

The success of antimicrobial therapy is determined by complex interactions between an administered drug, a host, and an infecting agent. In a clinical situation, the complexity of these interactions is usually reflected by a high variability in the dose-response relationship. Therefore, to minimize the dose-response variability, key characteristics of the drug, the infecting agent and the host have to be taken into account for selecting an appropriate antibiotic and an appropriate dose. Failure to do so may result in either therapeutic failure or emergence of resistant strains.
To date, dose and drug selection is mostly based on a static in vitro parameter, the MIC and on the drug′s serum concentration as a pharmacokinetic parameter. In practice, however, a pharmacodynamic effect in vivo is rather the result of a dynamic exposure of the infective agent to the unbound antibiotic drug fraction at the relevant effect site. Thus, static conditions in an in vitro setting hardly reflect a dynamic situation in a target organ under in vivo conditions. Furthermore, serum concentrations do not reflect the unbound concentrations at the target site.
In recent years substantial efforts were devoted to systematically elucidate the dynamic relationship between pharmacokinetic and pharmacodynamic variables. The main concept of this pharmacokinetic-pharmacodynamic approach is to use the concentration-effect relationship of the drug of interest in dosage adjustment and product development in a logical way and minimize trial-and-error approaches (29, 80). This approach can potentially result in substantial savings of time and expenses and may help to avoid unnecessary and, hence, unethical clinical studies (97). Thus, dosages and dosing intervals of antimicrobial agents should be designed with reference to dynamic pharmacokinetic and pharmacodynamic parameters. Accordingly, several efficacy indices or surrogate markers that take into account both pharmacokinetic and pharmacodynamic information have been defined and used by different authors to describe the antibacterial activity of various classes of antimicrobial agents (100, 106, 59).
Currently there are two main trends for antibiotic pharmacokinetic-pharmacodynamic models; those based on the MIC and those based on a kill-curve approach, both of which will be described in detail in the following.
MODELS BASED ON STATIC MICs
The most common approach to antibiotic dosing is to adjust the doses to obtain antibiotic plasma concentrations that are above the MIC for the respective pathogen throughout the dosing interval. The MIC is the lowest concentration that completely inhibits visible growth of the organism as detected by the unaided eye after a 18- to 24-h incubation period with a standard inoculum of approximately 105 CFU/ml (87).
In these approaches, the pharmacokinetic parameter is usually the serum concentration of the anti-infective agent, and the pharmacodynamic parameter is almost exclusively the MIC (104, 71, 105). The minimal bactericidal concentration (MBC), which is the lowest concentration of the antibiotic that causes complete destruction of the pathogen, has also been used for the same purpose (1, 4, 30, 51, 95, 108).
Figure 1 shows the most frequently used parameters, which are time above the MIC (t > MIC), ratio of peak concentration and MIC (Cmax/MIC), and ratio of 24-h area under the curve and MIC (AUC/MIC).
FIG. 1.

Currently used pharmacokinetic-pharmacodynamic parameters.
Another parameter similar to AUC/MIC is the area under the inhibitory curve (AUIC), which can be calculated from the area under the curve for the time period over 24 h where the serum concentration exceeds the MIC (AUCt > MIC/MIC over 24 h) (106, 25). However, AUC/MIC is much easier to calculate than AUIC which does not offer any significant advantage. Moreover, if the drug concentration remains above the MIC at all times, AUIC and AUC/MIC are identical.
Using the MIC approach, antibiotics are frequently divided into two major groups: those that exhibit time-dependent (concentration-independent) killing and minimally to moderately persistent effects and those that exhibit concentration-dependent killing and prolonged persistent effects (19).
For antibiotics that belong to the first group (beta-lactam antibiotics, vancomycin, macrolides), their effect depends on the length of time that the drug is in contact with the bacteria. Their effect will increase with increasing concentrations until a finite point (the maximum kill rate) is reached. After that point, increasing concentrations will not produce a corresponding increase in the effect; therefore, high peak concentration will not help. Maximum killing has been seen to occur at concentrations approximately four to five times the MIC (88). The parameter that has been most used to assess the efficacy of time-dependent antibiotics is the time that the antibiotic plasma concentration exceeds the MIC for a particular microorganism, or time above MIC (t > MIC) (33, 24, 115, 112).
The second group of antibiotics, which include the aminoglycosides and fluoroquinolones, exhibit a different killing pattern. In their case, the bacterial rate of killing is seen to increase with increasing concentrations of the antibiotic. The goal in this case is to maximize the drug concentration. The parameters that are most currently used are those which reflect an increase in drug concentration, i.e., Cmax/MIC (the ratio between the peak concentration and the MIC) and AUC > MIC. The area under the inhibitory curve (AUIC) is also popular and it can be easily calculated as the ratio of 24-h AUC (for time points with respective concentrations above the MIC) and the MIC (24, 115, 70, 83).
Time above MIC
This index measures the time during which antibiotic concentrations in plasma are above the MIC for a particular pathogen and it has been used to assess the bactericidal activity of β-lactam antibiotics (72, 52).
Craig has reviewed the data from the literature that use mortality as an endpoint and in which animals infected with Streptococcus pneumoniae were treated with penicillins or cephalosporins (20). A relationship was found between the duration of time that the serum level is above MIC and the efficacy, which was between 90 to 100% when serum levels were above the MIC for approximately half of the dosing interval (23).
Cmax/MIC Ratio
This parameter is the relationship between the Cmax reached in the patient at steady state and the MIC established for the pathogen responsible for the infection (99). Many studies have used it as a good predictor of clinical outcome for drugs showing concentration-dependent bactericidal effects, particularly for aminoglycosides (24, 99, 60, 78, 84, 10, 11, 56).
Parameters That Use AUC
The AUC24 h/MIC ratio relates the total AUC at steady state to the MIC, and has been found to correlate best with the efficacy of fluoroquinolones (106, 70, 56, 21). The term AUIC was introduced by Flaherty et al. for the area under the serum inhibitory concentration of clindamycin. It was calculated as the AUC of the reciprocal values of the serum inhibitory titer (SIT) versus time, similarly to that previously described for evaluating the area under the bactericidal curve (43, 44).
Later, Schentag et al. pointed out the equivalence between the area under the bactericidal curve and AUIC for several antibiotic classes, and proposed a method to calculate AUIC as the AUC for the period of time the concentrations are above the MIC divided by the MIC (106). However, today AUC24 h/MIC ratios are preferred since they are easier to calculate and are independent of the fact if concentrations exceed the MIC or not. However, the same AUC24 h/MIC, which implies equal potency, can be obtained with different dosage regimens that resulted in very different times above MIC. Particularly in the case of beta-lactams, the use of a constant AUC24 h/MIC can produce regimens that have significant time below the MIC and may result in negative therapeutic effects (25).
DISADVANTAGES OF STATIC MIC APPROACH
The MIC is a well-established laboratory parameter routinely determined in microbiology. It is currently by far the most commonly used pharmacodynamic parameter for the evaluation of efficacy of anti-infective agents. Once the “desirable” pharmacokinetic-pharmacodynamic parameters are determined, the drug concentrations are compared to the MIC to make dosing decisions.
However, although these parameters are useful predictors of the potency of the drug-microorganism interaction, this approach has both pharmacokinetic and pharmacodynamic disadvantages.
From the pharmacokinetic point of view, all of these models compare the MIC to measurements obtained from the concentration-time curve measured in plasma. Thus, two important factors are overlooked: protein binding and tissue distribution. After absorption most drugs are bound to plasma proteins. They can also leave the plasma and enter the tissues, where, again, they can be nonspecifically bound to either tissue proteins or structures (28). Protein binding is relevant because only the drug that is unbound from the plasma proteins will be available to exert a pharmacological effect. Tissue distribution also needs to be taken into account, given that most of the infections occur not in plasma, but in the interstitial space of the tissues.
From the pharmacodynamic point of view, the MIC approach provides only limited information on the kinetics of the drug action. For instance, the MIC does not provide information on the rate of bactericidal activity and whether increasing antimicrobial concentrations can enhance this rate. Since the MIC determination depends on the number of bacteria at a single time point, many different combinations of growth and kill rates can result in the same MIC. These different kill kinetics may be therapeutically relevant. Similarly, the MIC approach does not provide any information about the persistent activity of the antimicrobial agent that remains following exposure to the drug (56). These shortcomings are frequently compensated for by modifications of the MIC interpretation such as postantibiotic effects or sub-MIC effects.
An additional and important shortcoming is that MICs are conventionally measured for constant antibiotic concentrations and therefore represent threshold concentrations. This implies the existence of an all-or-nothing concentration-effect relationship. All concentrations below the MIC are treated equally and similarly no quantitative distinction is made for all concentrations above MIC. This is a very crude way of using antimicrobial information. Clearly, concentrations just below the MIC show some anti-infective activity and are different from those that are close to zero. Similarly, concentrations just above the MIC do not show the maximum effect that is only achieved with higher concentrations.
Thus, static MIC approaches do by no means reflect the in vivo scenario, where bacteria are not being exposed to constant but constantly changing antibiotic concentrations.
DYNAMIC IN VITRO MODELS BASED ON KILL CURVES
A different approach to assess the anti-infective efficacy of antibiotics is to use pharmacokinetic-pharmacodynamic models based on time-kill curves. Time-kill curves can follow microbial killing and growth as a function of both time and antibiotic concentration. Antibiotic concentration can either be held constant or changed to mimic an in vivo concentration profile, be it either in plasma or at the infection site. The resulting kill curves can be subsequently analyzed with appropriate pharmacokinetic-pharmacodynamic models. Finally these pharmacokinetic-pharmacodynamic models then aid to optimize dosage regimens based on a rational, scientific approach. The advantage of these in vitro models is that they allow direct comparison of the effects of various concentration profiles and provide for a much more detailed assessment of the pharmacokinetic-pharmacodynamic relationship than the simple use of MICs.
Kill curves can and have been used to study anti-infective effects both in animal and in vitro models, with the advantage of providing more detailed information about the time course of antibacterial effect.
Various types of in vitro models have been devised. the main classes are those with constant antibiotic concentrations, which study the effects of a constant concentration of drug against bacteria as a function of time; and those with variable antibiotic concentrations, in which the antibiotic concentrations fluctuate by dilution or diffusion.
Models with Constant Antibiotic Concentration
These models study the number of bacteria exposed to a constant antibiotic concentration. Early studies by Garrett et al. investigated the effect of constant concentrations of bacteriostatic and bactericidal drugs against bacteria as a function of time (48, 82, 49).
Curves in the presence (kill curves) and absence (growth curves) of antibiotic were compared. After an initial lag time, bacteria typically show a phase of logarithmic growth (log phase) that can be described by: equation 1
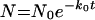 |
(1) |
where N is the number of bacteria at any given time point, N0 is the number of bacteria in the initial inoculum, and k0 is the first-order growth rate constant. N and N0 are usually expressed in CFU per milliliter. Exposure to an antibiotic while the bacteria are in log phase will produce a change in k0, resulting in a different growth rate constant (kapp).
The mathematical relationship between antibiotic concentration and kapp for bacteriostatic drugs was studied, and the interactions found were classified into four classes by Garrett (47). The classes were defined as follows. Class I interactions are defined by a linear relationship between kapp and concentration that can be expressed by: equation 2
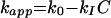 |
(2) |
where kI is a second-order inhibitory rate constant specific for the drug, C is the drug concentration, and k0 and kapp have been defined before.
Class II interactions, where the kapp decreases with increasing concentrations to approach zero asymptotically, are represented by the equation: equation 3
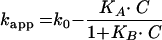 |
(3) |
where KA is an equilibration constant between the medium and the receptor sites at steady state, KB is the affinity constant between drug and receptor, and C, k0 and kapp have been defined before.
Class III interactions exhibit class I behavior at low concentrations, which turns into class II behavior at high concentrations. Class IV interactions show S-shaped plots of kapp versus concentration that may be due to binding of the drug to nutrients at low concentrations.
The main difference between bacteriostatic and bactericidal drugs is that for the latter the kapp may take negative values, as the bacteria are now being killed. There is a linear relationship between kapp and C that can be expressed mathematically by: equation 4
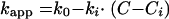 |
(4) |
where k0 is the growth rate constant in absence of drug, ki is the kill rate constant, C is the drug concentration, and Ci is the minimum drug concentration that will produce an effect (50).
Using a different mathematical approach, the growth curves for bacteria exposed to different antibiotic concentrations were described taking into account the effect lag time after initial drug exposure. Curves of growth in the presence of the antibiotic were expressed as quadratic functions of time, with initial growth rates and rates of change of growth as concentration dependent variables. Assuming k0 is a constant, kill rates would only depend on drug concentration (75, 76, 77, 114).
A disadvantage of these approaches is that they do not reflect the in vivo situation where drug concentrations fluctuate. Therefore, although they are good mathematical descriptors of antibacterial behavior, they have limited ability of predicting or correlating with clinical outcomes.
Models with changing antibiotic concentrations, on the other hand, try to simulate in vivo concentration-time profiles using human pharmacokinetic parameters in order to assess the antibacterial effect. Changing concentrations can be produced either by dilution or diffusion.
Models with Variable Antibiotic Concentrations
Dilution models.
A kinetic model that operated by pumping sterile broth into a flask (containing bacteria in log growth phase and antibiotic) at a constant rate resulted in antibiotic dilution (101, 102). The main limitation of this model was that the volume in the flask increased throughout the experiment, diluting also the inoculum and posing practical problems for large dilutions. A one compartment open model that used a system consisting of two flasks interconnected by a peristaltic pump to simulate first-order kinetics was developed by Grasso et al. (53). Sterile broth was pumped from the diluent reservoir flask to the flask containing the bacterial culture and the antibiotic. The volume in the culture flask could be regulated, resulting in exponentially decreasing antibiotic concentrations that simulated the human serum t1/2 of the antibiotic. An adjustment in the model enabled it to also simulate first-order absorption kinetics (53). Further modifications of this model enable to continuously monitor bacterial level by including a photometer to read turbidity levels, and to control the system using a computer (7, 6, 8, 69).
The main concern with these models is that the bacterial inoculum is diluted together with the antibiotic. This effect is particularly important when the dilution rate is higher than the bacterial growth rate, and the resulting increased bacterial clearance needs to be accounted for. Some equations to correct this effect for exponentially changing cultures have been proposed (117, 64). Löwdin et al. employed a 0.45-μm filter membrane in the culture flask to prevent bacterial outflow. By placing a stirrer above the filter, they avoided blockage of the membrane and ensured that the culture was homogeneously mixed. The culture flask had two arms, one of which was used to take samples through a silicon diaphragm. Medium was drawn from the culture vessel by a pump at a constant rate, causing a vacuum in the flask that in turn caused fresh sterile medium to be sucked into the flask through the other arm. The antibiotic was added through the first arm and followed first-order elimination kinetics (73). The original model contained the stirrer with a magnet covered in Teflon, however, bacteria were suspected to adhere to this surface. The Teflon-covered stirrer was substituted by a new stirrer in which the magnet was encased in glass (74).
A modification of this model allows to study the effects of antibiotics on intracellular pathogens (58). Instead of the culture flask, a glass chamber with a metal rack fitting Falcon cell culture inserts was used and connected to a pump. Cells cultures are grown in the inserts, infected and then transferred to the glass chamber, which contains medium with antibiotic. Half-lives of the antibiotics are simulated using a pump as described above, and samples are taken at appropriate time points. A magnet is placed in the bottom of the glass chamber to achieve an even concentration through the medium. After the experiments, cells are lysed and intracellular bacteria counted by plating (58).
Recently a simple one-compartment in vitro model was developed to study the pharmacodynamic effect of concentrations of piperacillin against E. coli, following administration of constant or fluctuating concentrations (90). The free interstitial concentrations of the drug reached in humans after different doses and dosing regimens were simulated. To simulate drug elimination in a stepwise fashion, broth solution containing antibiotic was withdrawn from the model at fixed time intervals and replaced with drug-free broth. The bacterial inoculum was not changed because the broth was both withdrawn and added through a filter. The number of cells was determined by colony count after overnight incubation. This model was also used in a separate study for the determination of the effect of different dosing regimens of piperacillin alone and in combination with tazobactam on Escherichia coli (26).
Diffusion models.
In diffusion models, the antibiotic concentrations are changed by applying concentration gradients. An in vitro model in which the compartments were separated by a hollow-fiber dialyser was used to simulate two-compartment kinetics. Drug dilution was achieved by pumping sterile broth from one compartment to the other. However, the model was reportedly too complex and impractical. Its use was recommended only when a very short drug t1/2 needed to be simulated or very flexible kinetics required (96). A different two-compartment model using a regenerated cellulose dialyser was developed by König et al. (66, 67). Dilution was accomplished by antibiotic diffusion from the culture compartment to the second compartment along a concentration gradient.
A model by Murakawa et al. uses bidirectional flow between the central and peripheral compartments to achieve a biexponential decline in concentrations. Bacteria and drug are placed initially in the central compartment, broth is pumped into the central compartment and bacteria are diluted out of the central compartment and the system, therefore necessitating mathematical adjustment for bacterial dilution (86). A model that integrates bidirectional flow with the use of a membrane to prevent bacterial dilution has been used by Palmer et al. (93).
A more complex model to simulate two-compartment pharmacokinetic was introduced by Blaser et al. (12). Serially placed bacterial compartments, representing extravascular infection sites, interface with a central compartment through artificial capillaries. Broth containing antibiotic is constantly pumped from the central compartment through the tubing. The porous capillary walls allow for bidirectional passage of antibiotics (<10 kDa) but are impermeable to bacteria. Sterile drug-free broth is continuously pumped to the central compartment from a diluent reservoir, thus displacing drug-containing broth from the system. An additional “absorption” compartment may be added between the diluent reservoir and the central compartment to simulate first-order absorption (10). The model can be used for simulation of both continuous and intermittent drug administration (13). Simultaneous first order elimination kinetics of two drugs with different half-lives were simulated to study antibacterial effects of drug combinations (120, 119, 14).
Models that mimic effect site pharmacokinetics.
Several in vitro models were developed that simulate in vivo conditions in specific infection sites or conditions, such as an bladder (54, 103), bacterial cystitis (55), otitis medium (113, 35), endocarditis (79), chronic pneumonia (68), tuberculosis (9), infected fibrin clots (94), and implant-related infections (27, 15). However, such attempts to more closely mimic an in vivo situation and to predict clinical response were hampered by the inability to measure pharmacokinetics (pharmacokinetic) at the site of infection. Measuring target site pharmacokinetics has recently become possible by microdialysis, a technique which allows for the on-line measurement of unbound drug concentrations in the interstitial space. Since microdialysis monitors free antibiotic concentrations in the fluid which directly surrounds the infective agents, the antimicrobial effect linked to the time versus drug concentration profile obtained by microdialysis may easily be simulated in an in vitro setting on bacterial cultures. This dynamic simulation may, thus, provide a rational approach to describing and predicting pharmacodynamics at the relevant targt site.
KILL CURVE DATA ANALYSIS
Although experimental kill curves enable a dynamic interpretation of drug-bacteria interactions, the strength of this approach is not fully exploited until the data are analyzed by means of mathematical models. Based on experimental data derived from kill curve experiments these models then serve as a basis to simulate different dosing scenarios. Several mathematical models have been proposed for this purpose, which may be divided into concentration-based, AUC-based, and dose-based approaches.
Concentration-Based Analysis
Zhi et al. published the mathematical solutions for. linear nonsaturable and nonlinear saturable possible pharmacodynamic interactions between β-lactam antibiotics and microorganisms (118). The equations, developed from a model originally proposed by Jusko et al. (63) were derived for different dosage regimens, such as single and multiple intravenous bolus and constant infusion at steady state. The authors applied the model to the activity of piperacillin against Pseudomonas aeruginosa in neutropenic mice and concluded that the saturable nonlinear model appeared to be appropriate.
A similar approach for concentration based pharmacokinetic-pharmacodynamic analysis is based on an Emax model. The concept of the mathematical method is briefly shown below (89). For a beta-lactam antibiotic, the rate of change in bacteria versus time (dN/dt) can be described by the following expression: equation 5
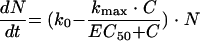 |
(5) |
where N is the number of bacteria in CFU/ml, k0 is the bacterial growth rate constant in the absence of drug, kmax is the maximum bacterial kill rate, C is the drug concentration, and EC50 is the drug concentration necessary to achieve half of the maximum effect.
If the bacteria are not in the logarithmic growth phase at time zero, an exponential correction factor (1 − e−Zt) may be necessary to compensate for this delay.
This model has been very useful to describe the effects of a number of beta lactam antibiotics (26, 89, 90). It could also be shown that the Emax model used for kill-curve fitting can be related to MICs.
Rearranging of equation 5 yields: equation 6
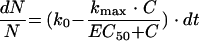 |
(6) |
which can be integrated on both sides: equation 7
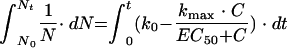 |
(7) |
where t is the incubation time necessary for measurement of the MIC, Nt is the number of bacteria at the end of the incubation period, and N0 is the initial bacterial inoculum. Solving and rearranging equation 7 yields: equation 8
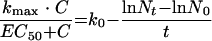 |
(8) |
where the right-hand term is a constant defined as d. Substituting the right-hand term with d and rearranging gives: equation 9
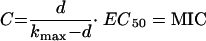 |
(9) |
Since this equation is true for any given concentration, it is also true for the MIC. The MIC was previously defined as the lowest concentration that completely inhibits visible growth of the organism as detected by the unaided eye after an 18- to 24-h incubation period (t) with a standard initial inoculum (N0) of approximately 105 CFU (87). Turbidity (visible growth) occurs at about 107 CFU/ml (Nt) (89).
AUC- and Dose-Based Analysis
Firsov et al. used a two-compartment in vitro dynamic model with antibiotic and bacterial dilution to study the effect of antibiotics and suggested two integral parameters to characterize antimicrobial effect duration (TE) and intensity (IE). The first parameter, TE is defined by the time from the moment of antibiotic administration to the moment when the bacterial count reaches its initial level again, and IE is the area between the microbial growth curves in the presence and absence of an antibiotic (36). These parameters were compared to 10 other indicators of bacterial killing and regrowth kinetics in terms of the respective AUC-response relationships as applied to the action of ciprofloxacin against E. coli. Comparisons were made on the basis of four criteria: relevance to be related to the AUC, sensitivity to the AUC, robustness and predictive ability in terms of IE. The authors conclude only IE and TE met all four criteria and propose IE as a more universal measure of the antimicrobial effect (37). Using the same model, commonly used predictors of antimicrobial effect (AUC/MIC, AUC > MIC, and t > MIC) were examined for pharmacokinetically different quinolones, trovafloxacin, and ciprofloxacin. Linear correlations were established between IE and log AUC/MIC, log AUC > MIC, and log t > MIC, the first two being specific for each drug and dosing regimen, allowing quantitative comparison of the effects (38, 39, 40, 41, 42).
A sigmoid dose-response model was used by Craig et al. to characterize the in vivo antimicrobial activity in an animal model: equation 10
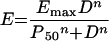 |
(10) |
where E is the observed effect at 24 h (measured as the reduction in log10 CFU/thigh or lung compared to untreated controls), D is the cumulative 24-h dose of amikacin, Emax is the maximum antimicrobial effect attributable to the drug, P50 is the 24-h dose producing 50% of Emax, and n is the slope of the dose-response relationship. Emax was calculated from the change in log10 CFU/thigh or lung over the 24 h of treatment, at the highest dose studied for each organism. P50 and n were calculated separately for each dosing interval using nonlinear regression.
A general shortcoming of AUC- and dose-based analyses is related to the fact that the parameters AUC and dose merely reflect single integrated parameters and do not reflect the dynamic profile of the drug concentration in vivo.
CONCLUSION: KILL CURVES VERSUS MIC
Traditionally dose and drug selection in antimicrobial therapy is based on a single static in vitro parameter, the MIC. In practice, however, an in vivo antimicrobial effect is rather the result of a dynamic exposure of the infective agent to the unbound antibiotic drug fraction at the relevant effect site. In light of this consideration, dynamic pharmacokinetic-pharmacodynamic approaches were developed to systematically elucidate the dynamic relationship between bacteria and anti-infectives. The concept of this pharmacokinetic-pharmacodynamic approach is to use the full concentration-profile versus effect rather than the MIC.
Few comparisons of the two approaches in the literature have been performed (18). It seems that the goal has been to minimize the available information and limit the pharmacodynamic input to the MIC. However, kill curve approaches and subsequent pharmacokinetic-pharmacodynamic analysis may provide more meaningful information about the interaction between bacteria and anti-infectives because these approaches describe this interaction in a more dimensional way by a dynamic integration of concentration and time and, hence, use the complete available information. In contrast, MIC measurements only provide a still photo of the effect of an antibiotic at a single concentration value. This value is regarded as an “all or nothing” threshold value, which would imply the antibiotic activity would be turned on or off above or below the MIC limit, respectively. However, this scenario does by no means reflect an in vivo setting where antibiotics actually also exert effects at very low concentrations.
It may therefore be concluded that a dynamic pharmacokinetic-pharmacodynamic approach based on kill curves is a more rational approach to describe drug-bacteria interactions than the classical MIC approach.
REFERENCES
- 1.Aeschlimann, J. R., and M. J. Rybak, 1998. Pharmacodynamic analysis of the activity of quinupristin-dalfopristin against vancomycin-resistant Enterococcus faecium with differing MBCs via time-kill-curve and postantibiotic effect methods. Antimicrob. Agents Chemother. 42:2188-2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Amyes, S. 1999. Ten years of ciprofloxacin: the past, present and future. Antimicrobial susceptibility. J. Antimicrob. Chemother. 43(Suppl. A):1-2. [DOI] [PubMed] [Google Scholar]
- 3.Andes, D. R., and W. A. Craig. 1998. Pharmacodynamics of fluoroquinolones in experimental models of endocarditis. Clin. Infect. Dis. 27:47-50. [DOI] [PubMed] [Google Scholar]
- 4.Andes, D. R., and W. A. Craig. 1999. Pharmacokinetics and pharmacodynamics of antibiotics in meningitis. Infect. Dis. Clin. North Am. 13:595-618. [DOI] [PubMed] [Google Scholar]
- 5.Bellido, F., J. C. Pechere, and E. W. Hancock. 1991. Novel method for measurement of outer membrane permeability to new b-lactams in intact Enterobacter cloacae cells. Antimicrob. Agents Chemother. 35:68-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bergan, T., I. B. Carlsen, and J. E. Fuglesang. 1980. An in vitro model for monitoring bacterial responses to antibiotic agents under simulated in vivo conditions. Infection. 8:S96-102. [DOI] [PubMed] [Google Scholar]
- 7.Bergan, T., and I. B. Carlsen. 1980. Bacterial kill rates of amoxycillin and ampicillin at exponentially diminishing concentrations simulating in vivo conditions. Infection. 8:S103-107. [DOI] [PubMed] [Google Scholar]
- 8.Bergan, T., and I. B. Carlsen. 1985. Effect of antibiotics eliminated by first order kinetics. J. Antimicrob. Chemother. 15:147-152. [DOI] [PubMed] [Google Scholar]
- 9.Birkness, K. A., M. Deslauriers, J. H. Bartlett, et al. 1999. An in vitro tissue culture bilayer model to examine early events in Mycobacterium tuberculosis infection. Infect. Immun. 67:653-658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Blaser, J., B. B. Stone, M. C. Groner, and S. H. Zinner. 1987. Comparative study with enoxacin and netilmicin in a pharmacodynamic model to determine importance of ratio of antibiotic peak concentration to MIC for bactericidal activity and emergence of resistance. Antimicrob. Agents Chemother. 31:1054-1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Blaser, J., and C. Konig. 1995. Once-daily dosing of aminoglycosides. Eur. J. Clin. Microbiol. Infect. Dis. 14:1029-1038. [DOI] [PubMed] [Google Scholar]
- 12.Blaser, J., B. B. Stone, and S. H. Zinner. 1985. Two compartment kinetic model with multiple artificial capillary units. J. Antimicrobial Chemotherapy. 15:131-137. [DOI] [PubMed] [Google Scholar]
- 13.Blaser, J., B. B. Stone, and S. H. Zinner. 1985. Efficacy of intermittent versus continuous administration of netilmicin in a two-compartment in vitro model. Antimicrob. Agents Chemother. 27:343-349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Blaser, J., B. Stone, M. C. Groner, and S. H. Zinner. 1985. Impact of netilmicin regimens on the activities of ceftazidime-netilmicin combinations against Pseudomonas aeruginosa in an in vitro pharmacokinetic model. Antimicrob. Agents Chemother. 28:64-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Blaser, J., P. Vergeres, A. F. Widmer, and W. Zimmerli. 1995. In vivo verification of in vitro model of antibiotic treatment of device-related infection. Antimicrob. Agents Chemother. 39:1134-1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chambers, H. F., and S. Kennedy. 1990. Effects of dosage, peak and trough concentrations in serum, protein binding, and bactericidal rate on efficacy of teicoplanin in a rabbit model of endocarditis. Antimicrob. Agents Chemother. 34:510-514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chambers, H. F., M. Sachdeva, and S. Kennedy. 1990. Binding affinity for penicillin-binding protein 2a correlates with in vivo activity of beta-lactam antibiotics against methicillin-resistant Staphylococcus aureus. J. Infect. Dis. 162:705-710. [DOI] [PubMed] [Google Scholar]
- 18.Corvaisier, S., P. H. Maire, X. Barbaut, N. Bleyzac, and R. W. Jelliffe. 1998. Comparisons between antimicrobial pharmacodynamic indices and bacterial killing as described by using the Zhi model. Antimicrob. Agents Chemother. 42:1731-1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Craig, W. A. 1998. Choosing an antibiotic on the basis of pharmacodynamics. Ear Nose Throat J. 77:7-11. [PubMed] [Google Scholar]
- 20.Craig, W. A. 1996. Antimicrobial resistance issues of the future. Diagn Microbiol Infect. Dis. 25:213-217. [DOI] [PubMed] [Google Scholar]
- 21.Craig, W. A. 1998. Pharmacokinetic/pharmacodynamic parameters: rationale for antibacterial dosing of mice and men. Clin. Infect. Dis. 26:1-10. [DOI] [PubMed] [Google Scholar]
- 22.Craig, W. A. 1995. Antibiotic selection factors and description of a hospital-based outpatient antibiotic therapy program in the USA Eur. J. Clin. Microbiol. Infect. Dis. 14:636-642. [DOI] [PubMed] [Google Scholar]
- 22a.Craig, W. A. 1995. Interrelationship between pharmacokinetics and pharmacodynamics in determining dosage regimens for broad-spectrum cephalosporins. Diagn Microbiol Infect. Dis. 22:89-96. [DOI] [PubMed] [Google Scholar]
- 23.Craig, W. A., and D. Andes. 1996. Pharmacokinetics and pharmacodynamics of antibiotics in otitis medium. Pediatr. Infect. Dis. J. 15:255-259. [DOI] [PubMed] [Google Scholar]
- 24.Craig, W. A., J. Redington, and S. C. Ebert. 1991. Pharmacodynamics of amikacin in vitro and in mouse thigh and lung infections. J. Antimicrob. Chemother. 27:29-40. [DOI] [PubMed] [Google Scholar]
- 25.Dalla Costa, T., and H. Derendorf. 1996. AUIC-a general target for the optimization of dosing regimens of antibiotics? Ann. Pharmacother. 30:1024-1028. [DOI] [PubMed] [Google Scholar]
- 26.Dalla Costa, T., A. Nolting, K. Rand, and H. Derendorf. 1997. Pharmacokinetic-pharmacodynamic modelling of the in vitro antiinfective effect of piperacillin-tazobactam combinations. Int. J. Clin. Pharmacol. Ther. 35:426-433. [PubMed] [Google Scholar]
- 27.Darouiche, R. O., A. Dhir, A. J. Miller, et al. 1994. Vancomycin penetration into biofilm covering infected prostheses and effect on bacteria. J. Infect. Dis. 170:720-723. [DOI] [PubMed] [Google Scholar]
- 28.Derendorf, H. 1989. Pharmacokinetic evaluation of beta-lactam antibiotics. J. Antimicrob. Chemother. 24:407-413. [DOI] [PubMed] [Google Scholar]
- 29.Derendorf, H., and B. Meibohm. 1999. Modeling of pharmacokinetic/pharmacodynamic (pharmacokinetic-pharmacodynamic) relationships: concepts and perspectives. Pharm. Res. 16:176-185. [DOI] [PubMed] [Google Scholar]
- 30.DiPiro, J. T., C. E. Edmiston, Jr., and J. M. Bohnen. 1996. Pharmacodynamics of antimicrobial therapy in surgery. Am. J. Surg. 171:615-622. [DOI] [PubMed] [Google Scholar]
- 31.Drake, T. A., W. M. Scheld, and M. A. Sande. 1985. Effects of sub-bactericidal concentrations of antibiotics in experimental models of endocarditis. J. Antimicrob. Chemother. 15:293-296. [DOI] [PubMed] [Google Scholar]
- 32.Drusano, G. L. 1998. Infection in the intensive care unit: beta-lactamase-mediated resistance among Enterobacteriaceae and optimal antimicrobial dosing. Clin. Infect. Dis. 27(Suppl. 1):S111-1116. [DOI] [PubMed] [Google Scholar]
- 33.Drusano, G. L. 1990. Human pharmacodynamics of beta-lactams, aminoglycosides and their combination. Scand. J. Infect. Dis. Suppl. 74:235-248. [PubMed] [Google Scholar]
- 34.Eagle, H., and A. D. Musselman. 1948. The rate of bacterial action of penicillin in vitro as a function of its concentration, and its paradoxically reduced activity at high concentrations against certain organisms. J. Exp. Med. 88:99-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Eden, T. 1985. Long-standing otitis medium with effusion-a convenient model for the study of antibiotic penetration to respiratory tract secretions. Scand. J. Infect. Dis. Suppl. 44:46-51. [PubMed] [Google Scholar]
- 36.Firsov, A. A., V. M. Chernykh, and S. M. Navashin. 1990. Quantitative analysis of antimicrobial effect kinetics in an in vitro dynamic model. Antimicrob. Agents Chemother. 34:1312-1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Firsov, A. A., S. N. Vostrov, A. A. Shevchenko, and G. Cornaglia. 1997. Parameters of bacterial killing and regrowth kinetics and antimicrobial effect examined in terms of area under the concentration-time curve relationships: action of ciprofloxacin against Escherichia coli in an in vitro dynamic model. Antimicrob. Agents Chemother. 41:1281-1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Firsov, A. A., A. A. Shevchenko, S. N. Vostrov, and S. H. Zinner. 1998. Inter- and intraquinolone predictors of antimicrobial effect in an in vitro dynamic model: new insight into a widely used concept. Antimicrob. Agents Chemother. 42:659-665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Firsov, A. A., S. N. Vostrov, A. A. Shevchenko, Y. A. Portnoy, and S. H. Zinner. 1998. A new approach to in vitro comparisons of antibiotics in dynamic models: equivalent area under the curve/MIC breakpoints and equiefficient doses of trovafloxacin and ciprofloxacin against bacteria of similar susceptibilities. Antimicrob. Agents Chemother. 42:2841-2847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Firsov, A. A., R. G. Vasilov, S. N. Vostrov, O. V. Kononenko, I. Y. Lubenko, and S. H. Zinner. 1999. Prediction of the antimicrobial effects of trovafloxacin and ciprofloxacin on staphylococci using an in-vitro dynamic model. J. Antimicrob. Chemother. 43:483-490. [DOI] [PubMed] [Google Scholar]
- 41.Firsov, A. A., S. N. Vostrov, O. V. Kononenko, S. H. Zinner, and Y. A. Portnoy. 1999. Prediction of the effects of inoculum size on the antimicrobial action of trovafloxacin and ciprofloxacin against Staphylococcus aureus and Escherichia coli in an in vitro dynamic model. Antimicrob. Agents Chemother. 43:498-502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Firsov, A. A., M. Ruble, D. Gilbert, et al. 1997. Net effect of inoculum size on antimicrobial action of ampicillin-sulbactam: studies using an in vitro dynamic model. Antimicrob. Agents Chemother. 41:7-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Flaherty, J. F., S. L. Barriere, J. Mordenti, J. G. Gambertoglio. 1987. Effect of Dose on Pharmacokinetics and Serum Bactericidal Activity of Mezlocillin. Antimicrob. Agents Chemother. 31:895-898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Flaherty, J. F., L. C. Rodondi, B. J. Guglielmo, J. C. Fleishaker, R. J. Townsend, and J. G. Gambertoglio. 1988. Comparative Pharmacokinetics and Serum Inhibitory Activity of Clindamycin in Different Dosing Regimens. Antimicrob. Agents Chemother. 32:1825-1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Francioli, P., and M. P. Glauser. 1985. Successful prophylaxis of experimental streptococcal endocarditis with single doses of sublethal concentrations of penicillin. J. Antimicrob. Chemother. 15:297-302. [DOI] [PubMed] [Google Scholar]
- 46.Frimodt-Moller, N., M. W. Bentzon, and V. F. Thomsen. 1986. Experimental infection with Streptococcus pneumoniae in mice: correlation of in vitro activity and pharmacokinetic parameters with in vivo effect for 14 cephalosporins. J. Infect. Dis. 154:511-517. [DOI] [PubMed] [Google Scholar]
- 47.Garrett, E. R. 1978. Kinetics of Antimicrobial Action. Scand. J. Infect. Dis. 14:54-85. [PMC free article] [PubMed] [Google Scholar]
- 48.Garrett, E. R., G. H. Miller, and M. R. Brown. 1966. Kinetics and mechanisms of action of antibiotics on microorganisms. V. Chloramphenicol and tetracycline affected Escherichia coli generation rates. J. Pharm. Sci. 55:593-600. [DOI] [PubMed] [Google Scholar]
- 49.Garrett, E. R., and H. Nolte. 1972. Kinetics and Mechanisms of Drug Action on Microorganisms. XIV. The Action of Fluorouracil, other Uracils and Derived Nucleosides on the Microbial Kinetics of Escherichia coli. Chemotherapy. 17:81-107. [DOI] [PubMed] [Google Scholar]
- 50.Garrett, E. R., and C. M. Won. 1973. Kinetics and Mechanisms of Drug Action on Microorganisms. XVII: Bactericidal Effects of Penicillin, Kanamicyn, and Rifampin with and without Organism Pretreatment with Bacteriostatic Chloramphenicol, Tetracycline, and Novobiocin. J. Pharm. Sci. 62:1666-1673. [DOI] [PubMed] [Google Scholar]
- 51.Gengo, F. M., T. W. Mannion, C. H. Nightingale, and J. J. Schentag. 1984. Integration of pharmacokinetics and pharmacodynamics of methicillin in curative treatment of experimental endocarditis. J. Antimicrob. Chemother. 14:619-631. [DOI] [PubMed] [Google Scholar]
- 52.Gerber, A. U., C. Feller, and H. P. Brugger. 1984. Time course of the pharmacological response to beta-lactam antibiotics in vitro and in vivo. Eur. J. Clin. Microbiol. 3:592-597. [DOI] [PubMed] [Google Scholar]
- 53.Grasso, S., G. Meinardi, de Carneri, I., and V. Tamassia. 1978. New in vitro model to study the effect of antibiotic concentration and rate of elimination on antibacterial activity. Antimicrob. Agents Chemother. 13:570-576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Greenwood, D., and F. O'Grady. 1978. An in vitro model of the urinary bladder. J. Antimicrob. Chemother. 4:113-120. [DOI] [PubMed] [Google Scholar]
- 55.Greenwood, D. 1985. An in-vitro model simulating the hydrokinetic aspects of the treatment of bacterial cystitis. J. Antimicrob. Chemother. 15:103-109. [DOI] [PubMed] [Google Scholar]
- 56.Hoffman, A., and D. Stepensky. 1999. Pharmacodynamic aspects of modes of drug administration for optimization of drug therapy. Crit Rev Ther Drug Carrier Syst. 16:571-639. [PubMed] [Google Scholar]
- 57.Hollenstein, U., M. Brunner, B. X. Mayer, et al. 2000. Target site concentrations after continuous infusion and bolus injection of cefpirome to healthy volunteers. Clin. Pharmacol. Ther. 67:229-236. [DOI] [PubMed] [Google Scholar]
- 58.Hulten, K., R. Rigo, I. Gustafsson, and L. Engstrand. 1996. New pharmacokinetic in vitro model for studies of antibiotic activity against intracellular microorganisms. Antimicrob. Agents Chemother. 40:2727-2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hyatt, J. M., P. S. McKinnon, G. S. Zimmer, and J. J. Schentag. 1995. The importance of pharmacokinetic/pharmacodynamic surrogate markers to outcome. Focus on antibacterial agents. Clin. Pharmacokinet. 28:143-160. [DOI] [PubMed] [Google Scholar]
- 60.Hyatt, J. M., D. E. Nix, and J. J. Schentag. 1994. Pharmacokinetic and pharmacodynamic activities of ciprofloxacin against strains of Streptococcus pneumoniae, Staphylococcus aureus, and Pseudomonas aeruginosa for which MICs are similar. Antimicrob. Agents Chemother. 38:2730-2737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hyatt, J. M., and J. J. Schentag. 2000. Pharmacodynamic modeling of risk factors for ciprofloxacin resistance in Pseudomonas aeruginosa. Infect. Control Hosp. Epidemiol. 21:S9-11. [DOI] [PubMed] [Google Scholar]
- 62.Hyatt, J. M., and J. J Schentag. 2000. Potential role of pharmacokinetics, pharmacodynamics, and computerized databases in controlling bacterial resistance. Infect. Control Hosp. Epidemiol. 21:S18-21. [DOI] [PubMed] [Google Scholar]
- 63.Jusko, W. J. 1971. Pharmacodynamics of chemotherapeutic effects: dose-time-response relationships for phase-nonspecific agents. J. Pharm. Sci. 60:892-895. [DOI] [PubMed] [Google Scholar]
- 64.Keil, S., and B. Wiedemann. 1995. Mathematical corrections for bacterial loss in pharmacodynamic in vitro dilution models. Antimicrob. Agents Chemother. 39:1054-1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Knudsen, J. D., K. Fuursted, S. Raber, F. Espersen, and Frimodt- N. Moller. 2000. Pharmacodynamics of glycopeptides in the mouse peritonitis model of streptococcus pneumoniae or staphylococcus aureus infection. Antimicrob. Agents Chemother. 44:1247-1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.König, P., J. P. Guggenbichler, E. Semenitz, and W. Foisner. 1986. Kill kinetics of bacteria under fluctuating concentrations of various antibiotics. I. Description of the model. Chemotherapy. 32:37-43. [DOI] [PubMed] [Google Scholar]
- 67.König, P., J. P. Guggenbichler, E. Semenitz, and W. Foisner. 1986. Kill Kinetics of bacteria under Fluctuating Concentrations of Various Antibiotics. II. Description of Experiments. Chemotherapy. 32:44-58. [DOI] [PubMed] [Google Scholar]
- 68.Kutlin, A., P. M. Roblin, and M. R. Hammerschlag. 1999. In vitro activities of azithromycin and ofloxacin against Chlamydia pneumoniae in a continuous-infection model. Antimicrob. Agents Chemother. 43:2268-2272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ledergerber, B., J. Blaser, and R. Luthy. 1985. Computer-controlled in-vitro simulation of multiple dosing regimens. J. Antimicrob. Chemother. 15:169-173. [DOI] [PubMed] [Google Scholar]
- 70.Leggett, J. E., B. Fantin, S. Ebert, et al. 1989. Comparative antibiotic dose-effect relations at several dosing intervals in murine pneumonitis and thigh-infection models. J. Infect. Dis. 159:281-292. [DOI] [PubMed] [Google Scholar]
- 71.Li, R. C., M. Zhu, and J. J. Schentag. 1999. Achieving an optimal outcome in the treatment of infections. The role of clinical pharmacokinetics and pharmacodynamics of antimicrobials. Clin. Pharmacokinet. 37:1-16. [DOI] [PubMed] [Google Scholar]
- 72.List, T. P. 1996. Continuous infusion of beta-lactam antibiotics: a potential strategy to improve parenteral antimicrobial therapy. A Consensus Document. University of Oklahoma, Oklahoma City, Okla.
- 73.Löwdin, E., I. Odenholt, S. Bengtsson, and O. Cars. 1996. Pharmacodynamic effects of sub-MICs of benzylpenicillin against Streptococcus pyogenes in a newly developed in vitro kinetic model. Antimicrob. Agents Chemother. 40:2478-2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Löwdin, E., I. Odenholt, and O. Cars. 1998. In vitro studies of pharmacodynamic properties of vancomycin against Staphylococcus aureus and Staphylococcus epidermidis. Antimicrob. Agents Chemother. 42:2739-2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mattie, H. 1981. Kinetics of antimicrobial action. Rev. Infect. Dis. 3:19-27. [DOI] [PubMed] [Google Scholar]
- 76.Mattie, H. 1978. A Mathematical Description of Short-Term Effects of Beta-lactam Antibiotics on Bacterial Growth In Vitro. Curr. Microbiol. 1:105-109. [Google Scholar]
- 77.Mattie, H., and van der Voet, G. B. 1979. Influence of aminopenicillins on bacterial growth kinetics in vitro in comparison with the antibacterial effect in vivo. Infection. 7:S434-437. [DOI] [PubMed] [Google Scholar]
- 78.McCormack, J. P., and J. J. Schentag. 1987. Potential Impact of Quantitative Susceptibility Tests on the Design of Aminoglycoside Dosing Regimens. Drug Intelligence Clin. Pharm. 21:187-191. [PubMed] [Google Scholar]
- 79.McGrath, B. J., S. L. Kang, G. W. Kaatz, and M. J. Rybak. 1994. Bactericidal activities of teicoplanin, vancomycin, and gentamicin alone and in combination against Staphylococcus aureus in an in vitro pharmacodynamic model of endocarditis. Antimicrob. Agents Chemother. 38:2034-2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Meibohm, B., and H. Derendorf. 1997. Basic concepts of pharmacokinetic/pharmacodynamic (pharmacokinetic-pharmacodynamic) modelling. Int. J. Clin. Pharmacol. Ther. 35:401-413. [PubMed] [Google Scholar]
- 81.Mentec, H., J. M. Vallois, A. Bure, et al. 1992. Piperacillin, tazobactam, and gentamicin alone or combined in an endocarditis model of infection by a TEM-3-producing strain of Klebsiella pneumoniae or its susceptible variant. Antimicrob. Agents Chemother. 36:1883-1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mielck, J. B., and E. R. Garrett. 1969. Kinetics and mechanisms of drug action on microorganisms. IX. Inhibitory action of lincomycin on Escherichia coli by microbial kinetics. Chemotherapy. 14:337-355. [DOI] [PubMed] [Google Scholar]
- 83.Moise, A., and J. Schentag. 1998. Pharmacokinetic and pharmacodynamic modelling of antibiotic therapy. Curr. opinion in Infectious Diseases. 11:673-680. [DOI] [PubMed] [Google Scholar]
- 84.Moore, R. D., P. S. Lietman, and C. R. Smith. 1987. Clinical response to aminoglycoside therapy: importance of the ratio of peak concentration to minimal inhibitory concentration. J. Infect. Dis. 155:93-99. [DOI] [PubMed] [Google Scholar]
- 85.Mordenti, J. J., R. Quintiliani, and C. H. Nightingale. 1985. Combination antibiotic therapy: comparison of constant infusion and intermittent bolus dosing in an experimental animal model. J. Antimicrob. Chemother. 15:313-321. [DOI] [PubMed] [Google Scholar]
- 86.Murakawa, T., H. Sakamoto, T. Hirose, and M. Nishida. 1980. New in vitro kinetic model for evaluating bactericidal efficacy of antibiotics. Antimicrob. Agents Chemother. 18:377-381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.National Committee for Clinical Laboratory Standards. 1997. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria that grow aerobically. 4th. ed. Approved Standard. NCCLS Publication No. M7-A4. National Committee for Clinical Laboratory Standards, Villanova, Pa.
- 88.Nightingale, C. 1980. Pharmacokinetics of the oral cephalosporins in adults. J. Int. Med. Res. 8:2-8. [PubMed] [Google Scholar]
- 89.Nolting, A. 1994. Pharmacokinetic-pharmacodynamic modeling of the antibacterial effects of piperacillin. University of Florida, Gainesville, Fla.
- 90.Nolting, A., Dalla Costa, T., K. H. Rand, and H. Derendorf. 1996. Pharmacokinetic-pharmacodynamic modeling of the antibiotic effect of piperacillin in vitro. Pharm. Res. 13:91-96. [DOI] [PubMed] [Google Scholar]
- 91.Odenholt, T. I. 1989. Pharmacodynamics of beta-lactam antibiotics. Studies on the paradoxical and postantibiotic effects in vitro and in an animal model. Scand. J. Infect. Dis. Suppl. 58:1-55. [DOI] [PubMed] [Google Scholar]
- 92.Onyeji, C. O., D. P. Nicolau, C. H. Nightingale, and R. Quintiliani. 1994. Optimal times above MICs of ceftibuten and cefaclor in experimental intra-abdominal infections. Antimicrob. Agents Chemother. 38:1112-1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Palmer, S. M., S. L. Kang, D. M. Cappelletty, and M. J. Rybak. 1995. Bactericidal killing activities of cefepime, ceftazidime, cefotaxime, and ceftriaxone against Staphylococcus aureus and beta-lactamase-producing strains of Enterobacter aerogenes and Klebsiella pneumoniae in an in vitro infection model. Antimicrob. Agents Chemother. 39:1764-1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Palmer, S. M., and M. J. Rybak. 1997. An evaluation of the bactericidal activity of ampicillin/sulbactam, piperacillin/tazobactam, imipenem or nafcillin alone and in combination with vancomycin against methicillin-resistant Staphylococcus aureus (MRSA) in time-kill curves with infected fibrin clots. J. Antimicrob. Chemother. 39:515-518. [DOI] [PubMed] [Google Scholar]
- 95.Peterson, L. R., J. A. Moody, C. E. Fasching, and D. N. Gerding. 1989. Influence of protein binding on therapeutic efficacy of cefoperazone. Antimicrob. Agents Chemother. 33:566-568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Reeves, D. S. 1985. Advantages and disadvantages of an in-vitro model with two compartments connected by a dialyser: results of experiments with ciprofloxacin. J. Antimicrob. Chemother. 15:159-167. [DOI] [PubMed] [Google Scholar]
- 97.Reigner, B. G., P. E. Williams, I. H. Patel, et al. 1997. An evaluation of the integration of pharmacokinetic and pharmacodynamic principles in clinical drug development. Experience within Hoffmann La Roche. Clin. Pharmacokinet. 33:142-152. [DOI] [PubMed] [Google Scholar]
- 98.Renard, L., P. Sanders, M. Laurentie, and J. M. Delmas. 1996. Pharmacokinetic-pharmacodynamic model for spiramycin in staphylococcal mastitis. J. Vet. Pharmacol. Ther. 19:95-103. [DOI] [PubMed] [Google Scholar]
- 99.Sanchez-Navarro, A., and Sanchez Recio, M. M. 1999. Basis of anti-infective therapy: pharmacokinetic-pharmacodynamic criteria and methodology for dual dosage individualisation. Clin. Pharmacokinet. 37:289-304. [DOI] [PubMed] [Google Scholar]
- 100.Sanchez-Recio, M. M., C. I. Colino, and Sanchez-Navarro, A. 2000. A retrospective analysis of pharmacokinetic/pharmacodynamic indices as indicators of the clinical efficacy of ciprofloxacin. J. Antimicrob. Chemother. 45:321-328. [DOI] [PubMed] [Google Scholar]
- 101.Sanfilippo, A., and E. Morvillo. 1968. An Experimental Model for the Study of the Antibacterial Activity of the Sulfonamides. Chemotherapy. 13:54-60. [DOI] [PubMed] [Google Scholar]
- 102.Sanfilippo, A., and G. Schioppacassi. 1973. New approach to the evaluation of antibacterial activity of aminosidine. Chemotherapy. 18:297-303. [DOI] [PubMed] [Google Scholar]
- 103.Sano, M., Y. Kumamoto, M. Nishimura, T. Tsukamoto, T. Hirose, and S. Ohya. 1994. Inhibition of biofilm formation by clarithromycin (CAM) in an experimental model of complicated bladder infection-in vitro study using automated simulation of urinary antimicrobial concentration. Kansenshogaku Zasshi. 68:1306-1317. [DOI] [PubMed] [Google Scholar]
- 104.Schentag, J. J. 1999. Pharmacokinetic and pharmacodynamic surrogate markers: studies with fluoroquinolones in patients. Am. J. Health Syst Pharm. 56:S21-4. [DOI] [PubMed] [Google Scholar]
- 105.Schentag, J. J. 1999. Antimicrobial action and pharmacokinetics/pharmacodynamics: the use of AUIC to improve efficacy and avoid resistance. J. Chemother. 11:426-439. [DOI] [PubMed] [Google Scholar]
- 106.Schentag, J. J., D. E. Nix, and M. H. Adelman. 1991. Mathematical examination of dual individualization principles (I): Relationships between AUC above MIC and area under the inhibitory curve for cefmenoxime, ciprofloxacin, and tobramycin. Ann. Pharmacother. Dicp. 25:1050-1057. [DOI] [PubMed] [Google Scholar]
- 107.Schentag, J. J., Strenkoski-Nix, L. C., D. E. Nix, and A. Forrest. 1998. Pharmacodynamic interactions of antibiotics alone and in combination. Clin. Infect. Dis. 27:40-46. [DOI] [PubMed] [Google Scholar]
- 108.Stratton, C. W., M. P. Weinstein, and L. B. Reller. 1982. Correlation of serum bactericidal activity with antimicrobial agent level and minimal bactericidal concentration. J. Infect. Dis. 154:160-168. [DOI] [PubMed] [Google Scholar]
- 109.Thauvin, C., G. M. Eliopoulos, S. Willey, C. Wennersten, and R. C. Moellering, Jr. 1974. Continuous-infusion ampicillin therapy of enterococcal endocarditis in rats. Antimicrob. Agents Chemother. 31:139-143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Thomas, J. K., A. Forrest, S. M. Bhavnani, et al. 1998. Pharmacodynamic evaluation of factors associated with the development of bacterial resistance in acutely ill patients during therapy. Antimicrob. Agents Chemother. 42:521-527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Tran, J. Q., C. H. Ballow, A. Forrest, et al. 2000. Comparison of the abilities of grepafloxacin and clarithromycin to eradicate potential bacterial pathogens from the sputa of patients with chronic bronchitis: influence of pharmacokinetic and pharmacodynamic variables. J. Antimicrob. Chemother. 45(Suppl.)B:9-17. [DOI] [PubMed] [Google Scholar]
- 112.Turnidge, J. D. 1998. The pharmacodynamics of beta-lactams. Clin. Infect. Dis. 27:10-22. [DOI] [PubMed] [Google Scholar]
- 113.Vance-Bryan, K., T. A. Larson, M. W. Garrison, et al. 1992. An in vitro pharmacodynamic model to simulate antibiotic behavior of acute otitis medium with effusion. Pharmaceutical Research. 9:920-924. [DOI] [PubMed] [Google Scholar]
- 114.van der Voet, G. B., H. Mattie, and van Furth, R. 1985. Comparison of the antibacterial activity of azlocillin and ticarcillin in vitro and in irradiated neutropenic mice. J. Antimicrob. Chemother. 16:605-613. [DOI] [PubMed] [Google Scholar]
- 115.Vogelman, B., S. Gudmundsson, J. Leggett, J. Turnidge, S. Ebert, and W. A. Craig. 1988. Correlation of antimicrobial pharmacokinetic parameters with therapeutic efficacy in an animal model. J. Infect. Dis. 158:831-847. [DOI] [PubMed] [Google Scholar]
- 116.Vondracek, T. G. 1995. Beta-lactam antibiotics: is continuous infusion the preferred method of administration? Ann. Pharmacother. 29:415-424. [DOI] [PubMed] [Google Scholar]
- 117.White, C. A., R. D. Toothaker, A. L. Smith, and J. T. Slattery. 1987. Correction for bacterial loss in in vitro dilution models. Antimicrob. Agents Chemother. 31:1859-1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zhi, J., C. H. Nightingale, and R. Quintiliani. 1988. Microbial pharmacodynamics of piperacillin in neutropenic mice of systematic infection due to Pseudomonas aeruginosa. J. Pharmacokinet. Biopharm. 16:355-375. [DOI] [PubMed] [Google Scholar]
- 119.Zinner, S. H., and J. Blaser. 1986. In-vitro studies of antibiotic combinations with special emphasis on the evaluation of newly developed methods. J. Antimicrob. Chemother. 17:1-5. [DOI] [PubMed] [Google Scholar]
- 120.Zinner, S. H., J. Blaser, B. B. Stone, and M. C. Groner. 1985. Use of an in-vitro kinetic model to study antibiotic combinations. J. Antimicrob. Chemother. 15:221-226. [DOI] [PubMed] [Google Scholar]