpression of NS reduces the intensity of the TRF1-DNA complex by 40% (lanes 9 and 10). NS does not bind telomeric DNAs by itself (lanes 7 and 8) or through TRF1 (lane 10). (A2) Western blots show input HEK293 nuclear extracts expressing the indicated proteins used for EMSA experiments. (B) ChIP assays showed that immunoprecipitation of HA-tagged NS (NS/α-HA) does not co-purify telomeric DNAs (Tel-ChIP) more than the vector/α-HA (Ctrl/α-HA), NS/IgG, or Alu sequence controls (left). NS coexpression does not change the amount of telomere-bound TRF1 (TRF1+NS/α-Myc) in ChIP (right).
NS reduces the dynamic telomere association of TRF1
Because NS does not affect the static amount of telomere-bound TRF1, we decided to use the FLIP (fluorescence loss in photobleaching) approach to investigate whether NS regulates the dynamic telomere association of TRF1 in live cells. We chose U2OS cells for the dynamic study because their TRF1–GFP signals give the best telomere-to-nucleoplasmic ratios compared with other cell lines we tested. Consistent with the ChIP result, we found that NS overexpression did not change the static distribution pattern of TRF1 as detected by immunofluorescence (Fig. 6A). To study the dynamic telomere association of TRF1, a FLIP paradigm was designed to measure the telomeric retention time of TRF1–GFP proteins. In the FLIP experiments, a 17×4.6 μm rectangular region in the nucleoplasm was repetitively bleached and the loss of the TRF1–GFP fluorescent signals at non-bleached telomeres was recorded over a period of 136 seconds (Fig. 6B). The validity of using the C-terminally GFP-fused TRF1 to track the distribution of endogenous TRF1 proteins was previously verified (Zhu et al., 2009). The time to reach half-maximal loss of TRF1 signals (mean decay half-time [T1/2]) was used as a measurement of its telomeric retention time. Our FLIP results showed that NS decreases the telomeric retention of TRF1 (Fig. 6C). The average T1/2 of NS-overexpressing cells (22.6 seconds) was significantly shorter than the T1/2 of control cells (33.8 seconds) (P<0.0001, Repeated Measures ANOVA). Together, our data demonstrate that NS regulates the dynamic telomere association of TRF1, but not the static amount of telomere-bound TRF1 proteins.
Fig. 6.

NS shortens the telomeric retention time of TRF1. (A) Confocal studies show that the static distribution of TRF1–GFP is not changed by overexpression of HA-tagged NS (NS-OE) in U2OS cells. Right and bottom panels show enlarged areas indicated by the small rectangles in the left upper panels. Dashed lines demarcate the nucleo-cytoplasmic boundary. (B) The dynamic telomere association of TRF1 was measured its telomeric retention time using the fluorescence loss in photobleaching (FLIP) approach in U2OS cells. Time-sequenced images with labels indicating the bleached areas (red rectangles), measured telomeres (green arrows) and intervals between image acquisition and the first bleaching pulse (in seconds) are shown. (C) NS-OE significantly decreases the telomeric retention time of TRF1 (P<0.0001, by Repeated Measures ANOVA). Error bars represent s.e.m., shown on one side (indicated by arrows) of the curves. Y-axis represents the relative fluorescence index (RFI). Top arrows indicate bleaching pulses. Scale bars: 10 μm (A) and 5 μm (B).
NS decreases the formation of telomere-dysfunction-induced foci
Because overexpressed TRF1 reduces the telomere length presumably by blocking the telomerase from gaining access to the telomere (van Steensel and de Lange, 1997), we speculated that NS might help the repair of damaged telomeres by reducing the telomeric retention of TRF1. To test this possibility, we measured the number of telomere-dysfunction-induced (TIF) foci per cell by confocal colocalization of 53BP1 (p53 binding protein 1) and TRF1–GFP signals in TRF2ΔBΔM-transfected U2OS cells (Fig. 7A). 53BP1 was originally identified as a p53 binding partner (Iwabuchi et al., 1994) and later shown to be a DNA damage response protein that is rapidly recruited to sites of DNA damage (Anderson et al., 2001; Schultz et al., 2000). Notably, we found that NS had a significant effect in protecting against TIF formation in TRF2ΔBΔM-transfected cells (Fig. 7B). NS overexpression (NS-OE) reduced the number of TRF2ΔBΔM-induced TIF per cell from 4.3 (±0.2) down to 1.8 (±0.2) (P<0.0001, t-test). This effect was TIF-specific, because NS did not change the number of non-telomere-related DNA damage foci per cell (non-TIF, defined by the 53BP1+/TRF1–GFP− signal). The numbers of non-TIF foci per cell are 6.5 (±0.5) and 6.2 (±0.4) in the control (Ctrl) and NS-OE groups, respectively (P=0.56). It is also not caused by changing the number of TRF1–GFP foci per cell (36.9±1.1 versus 35.5±1.0 in the control and NS-OE groups, respectively, P=0.38). To check whether this effect of NS correlates with its ability to decrease the telomeric retention of TRF1, we measured the FLIP rate of TRF1 in control and NS-overexpressing cells when telomeres were damaged (Fig. 7C). The FLIP results showed that the telomere-associated TRF1 was more dynamic under the TRF2ΔBΔM-transfected condition (T1/2=26.3 seconds, black line) compared with the control condition (T1/2=33.8 seconds, blue line) (P<0.01, Repeated Measures ANOVA) (Fig. 7D). Upon telomere damage, NS was capable of further lowering the telomeric retention time of TRF1 (T1/2=18.8 seconds, red line) (P<0.05, Repeated Measures ANOVA). These results show that NS can shorten the dynamic association of TRF1 with the telomere and prevent the formation of TIF in TRF2ΔBΔM-transfected cells.
Fig. 7.

NS decreases formation of telomere-dysfunction-induced foci and the telomeric retention time of TRF1 in TRF2ΔBΔM-transfected cells. (A) TIF formation was induced by TRF2ΔBΔM transfection in U2OS cells. The number of TIF per cell was measured by confocal colocalization of 53BP1 and TRF1–GFP signals in control (A1) and NS-OE (A2) cells. (B) NS shows a significant effect in reducing the number of TIF per cell (left), but has no effect on the number of non-telomere-related DNA damage foci (non-TIF, middle), as defined by the 53BP1+/TRF1–GFP− signal, or the number of total TRF1–GFP foci per cell (right). (C) Time-sequenced FLIP images of TRF2ΔBΔM-transfected U2OS cells (see Fig. 6B for label description). (D) The FLIP results show that TRF2ΔBΔM expression decreases the telomeric retention time of TRF1 (black line) compared with the control-transfected cells (blue line) (P<0.01), which supports the idea that TRF2ΔBΔM destabilizes the telomere-capping complex. Overexpression of NS further accelerates the exchange rate between telomere-bound and unbound TRF1 proteins (red line) (P=0.02). Scale bars: 5 μm.
Discussion
NS and GNL3L regulate TRF1 dimerization in opposite directions
It is known that TRF1 binds telomeric DNAs as homodimers, but it is not clear whether non-telomere-bound TRF1 dimerizes first in the nucleoplasm or does so during its binding to the telomere. In an attempt to determine the differential activities of NS and GNL3L on TRF1 regulation, we uncovered an extrachromosomal mechanism that operates bidirectionally to inhibit or promote TRF1 dimerization (Fig. 8). Because NS and GNL3L do not bind telomeric DNAs by themselves or through TRF1 or TRF2, such activities most likely occur outside the telomere. In a previous report, we found that GNL3L can promote the dimerization and telomeric association of TRF1 (Zhu et al., 2009), Here, we discover that NS binding decreases the GNL3L binding and homodimerization of TRF1. The ability of NS and GNL3L to compete against each other for TRF1 binding is probably caused by a steric hindrance effect, because all NS and GNL3L mutants with the ability to bind TRF1 are capable of doing so. NS and GNL3L interact with separate domains of TRF1, that is, the undefined domain and homodimerization domain (HBD, also known as TRF homology or TRFH), respectively. Because the HBD domain of TRF1 also serves as its binding interface for TIN2 (Chen et al., 2008), this might explain why GNL3L can compete against TIN2 for TRF1 binding but NS cannot. Mutant analyses showed that NSΔB and NSΔG have the ability to block the GNL3L binding but not the dimerization of TRF1, indicating that the NS ability to perturb TRF1 dimerization is not determined by its ability to displace TRF1-bound GNL3L.
Fig. 8.

Opposing regulation of TRF1 dimerization and telomere association by NS and GNL3L. We propose that the dimerization of TRF1 is oppositely regulated by NS and GNL3L before it binds to the telomere. In this pre-telomeric phase, GNL3L promotes and NS inhibits the dimerization of non-telomere-bound TRF1. The relative amount of TRF1 monomers versus dimers in the nucleoplasm determines the telomeric retention time of a dynamic pool of telomere-bound TRF1, which might serve the function of blocking the telomeric access of telomere remodelling and repair proteins and as the precursor for the stably bound TRF1 that protects the integrity of the telomere.
NS reduces the telomeric retention time of TRF1
Since TRF1 binds telomeric DNAs as dimers, the NS-mediated inhibition on TRF1 dimerization may decrease its telomeric association. Even though this idea is consistent with our EMSA result, it is not supported by the telomere ChIP data, which show that the amount of telomere-bound TRF1 in vivo is not affected by NS perturbation. Instead, the NS effect is mainly on the dynamic association between TRF1 and the telomere. Based on these findings, we speculate that NS might have a function in promoting the dynamic exchange between the telomere-bound and unbound TRF1 proteins by shifting the nucleoplasmic TRF1 from the dimer pool to the monomer pool.
Biological implications of NS-regulated TRF1 dynamics
It has been shown that one of the functions of TRF1 is to block the telomere access by the telomerase, which negatively affects the telomere length (van Steensel and de Lange, 1997). At the same time, TRF1 also has a crucial role in protecting the integrity of the telomere (Martinez et al., 2009). How these two activities of TRF1 work in sync at the cellular level needs further clarification. We propose that the telomere-bound TRF1 consists of two dynamically distinct pools of proteins. The dynamic TRF1 population is constantly shifting between the telomere-bound state and the non-telomere-bound state, whereas the stabilized TRF1 population is held in place by the formation of the telomere-capping complex and undergoes little exchange with the non-telomere-bound TRF1. We speculate that the fast-exchanging component of the telomere-bound TRF1 serves as the precursor of the highly organized telomere complex on one hand and prevent the telomere access by the telomere remodelling or repair machinery on the other hand. The NS function described in this study primarily affects the retention time of the dynamic (or destabilized) pool of telomere-bound TRF1, and, therefore, might allow the remodelling and repair proteins better access to the telomeric DNAs. Given the preferential expression of NS and GNL3L in the undifferentiated and differentiated cells, respectively, and their opposite roles in regulating TRF1 dimerization, one might infer that the telomere-remodeling capability differs from cell to cell. Undifferentiated cells, which express high levels of NS and low levels of GNL3L, might be more active in telomere remodeling and repair than differentiated cells, which express low levels of NS and high levels of GNL3L. Because NS and GNL3L are separate genes only in vertebrates (Tsai and Meng, 2009), such expansion of the functional repertoire in telomere regulation probably arose during the evolution of the vertebrate lineage. Of note, the NS effect on telomere damage prevention was determined in U2OS cells, chosen for their superior imaging quality of TRF1–GFP and 53BP1 signals for both static and dynamic studies. U2OS cells belong to the alternative lengthening of telomeres (ALT) cell type that uses the homologous recombination (HR) instead of the telomerase-dependent mechanism for telomere remodeling and repair (Bryan et al., 1997; Bryan et al., 1995). Therefore, it is possible that if TRF1 differentially affects the telomere access of telomerase and HR proteins in telomerase-active and ALT cells, respectively, the function of NS on telomere damage prevention might also differ in these two cell types.
In conclusion, this study shows that NS and GNL3L act as chaperone proteins that regulate the monomeric versus dimeric forms of non-telomere-bound TRF1. Even though NS and GNL3L do not take part in the formation of the telomere-capping complex, they modulate the dynamics of telomere-bound TRF1, and NS, in particular, has the ability to prevent telomere damage in TRF2ΔBΔM-transfected cells.
Materials and Methods
cDNA constructs and antibodies
Deletions, point mutations and epitope tagging were introduced using the stitching PCR strategy as described previously (Meng et al., 2007; Tsai and McKay, 2005). The N-terminal FLAG-tagged TRF2ΔBΔM construct (containing aa 45–454) was obtained from Zhou Songyang (Baylor College of Medicine, Houston, TX). Primary antibodies used here include those against HA (HA.11, Covance), Myc (9E10, Covance), FLAG (M2, Sigma), TRF1 (TRF-78, Chemicon), 53BP1 (4937, Cell Signaling), NS (Ab2438, Ab1164 and Ab138) and GNL3L (Ab134).
Cell culture
Procedures for cell culture and plasmid transfection were described in our previous work (Meng et al., 2007). All biochemical studies were performed in vitro or in HEK293 cells (Figs 1, 2, 3, 4 and 5). Dynamic studies were conducted in U2OS cells (Figs 6, 7).
GST pull-down assays and protein purification
For pull-down experiments, GST fusion proteins were expressed using the pGEX 4T-2 vector in BL21/DE3 cells (Tsai and McKay, 2002). Epitope-tagged proteins were expressed in HEK293 cells and extracted in phosphate-buffered saline (PBS) with Triton X-100 (1%). Sepharose-bound GST fusion proteins (1–2 μg) were used for each reaction. To purify NS and GNL3L proteins, GST-fused recombinant proteins were expressed in BL21/DE3 bacteria, purified by glutathione beads, and released from the GST backbone by thrombin cleavage.
Coimmunoprecipitation
Protein lysates were extracted in NTEN buffer and incubated with primary antibody and Protein-G–Sepharose beads (Amersham) for 4 hours at 4°C. Immunoprecipitates were washed with RIPA buffer before SDS-PAGE. For two-step coimmunoprecipitation, the TRF1 (FLAG-tagged) complex was precipitated using anti-FLAG antibody and eluted by FLAG peptide incubation. The co-complex of TRF1 (FLAG-tagged) and GNL3L (HA-tagged) was subsequently immunoprecipitated from the TRF1 complex using the anti-HA antibody.
Electrophoretic mobility shift assay
EMSA experiments were carried out based on the protocol described previously. Nuclear extracts were prepared from HEK293 cells transfected with the indicated constructs. To generate EMSA probes, the (TTAGGG)6 primer was radiolabeled with [γ-32P]ATP in a T4 kinase reaction, annealed with excess amounts of the (CCCTAA)6 primer, and purified through QIAquick nucleotide removal columns (Qiagen). Reaction and gel electrophoresis conditions were described previously (Yasumoto et al., 2007; Zhu et al., 2009).
Chromatin immunoprecipitation
Cells were crosslinked using 1% formaldehyde, lysed in 50 mM Tris buffer with 1% SDS and sonicated to obtain chromatin fragments from 300 bp to 1 kb. The resulting lysates were incubated with antibodies and Protein-G–Sepharose. Chromosomal DNA was extracted from the immunoprecipitates by RNase-A and proteinase-K treatment and reverse crosslinking. DNA samples were dot blotted and hybridized with a 32P-labeled (TTAGGG)4 or Alu (5′-GGC CGG GCG CGG TGG CTC ACG CCT GTA ATC CCA GCA-3′) oligonucleotide probe. All lysates were normalized based on protein concentrations. A part of the lysate (1/20th) was used to measure the input DNA amount.
Fluorescence loss in photobleaching
Bleaching experiments were performed in U2OS cells grown on Nalgene Lab Tek II chamber slides by using a Zeiss LSM510 confocal microscope equipped with a 63× plan-apochromat oil objective, as described previously (Meng et al., 2007). The FLIP paradigm was modified based on a previously published method (Mattern et al., 2004). Specifically, we measured the dynamic telomere association of TRF1 by the rate of fluorescence loss of TRF1–GFP signals on unbleached telomeres while bleaching a 17×4.6 μm rectangular region within the nucleoplasm with repetitive bleaching pulses of 150 msecond duration and 787 msecond interval. Images were acquired with 3× confocal zoom. The telomeric relative fluorescence index (RFI) in bleached cells was normalized to the telomeric intensity of neighboring non-bleached cells after background subtraction using the calculation: RFI=(It/I0)*(C0/Ct), where It and I0 are the background-subtracted intensities of the telomere in the bleached cell at time-point t and before photobleaching, respectively (Phair and Misteli, 2000). Ct and C0 are the background-subtracted intensities of the telomere in the neighboring non-bleached cell at time-point t and before photobleaching, respectively. The telomeric RFI were measured from 24 cells collected from two independent experiments. For every cell, the RFI represents the average of six telomeres in random positions.
Confocal quantification of telomere dysfunction-induced foci
TIF were determined by the colocalization of 53BP1 and TRF1–GFP signals. Images were acquired using a Zeiss LSM510 confocal microscope using a 63× plan-apochromat oil objective (1.4 NA). Scanning was set with a 512×512 frame size, 3× zoom, and <1.0 μm optical thickness. Stacked images of 60 cells were collected at 0.5 μm intervals from three independent experimental repeats, and using ImageJ 1.36b software.
Acknowledgements
We gratefully thank Zhou Songyang (Baylor College of Medicine, Houston, TX) for his gift of the TRF2ΔBΔM construct. Special thanks go to His-Wen Tsai for his unwavering support of our work.
Footnotes
Funding
This work was supported by the National Institutes of Health [grant number RO1 CA113750]. Deposited in PMC for release after 12 months.
References
- Anderson L., Henderson C., Adachi Y. (2001). Phosphorylation and rapid relocalization of 53BP1 to nuclear foci upon DNA damage. Mol. Cell Biol. 21, 1719-1729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baddoo M., Hill K., Wilkinson R., Gaupp D., Hughes C., Kopen G. C., Phinney D. G. (2003). Characterization of mesenchymal stem cells isolated from murine bone marrow by negative selection. J. Cell Biochem. 89, 1235-1249 [DOI] [PubMed] [Google Scholar]
- Bianchi A., Smith S., Chong L., Elias P., de Lange T. (1997). TRF1 is a dimer and bends telomeric DNA. EMBO. J. 16, 1785-1794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broccoli D., Smogorzewska A., Chong L., de Lange T. (1997). Human telomeres contain two distinct Myb-related proteins, TRF1 and TRF2. Nat. Genet 17, 231-235 [DOI] [PubMed] [Google Scholar]
- Bryan T. M., Englezou A., Gupta J., Bacchetti S., Reddel R. R. (1995). Telomere elongation in immortal human cells without detectable telomerase activity. EMBO. J. 14, 4240-4248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryan T. M., Englezou A., Dalla-Pozza L., Dunham M. A., Reddel R. R. (1997). Evidence for an alternative mechanism for maintaining telomere length in human tumors and tumor-derived cell lines. Nat. Med. 3, 1271-1274 [DOI] [PubMed] [Google Scholar]
- Chang W., Dynek J. N., Smith S. (2003). TRF1 is degraded by ubiquitin-mediated proteolysis after release from telomeres. Genes Dev. 17, 1328-1333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y., Yang Y., van Overbeek M., Donigian J. R., Baciu P., de Lange T., Lei M. (2008). A shared docking motif in TRF1 and TRF2 used for differential recruitment of telomeric proteins. Science 319, 1092-1096 [DOI] [PubMed] [Google Scholar]
- Chong L., van Steensel B., Broccoli D., Erdjument-Bromage H., Hanish J., Tempst P., de Lange T. (1995). A human telomeric protein. Science 270, 1663-1667 [DOI] [PubMed] [Google Scholar]
- de Lange T. (2005). Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev. 19, 2100-2110 [DOI] [PubMed] [Google Scholar]
- Iwabuchi K., Bartel P. L., Li B., Marraccino R., Fields S. (1994). Two cellular proteins that bind to wild-type but not mutant p53. Proc. Natl. Acad. Sci. USA 91, 6098-6102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang W. Q., Zhong Z. H., Henson J. D., Reddel R. R. (2007). Identification of candidate alternative lengthening of telomeres genes by methionine restriction and RNA interference. Oncogene 26, 4635-4647 [DOI] [PubMed] [Google Scholar]
- Kim M. K., Kang M. R., Nam H. W., Bae Y. S., Kim Y. S., Chung I. K. (2008). Regulation of telomeric repeat binding factor 1 binding to telomeres by casein kinase 2-mediated phosphorylation. J. Biol. Chem. 283, 14144-14152 [DOI] [PubMed] [Google Scholar]
- Lee T. H., Perrem K., Harper J. W., Lu K. P., Zhou X. Z. (2006). The F-box protein FBX4 targets PIN2/TRF1 for ubiquitin-mediated degradation and regulates telomere maintenance. J. Biol. Chem. 281, 759-768 [DOI] [PubMed] [Google Scholar]
- Maki N., Takechi K., Sano S., Tarui H., Sasai Y., Agata K. (2007). Rapid accumulation of nucleostemin in nucleolus during newt regeneration. Dev. Dyn. 236, 941-950 [DOI] [PubMed] [Google Scholar]
- Martinez P., Thanasoula M., Munoz P., Liao C., Tejera A., McNees C., Flores J. M., Fernández-Capetillo O., Tarsounas M., Blasco M. A. (2009). Increased telomere fragility and fusions resulting from TRF1 deficiency lead to degenerative pathologies and increased cancer in mice. Genes Dev. 23, 2060-2075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattern K. A., Swiggers S. J., Nigg A. L., Lowenberg B., Houtsmuller A. B., Zijlmans J. M. (2004). Dynamics of protein binding to telomeres in living cells: implications for telomere structure and function. Mol. Cell Biol. 24, 5587-5594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng L., Zhu Q., Tsai R. Y. (2007). Nucleolar trafficking of nucleostemin family proteins: common versus protein-specific mechanisms. Mol. Cell Biol. 27, 8670-8682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng L., Lin T., Tsai R. Y. (2008). Nucloplasmic mobilization of nucleostemin stabilizes MDM2 and promotes G2-M progression and cell survival. J. Cell Sci. 121, 4037-4046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohmura M., Naka K., Hoshii T., Muraguchi T., Shugo H., Tamase A., et al. (2008). Identification of stem cells during prepubertal spermatogenesis via monitoring of nucleostemin promoter activity. Stem. Cells 26, 3237-3246 [DOI] [PubMed] [Google Scholar]
- Phair R. D., Misteli T. (2000). High mobility of proteins in the mammalian cell nucleus. Nature 404, 604-609 [DOI] [PubMed] [Google Scholar]
- Potts P. R., Yu H. (2005). Human MMS21/NSE2 is a SUMO ligase required for DNA repair. Mol. Cell Biol. 25, 7021-7032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz L. B., Chehab N. H., Malikzay A., Halazonetis T. D. (2000). p53 binding protein 1 (53BP1) is an early participant in the cellular response to DNA double-strand breaks. J. Cell Biol. 151, 1381-1390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddiqi S., Gude N., Hosoda T., Muraski J., Rubio M., Emmanuel G., Fransioli J., Vitale S., Parolin C., D'Amario D., et al. (2008). Myocardial induction of nucleostemin in response to postnatal growth and pathological challenge. Circ. Res. 103, 89-97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith S., Giriat I., Schmitt A., de Lange T. (1998). Tankyrase, a poly(ADP-ribose) polymerase at human telomeres. Science 282, 1484-1487 [DOI] [PubMed] [Google Scholar]
- Songyang Z., Liu D. (2006). Inside the mammalian telomere interactome: regulation and regulatory activities of telomeres. Crit. Rev. Eukaryot. Gene Expr. 16, 103-118 [DOI] [PubMed] [Google Scholar]
- Tsai R. Y. (2009). Nucleolar modulation of TRF1: a dynamic way to regulate telomere and cell cycle by nucleostemin and GNL3L. Cell Cycle. 8, 2912-2916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai R. Y., McKay R. D. (2002). A nucleolar mechanism controlling cell proliferation in stem cells and cancer cells. Genes Dev. 16, 2991-3003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai R. Y., McKay R. D. (2005). A multistep, GTP-driven mechanism controlling the dynamic cycling of nucleostemin. J. Cell Biol. 168, 179-184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai R. Y., Meng L. (2009). Nucleostemin: A latecomer with new tricks. Int. J.Biochem. Cell Biol. 41, 2122-2124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Steensel B., de Lange T. (1997). Control of telomere length by the human telomeric protein TRF1. Nature 385, 740-743 [DOI] [PubMed] [Google Scholar]
- van Steensel B., Smogorzewska A., de Lange T. (1998). TRF2 protects human telomeres from end-to-end fusions. Cell 92, 401-413 [DOI] [PubMed] [Google Scholar]
- Wu Z. Q., Yang X., Weber G., Liu X. (2008). Plk1 phosphorylation of TRF1 is essential for its binding to telomeres. J. Biol. Chem. 283, 25503-25513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasumoto H., Meng L., Lin T., Zhu Q., Tsai R. Y. (2007). GNL3L inhibits activity of estrogen-related receptor {gamma} by competing for coactivator binding. J. Cell Sci. 120, 2532-2543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X. Z., Perrem K., Lu K. P. (2003). Role of Pin2/TRF1 in telomere maintenance and cell cycle control. J. Cell Biochem. 89, 19-37 [DOI] [PubMed] [Google Scholar]
- Zhu Q., Yasumoto H., Tsai R. Y. (2006). Nucleostemin delays cellular senescence and negatively regulates TRF1 protein stability. Mol. Cell Biol. 26, 9279-9290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Q., Meng L., Hsu J. K., Lin T., Teishima J., Tsai R. Y. (2009). GNL3L stabilizes the TRF1 complex and promotes mitotic transition. J. Cell Biol. 185, 827-839 [DOI] [PMC free article] [PubMed] [Google Scholar]