

Abstract
Adhesion of epithelium to the extracellular matrix is crucial for the maintenance of systemic and oral health. In the oral cavity, teeth or artificial dental implants penetrate the soft tissue of the gingiva. In this interface, gingival soft tissue needs to be well attached via the epithelial seal to the tooth or implant surface to maintain health. After injury or wounding, epithelial tissue rapidly migrates to form the initial epithelial cover to restore the barrier against infection. These events are crucially dependent on deposition of extracellular matrix and proper activation and function of integrin receptors in the epithelial cells. Recent experimental evidence suggests that epithelial integrins also participate in the regulation of periodontal inflammation. In this review, we will discuss the structure and function of epithelial integrins and their extracellular ligands and elaborate on their potential role in disease and repair processes in the oral cavity.
Keywords: wound healing, receptors, extracellular matrix (ECM), cell-matrix interactions, gingiva, keratinocyte(s)
Integrins
Integrins are cell adhesion receptors that bind to extracellular matrix ligands, such as fibronectin and collagens. An overview of integrins is presented in the Appendix. Briefly, all integrins are products of two separate genes encoding specific α and ß subunits (Fig. 1). Inside the cell, the cytoplasmic domains of integrins associate with cytoskeletal proteins (Fig. 2). Integrins can be activated by ligand binding or via intracellular processes (Fig. 2). Integrins mediate information from the extracellular matrix (ECM) into the cell in a two-way process that regulates gene expression, cell proliferation, and cell migration (Fig. 3).
Figure 1.
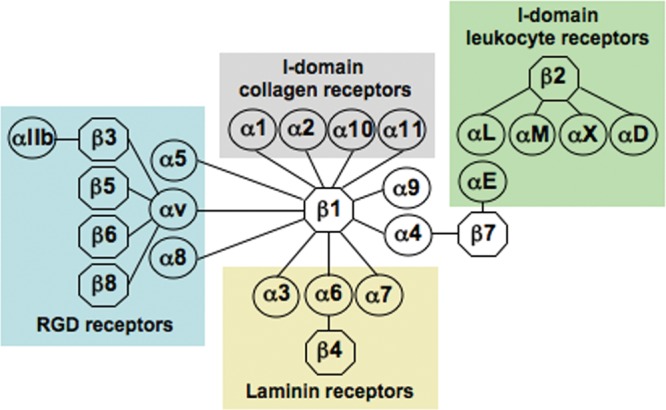
Schematic presentation of the integrin family. Modified from Hynes, 2002.
Figure 2.
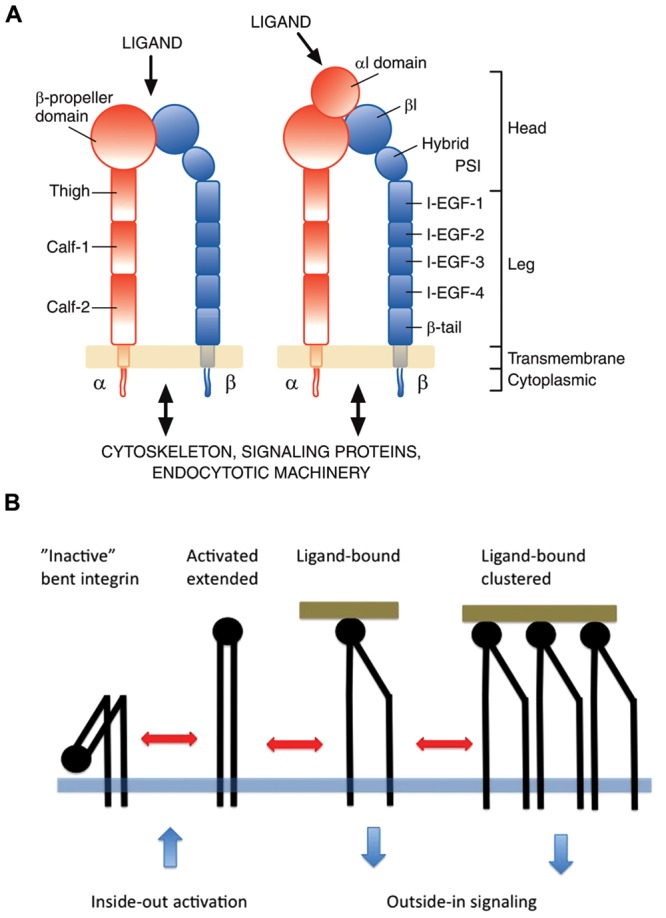
Basic domain structure of integrins (A) and integrin I-domain conformations (B). (A) The domain structure of an integrin. Nine out of 18 human integrin alpha subunits have an inserted domain (alphaI or alphaA domain) (on right) that is missing in the other 9 alpha subunits (on left). Otherwise the extracellular parts of all human integrins have identical domain structures. In the alphaI domain receptors, this domain forms the ligand-binding site. In the other integrins, ligands bind to the beta-propeller (alpha subunit)–betaI domain (beta subunit) interface. Both subunits have single transmembrane domains and short cytoplasmic domains (with the exception of the beta4 subunit, not shown in the Fig.). Cytoplasmic domains mediate the interaction of integrins with cytoskeletal proteins, signaling networks, and endocytotic machinery. I-EGF1-4, integrin epidermal growth factor-like domains 1-4; PSI/Hybrid, plexin-semaphorin-integrin domain/hybrid domain. (B) In an “inactive”, “bent” conformation, the ligand-binding head domain of an integrin points toward plasma membrane. Intracellular signals can activate the integrins and force them to “stand up”. The “extended” integrin binds to a ligand, which causes further changes in the conformation, e.g., the separation of the leg parts and the intracellular domains. Multivalent ligands can cluster integrins, which also modifies signaling.
Figure 3.
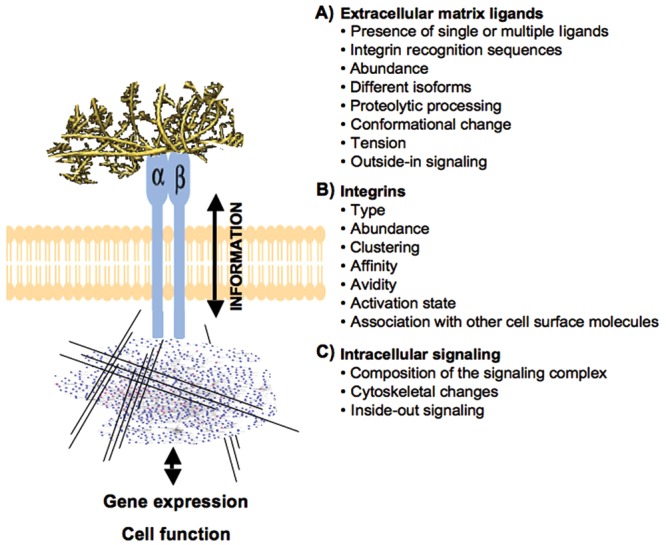
Summary of the key factors regulating integrin-mediated interactions and information exchange between the cell and extracellular matrix (ECM). Integrins mediate cell adhesion and migration on the ECM and function as two-way mediators of information between the ECM and cells. (A) While attaching the cells to the pericellular matrix, integrins also sense changes in the properties (e.g., tension, proteolytic processing, conformational change) and composition of the ECM and relay this information outside-in to the cell. (B) The information exchange through integrins is modulated by and depends on several integrin-related factors, including integrin type, abundance, clustering, etc. (C) Interaction with the matrix triggers distinct signaling cascades and cytoskeletal changes allowing the cell to adapt its functions appropriately. Reciprocally, changes in the function of intracellular signaling networks or cytoskeleton modulate integrin activity that can then lead to appropriate changes in the cells’ interactions with the ECM.
Adhesion Mechanisms of Junctional Epithelium to Tooth Surface
Junctional epithelium (JE) forms a non-keratinized thin structure that attaches the gingival soft tissue to tooth enamel or cementum (reviewed in Bosshardt and Lang, 2005). JE undergoes continuous renewal by active cell proliferation of basal epithelial cells (keratinocytes) both on the connective tissue side and against the hard tissue. Because of its unique location between hard and soft tissue, JE serves a crucial protective role against bacterial and physical insults. Intercellular junctions are relatively loose in JE that contains only a few desmosomes, adherens junctions, and gap junctions, thus allowing tissue exudate and inflammatory cells to penetrate toward the gingival sulcus (Bosshardt and Lang, 2005).
Unique to JE, it has a true basement membrane toward the connective tissue of gingiva (called the external basal lamina, EBL) and a simple ECM (called the internal basal lamina, IBL) against the enamel. The EBL contains the very same structures seen in typical basement membranes, namely, lamina lucida against the basal keratinocytes and lamina densa toward the connective tissue stroma. The IBL differs significantly from a typical basement membrane in terms of its protein composition (Table, A). All classic basement membrane zone proteins, including laminin 111, laminin 511, type IV and VII collagens, and perlecan, are absent from the IBL (Hormia et al., 1998). The main cell adhesion protein identified so far in the IBL is laminin 332 (previously called laminin 5), which is also present in the EBL (Hormia et al., 1998, 2001; Oksonen et al., 2001). Curiously, 2 proteins that are not commonly found in other epithelial basement membranes, namely, type VIII collagen and versican, have also been reported to be present at the JE-tooth interface (Salonen et al., 1991; Abiko et al., 2001). In addition, other proteins may also be present, such as tenascin-C (Ghannad et al., 2008). Likely more proteins will be found with emerging proteomics techniques.
Table.
(A) Molecular Composition of the External (EBL) and Internal (IBL) Basal Lamina of the Junctional Epithelium; (B) Integrin Expression in the Junctional Epithelium (JE), Oral Keratinized Gingival Epithelium (GE), and Wound Epithelium of Keratinized Gingiva (WGE) (See text for references.)
| A. | Basal Lamina Component | EBL | IBL | B. | Integrin | JE | GE | WGE |
|---|---|---|---|---|---|---|---|---|
| LM111 | √ | – | α2ß1 | √ | √ | √ | ||
| LM332 | √ | √ | α3ß1 | √ | √ | √ | ||
| LM511 | √ | – | α5ß1 | √* | – | √ | ||
| Type IV collagen | √ | – | α9ß1 | ? | √ | √ | ||
| Type VIII collagen | ? | √ | α6ß4 | √ | √ | √ | ||
| Perlecan | √ | – | αvß1 | ? | – | √ | ||
| Versican | ? | √ | αvß6 | √ | – | √ | ||
| Tenascin-C | √ | √* |
Molecule present; √*Variable expression (unpublished results); –Molecule not present; ?Not reported.
Basal keratinocytes adhere to the IBL via hemidesmosomes (reviewed in Bosshardt and Lang, 2005). At the most apical aspect of the JE, basal cells synthesizing both IBL and EBL are very close together, and it is unlikely that soluble mediators would be sufficiently different to regulate such dissimilar gene expression profiles. Although molecular cues from the mineralized matrix of the tooth may also play some role, it is more likely that lack of fibroblast influence (cross-talk) during the formation of IBL limits basal keratinocyte gene expression to a simpler variety. Consistent with this hypothesis is the fact that normal basement membranes are jointly produced by basal keratinocytes and fibroblasts, which have extensive cross-talk through paracrine-soluble mediators (Smola et al., 1998). Without the presence of fibroblasts, keratinocytes continue to express laminin 332, but fail to deposit laminin 111 and type IV collagen (Smola et al., 1998), mimicking the situation at the IBL zone. Recent investigations have demonstrated that, in murine JE, basal cells at the IBL express more than 10 times the laminin 332 transcript found in EBL or in oral epithelium (Kinumatsu et al., 2009). Laminin 332 expression is stimulated by several growth factors and cytokines, including TGFß1, tumor necrosis factor-α, keratinocyte growth factor, EGF, and interferon-γ (Kainulainen et al., 1998; Amano et al., 2004). Many of these factors are constitutively expressed at the JE and may, therefore, be responsible for the abundance of laminin 332 in the IBL (Li et al., 2005; Ghannad et al., 2008). The function of laminin 332 in the IBL may vary depending on the processing of the molecule (see Appendix). It may also regulate granulation tissue formation (see Appendix).
Expression of integrins in normal JE differs from that of basal cells in the intact oral gingival epithelium (Table, B). Interestingly, healthy JE appears to express integrins that are similar to the ones present in keratinocytes in the oral mucosa and skin during wound re-epithelialization, including expression of αvß6 integrin (see below). This suggests that JE cells are phenotypically unique and/or that they remain constantly in an activated state similar to wound healing (Table, B). In general, basal keratinocytes, including epithelial cells of the JE, interact with the C-terminal LG domains of the α3 chain of laminin 332 via α3ß1 and α6ß4 integrins (Fig. 4A; Aumailley et al., 2003; see below). Integrin α6ß4 is a crucial part of the hemidesmosome, where it binds to processed laminin 332 (reviewed in Litjens et al., 2006). This binding not only supports the firm adhesion of basal keratinocytes but also maintains cell proliferation (Murgia et al., 1998). Individuals with mutations in either α6 or ß4 integrins have junctional epidermolysis bullosa similar to patients with mutations in laminin 332 (Litjens et al., 2006), but it is unclear if periodontal tissues are also affected in these patients. At the IBL, α6ß4 integrin co-localizes with laminin 332 (Hormia et al., 1992, 2001), suggesting that their interaction is the main mechanism holding the JE attached to the mineralized tissue. Intracellularly, α6ß4 integrin binds plectin to a complex that accumulates BP180 and BP230 (Hormia et al., 2001; Litjens et al., 2006). These interactions support the mechanical stability of hemidesmosomes.
Figure 4.
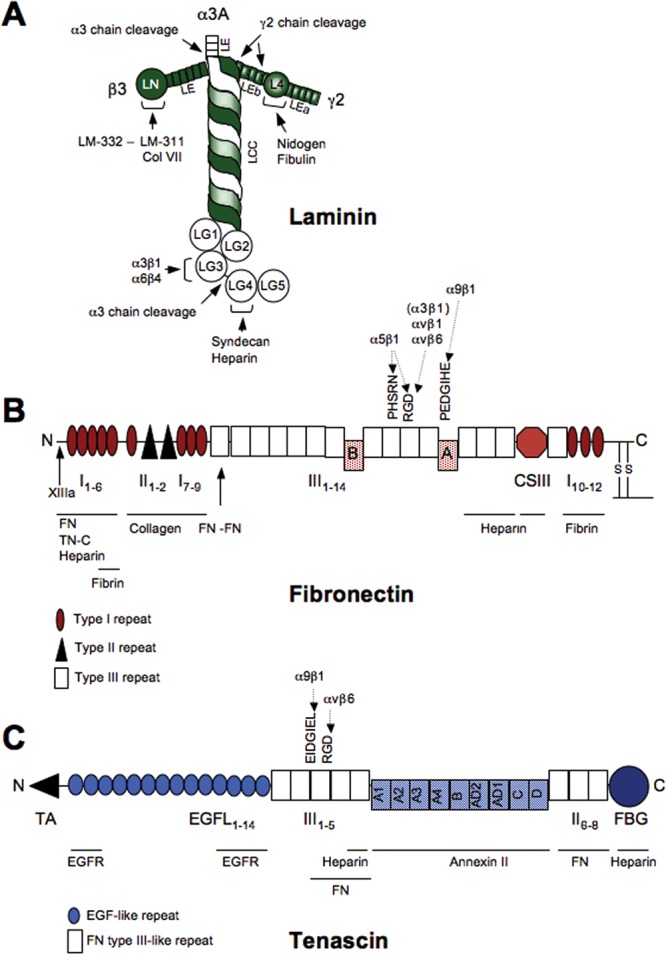
Structural and functional domains, major matrix-binding sites, and adhesion sites for epithelial integrins in laminin 332, fibronectin, and tenascin-C. Additional binding sites for non-epithelial integrins exist. (A) Laminin-332 is a T-shaped molecule consisting of 3 polypeptide chains, α3A (or long isoform α3B), ß3, and γ2. (B) Fibronectin protomer consists of 2 similar subunits linked in an antiparallel orientation by 2 disulphide bridges at their C-termini. It has 3 sites of alternative splicing: Type III repeats A and B can be independently included or excluded to form cellular fibronectin isoforms EDA and EDB, respectively. Splicing within the CSIII segment can produce several variations. (C) The N-termini of 3 tenascin-C monomers are joined via their TA domains to form a trimer. Two trimers are further linked via a disulfide bond to form a hexamer. Nine type III repeats (A-D) can be independently included or excluded to produce different isoforms.
During coronal migration of the DAT cells (see Appendix), hemidesmosomes are disassembled to allow for cell movement. Although the regulation of this process in JE is not fully understood, it is believed to start with phosphorylation of the ß4 integrin cytoplasmic domain, which leads to a disassociation between ß4 integrin and plectin (Litjens et al, 2006; Wilhelmsen et al., 2007). There are some indications that at least α5ß1 integrin and tenascin-C may also be present in JE (see Table). Thus, further research is needed to clarify the exact roles of α6ß4, α3ß1, and other integrins in keratinocyte migration in the JE and during periodontal pocket formation. Understanding how keratinocytes migrate on the IBL is also challenging, because the only “certified” ECM ligand consistently present at that location is laminin 332, whose proteolytic processing stage remains unknown.
Adhesion of Peri-Implant Epithelium
The dimension of the dento-gingival complex (distance from the gingival margin to bone) has been reported to be slightly greater for oral implants (2.85-3.80 mm) than the corresponding dimension of this complex around teeth (2.73-3.25 mm), regardless of whether the implants have been submerged (Buser et al., 1992; Abrahamsson et al., 1996, 1999; Berglundh and Lindhe, 1996; Cochran et al., 1997; Hermann et al., 2001; Fig. 5). The biological width around natural teeth has been reported to be about 2 mm, composed of 1 mm of epithelial attachment mediated by the JE and 1 mm of gingival connective tissue attachment (Gargiulo et al., 1961; Vacek et al., 1994). Many studies have reported that the peri-implant JE is about 2 mm long (Myshin and Wiens, 2005; Rompen et al., 2006; references above). Thus, the increase in the dimension of the dento-gingival complex around oral implants is largely due to the increase in the length of the peri-implant JE, suggesting that conventional implant surfaces cannot deter the formation of “long epithelial attachment” (Fig. 5). Many of these studies have demonstrated the presence of JE with hemidesmosomal attachments to the implant surface, and at the light-microscopic level, the epithelium appeared to be “attached” (see e.g., Gould et al., 1984; reviewed in Rompen et al., 2006). More recent animal studies, however, challenge the presence of a true JE on dental implants (Ikeda et al., 2000, 2002; Fujiseki et al., 2003; Atsuta et al., 2005a,b). In a fairly recent study, peri-implant epithelium (PIE) appeared to “lean” on the implant, but was structurally very different from JE, displaying slower cell proliferation, weaker expression of JE differentiation marker cytokeratin 19, and no evidence of direct adhesion of the PIE on the implant surface (Fujiseki et al., 2003). Earlier studies have also shown slower proliferation of PIE (Inoue et al., 1997). Other studies indicate that only the bottom third of the PIE is actually attached to the implant (Ikeda et al., 2000). Consistent with the latter paper, laminin 332 expression between PIE and the implant surface also appears to be limited to the lower part of the the PIE (Atsuta et al., 2005a,b; Fig. 5), but very little is known about the expression of integrins in the PIE. Suboptimal attachment of the PIE may contribute to the formation of inflammatory lesions and bone loss around the implants, which has become a common clinical problem (Roos-Jansåker et al., 2006; Máximo et al., 2008; Koldsland et al., 2010). It is possible that poor PIE adhesion allows for apical migration of plaque biofilm and could, therefore, directly explain inflammation and bone loss around bone-level dental implants. Future research should focus on improving epithelial attachment on implants and especially on different abutments, which mediate soft-tissue adhesion in 2-piece implant systems.
Figure 5.
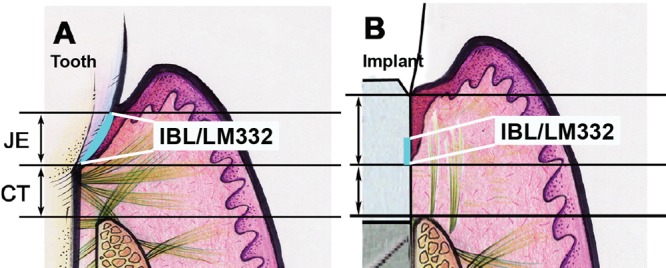
Comparison of epithelial adhesion to the natural tooth (A) and dental implant surface (B). Epithelium attaches to the implant by a longer junctional peri-implant epithelium (JE) as compared with the tooth. Internal basal lamina (IBL) containing laminin 332 (LM332) may be present only in the apical third of the implant-JE interface. CT, connective tissue.
Failure to Activate Integrins is Linked to Periodontal Disease
Kindlins (kindlin-1, -2, and -3) comprise a family of 3 related proteins that are involved in integrin activation inside cells (Larjava et al., 2008; Meves et al., 2009). Mutations in kindlin-1 and kindlin-3 are associated with human clinical syndromes (Larjava et al., 2008). Loss of kindlin-1 causes Kindler syndrome, which is a rare skin-blistering disorder with oral manifestations that include development of early-onset aggressive periodontitis (reviewed in Wiebe et al., 2008). In healthy individuals, kindlin-1 is localized in basal keratinocytes of oral epithelia, while it is absent in patients with the Kindler syndrome (Petricca et al., 2009). Functional studies using cultured keratinocytes have shown that kindlin-1 deficiency leads to reduced cell adhesion, migration, and proliferation due to deficient integrin activation (Herz et al., 2006; Lai-Cheong et al., 2008; Has et al., 2009; Petricca et al., 2009). Interestingly, analysis of histological data from a case report suggests that JE indeed fails to attach firmly to the tooth surface (Wiebe et al., 2008). Kindlin-1 binds to ß1 integrin in keratinocytes, and its deficiency does not affect hemidesmosome formation, suggesting that inter-hemidesmosomal ß1 integrin-mediated cell adhesion makes a significant contribution in the formation of firm adhesion of JE to tooth structure.
Regulation of Epithelial Cell Proliferation and Inflammation via αvß6 Integrin
Integrin αvß6 is an exclusively epithelial adhesion protein that is absent from most parts of normal healthy epidermis and oral mucosa (Breuss et al., 1993). However, αvß6 integrin is constitutively expressed in the JE and oral epithelium of the gingival papilla (Csiszar et al., 2007; Ghannad et al., 2008). In vitro, αvß6 integrin binds to the RGD-containing ECM ligands, including fibronectin, tenascin, vitronectin, and the latent TGFß1 (Huang et al., 1996; Koivisto et al., 1999; Munger et al., 1999). Expression of αvß6 integrin is induced during wound healing, in cancer, and in certain inflammatory conditions (Clark et al., 1996; Haapasalmi et al., 1996; Hamidi et al., 2000; Impola et al., 2004; Hahm et al., 2007). The function of αvß6 integrin in the progression of oral squamous cell carcinoma has been recently reviewed and will not be a subject of this review (Thomas et al., 2006). Interestingly, the major function of αvß6 integrin in vivo may not relate to cell adhesion per se but to its ability to activate latent TGFß1. The first evidence of this function came from findings showing that inactivation of the ß6 integrin gene results in mild inflammatory changes in the skin and lungs that are associated with altered TGFß1 signaling (Huang et al., 1996). TGFß1 belongs to a family of polypeptides that have multiple regulatory functions in tissue repair and the immune system (for reviews, see Chang et al., 2002 and Verrecchia and Mauviel, 2002). TGFß1 is synthesized as a latent precursor molecule containing latency-associated peptide (ß1-LAP) that associates with latent TGFß1-binding protein (LTBP1), a component of the ECM (Taipale et al., 1994). Activation of latent TGFß1 is a complex process that may involve proteolytic cleavage, conformational changes caused, e.g., by transglutaminase, thrombospondin-1 (TSP-1), or αvß6 integrin. Fairly recent findings have shown that αvß6 integrin mediates TGFß1 activation by binding to the RGD sequence of the ß1-LAP of the TGFß1 protein complex that is fixed to the ECM by LTBP-1 (Annes et al., 2004). This binding is believed to generate a retractile force, which introduces a conformational change in the LAP and subsequent activation of TGFß1 (Annes et al., 2004).
TGFß1 inhibits epithelial cell proliferation via up-regulation of cyclin-dependent kinase inhibitors p15 and p21 (Kane et al., 1990; Glick et al., 1993; Robson et al., 1999). One of the most recognized functions of TGFß1, however, is in immunoregulation, where it can either act as a pro-inflammatory cytokine or induce an anti-inflammatory response (AIR), depending on the biological context and cell types (Wahl et al., 2004; Li et al., 2006). The TGFß1 AIR is evidenced by the fact that TGFß1 knockout animals die a few weeks after birth from massive infiltration of lymphocytes and macrophages in many organs (Shull et al., 1992; Kulkarni et al., 1993). TGFß1 mediates the AIR through its immunosuppressive action on T-cells and macrophages. Involvement of αvß6 integrin-mediated activation of TGFß1 in the regulation of lung and skin inflammation has been demonstrated in ß6 integrin-null and ß6/TSP-1 double-null animals whose phenotype resembles, in a milder form, that of TGFß1-null mice (Huang et al., 1996; Ludlow et al., 2005). Recent evidence indicates that integrin-mediated activation of TGFß1 plays a major role in the AIR in vivo (Yang et al., 2007). Thus, it has become evident that integrin-mediated activation of TGFß1 regulates the AIR in many tissues, including soft tissues of the oral cavity.
As indicated above, αvß6 integrin is constitutively expressed in JE (Table, B) together with TGFß1 (Ghannad et al., 2008). No other αvß6 integrin ligands have been convincingly identified at the JE, although tenascin-C might be present (Table, A). Mice deficient in ß6 integrin develop all the classic signs of chronic periodontal disease, including inflammation, periodontal pocket formation, and bone loss (Ghannad et al., 2008). Thus, presence of αvß6 integrin in the JE plays an active protective role in periodontal tissues. Interestingly, expression of αvß6 integrin was markedly down-regulated in the pocket epithelium of chronic periodontitis patients (Ghannad et al., 2008). The current understanding of the role of αvß6 integrin in periodontal protection points to the AIR of TGFß1: The constitutive expression of αvß6 integrin in JE activates TGFß1, which controls inflammation at the site. During periodontal disease, increased proliferation of keratinocytes at the JE may also contribute to pocket formation, since reduced expression of αvß6 integrin would result in reduced TGFß1 activation and increased proliferation of JE cells. In fact, lack of TGFß-responsive cyclin-dependent kinase inhibitors in the JE has been shown to increase cell proliferation, suggesting that TGFß1 can play an important role in the regulation of JE proliferation (Watanabe et al., 2004). Consistent with the notion that TGFß1 is important in protecting the periodontium from inflammation, significantly increased levels of TGFß1 expression are found in non-active periodontal sites (Dutzan et al., 2009). Interestingly, mice deficient in the matricellular protein periostin also develop signs of periodontal disease (Rios et al., 2005). Periostin serves as a ligand for αvß3 and αvß5 integrins (Gillan et al., 2002). Periostin is strongly expressed in the periodontal ligament, but it is not clear whether periostin or αvß5 is expressed in the JE. Nevertheless, epithelial periostin can also regulate TGFß1 activation that could partially contribute to the immunoprotection of the periodontium (Sidhu et al., 2010). Thus, for periodontal health to be maintained, controlling inflammatory response may prove to be equally as important as the elimination of bacterial biofilm (Van Dyke and Serhan, 2003; Hasturk et al., 2006).
Role of Epithelial Cell Adhesion Molecules During Oral Mucosal Wound Healing
Few studies specifically explore the mechanisms of oral mucosal wound re-epithelialization. Therefore, much of the presented data draw from findings from skin wound healing and in vitro experiments, with an assumption that oral wounds heal largely in a similar manner.
After wounding occurs, epithelial cells come into contact with proteins from the underlying connective tissue at the wound edge, including type I collagen. In addition, they encounter the proteins present in the wound blood clot, consisting of polymerized fibrils of plasma fibronectin that are cross-linked to fibrin (Figs. 4B, 6). This fibrin-fibronectin matrix acts as a scaffold for further accumulation of ECM molecules such as heparin, denatured collagen, and tenascin-C (Gailit and Clark, 1994; Pankov and Yamada, 2002; Figs. 4, 6). Wounding also induces the expression of novel matrix molecules underneath the migrating keratinocytes, such as the EDA fibronectin (extra domain A or EIIIA), tenascin-C, and the unprocessed laminin 332 (Ffrench-Constant et al., 1989; Larjava et al., 1993; Häkkinen et al., 2000; Singh et al., 2004; Figs. 4, 6). Therefore, in wounds, keratinocytes encounter an environment with a complicated composite matrix of novel substances. Of these molecules, especially EDA fibronectin and laminin 332 appear essential for keratinocyte migration and re-epithelialization during wound healing (Muro et al., 2003; Hartwig et al., 2007). Tenascin-C regulates fibronectin deposition in wounds but does not seem to play a critical role in wound re-epithelialization, at least in the skin, although its elimination interferes with corneal wound healing (Mackie and Tucker, 1999). Its increased expression in the subepithelial connective tissue in oral mucosal wounds, however, may be associated with significantly reduced scar formation in these wounds compared with skin (Wong et al., 2009).
Figure 6.

Structural organization (hematoxylin-eosin staining; A-C) and schematic presentation (D-F) of the expression of keratinocyte integrins and matrix molecules in healthy (A, D) and wounded (B, C, E, F) human gingival mucosa. Healthy epithelium (A, D), 3-day wound (early wound; B, E), and 7-day wound (migrating epithelial fronts have just joined; C, F). E, epithelium; CT, connective tissue, FC, fibrin clot; GT, granulation tissue; BM, basement membrane. Arrows mark the wound margin. 1Expression shown in skin wounds, presence in oral epithelia unknown. 2Expression has not been studied during re-epithelialization. 3Induction of expression is based on indirect evidence from immunostaining experiments.
Because wounding alters the composition of ECM around keratinocytes, they need to adjust their cell adhesion receptors to interact with it. The first major change in integrin expression happens shortly after wounding, when wound edge keratinocytes dissolve their hemidesmosomal connections with basement membrane, and the distribution of α6ß4 integrin becomes diffuse around the basal keratinocytes (Borradori and Sonnenberg, 1999). At the same time, the expression of α2ß1, α3ß1, and α9ß1 integrins increases (Cavani et al., 1993; Juhasz et al., 1993; Larjava et al., 1993; Häkkinen et al., 2000; Singh et al., 2004). Concurrently, wounding induces the expression of 3 new fibronectin receptors, namely, α5ß1, αvß1, and αvß6 integrins, in the wound keratinocytes (Cavani et al., 1993; Larjava et al., 1993; Haapasalmi et al., 1996; Fig. 6). The process of re-epithelialization is well protected by collaboration between these different integrins and other receptor systems (Fig. 6). To this end, many of the integrins expressed by wound keratinocytes can bind multiple ligands present in the wound matrix, and, conversely, the same ligand can be recognized by several different integrins. For example, wound keratinocytes may use α9ß1 and αvß6 integrins for tenascin-C binding, and laminin 332 is recognized by α2ß1, α3ß1, and α6ß4 integrins, whereas α5ß1, αvß1, and αvß6 integrins serve as receptors for both plasma and cellular EDA fibronectin (Carter et al., 1991; Prieto et al., 1993; Yokosaki et al., 1994; Johansson et al., 1997; Décline and Rousselle, 2001; Fig. 4). In addition, α9ß1 integrin can serve as a receptor for EDA fibronectin and regulate keratinocyte proliferation at the wound edge (Liao et al., 2002; Singh et al., 2009; Fig. 4). The overlapping functions of keratinocyte adhesion molecules help to explain why elimination of individual cell adhesion molecules often produces amazingly mild effects in animal wound-healing models. For example, cultured keratinocytes can adaptively use at least αvß6, αvß1, α5ß1, and α3ß1 integrins for fibronectin binding (Koivisto et al., 1999). However, it appears that ß1 integrins as a group are fundamental for wound re-epithelialization, since keratinocyte migration and re-epithelialization are severely compromised in mice with keratinocyte-specific knockout of the ß1 integrin subunit (Raghavan et al., 2000; Grose et al., 2002).
Since intermediate adhesiveness to matrix proteins favors cell motility, utilization of intermediate-strength integrin-matrix interactions in co-operation is required for re-epithelialization. This may be achieved by focalized denaturation of collagens, assembly of composite matrices with reduced adhesiveness, reducing the strength of high-affinity integrin binding, and robust expression of low-affinity integrins. For example, the high-affinity interaction of α2ß1 integrin with fibrillar collagens induces the expression of MMP-1 at the migrating epithelial front, resulting in focalized denaturation of the collagen matrix and dissociation of these high-affinity contacts (Saarialho-Kere et al., 1993; Pilcher et al., 1997). Notably, migration of human keratinocytes on type I collagen in vitro requires both α2ß1 integrin and MMP-1 (Pilcher et al., 1997). Denatured collagen may also indirectly influence keratinocyte migration through binding of fibronectin and growth factors (Davis et al., 2000).
Strong expression of αvß1 integrin, a low-affinity fibronectin receptor, may also facilitate keratinocyte migration by supporting cell attachment without decelerating the migration speed (Zhang et al., 1993; Koivisto et al., 1999). Additionally, it appears that the interplay between and among fibronectin, tenascin-C, and their integrin receptors regulates wound re- epithelialization, since binding of tenascin-C to fibronectin reduces the strength of the high-affinity α5ß1 integrin-fibronectin interaction to facilitate migration (Kim et al., 1992a; Hauzenberger et al., 1999; Ingham et al., 2004).
The role of α3ß1 integrin in re-epithelialization is complex, and its exact functions in re-epithelialization have not yet been conclusively established. Curiously, α3ß1 integrin has been reported to either mediate the migration of cultured keratinocytes or inhibit it (Kim et al., 1992b; Zhang and Kramer, 1996; Goldfinger et al., 1999; Décline and Rousselle, 2001; deHart et al., 2003). Similarly, in vivo re-epithelialization studies have yielded conflicting results. In two recent studies, re-epithelialization was either slightly accelerated or not negatively affected in skin wounds of mice with keratinocyte-targeted knockout of the α3 integrin subunit (Margadant et al., 2009; Mitchell et al., 2009). However, results of another recent study suggested that α3ß1 integrin facilitates re-epithelialization by modulating TGFß1-mediated responses in the wound (Reynolds et al., 2008). In addition, α3ß1 integrin can function as a trans-dominant inhibitor of other ß1 integrins, including α2ß1 and α5ß1 (Hodivala-Dilke et al., 1998), again reducing the strength of keratinocyte attachment during re-epithelialization. Interestingly, α6ß4 integrin facilitates re-epithelialization by supporting EGF signaling even when it is not bound to its ligand, laminin 332, and deletion of the signaling domain of the ß4 subunit causes decelerated wound re-epithelialization (Russell et al., 2003; Nikolopoulos et al., 2005). Further studies are needed for a more detailed understanding of the roles of the laminin receptors α3ß1 and α6ß4 integrins in wound re-epithelialization.
The re-epithelialization phase of oral mucosal wound healing comes to an end when the migrating epithelial fronts originating from the wound edges have joined and cover the wound surface. At this stage, the expression of ß1 integrins is down-regulated (Fig. 6), and α6ß4 integrin binding to the proteolytically cleaved laminin 332 is restored (Larjava et al., 1993; Goldfinger et al., 1999). As a consequence, hemidesmosomal adhesions provide nucleation sites for complete basement membrane restoration, allowing keratinocytes to resume their normal differentiation process (Jones et al., 1994; Litjens et al., 2006). In small oral mucosal wounds, the nucleation of the basement membrane occurs simultaneously in several places along the wound epithelium (Larjava et al., 1993).
During this phase of oral mucosal wound healing, αvß6 integrin expression is significantly up-regulated in the basal and several suprabasal keratinocyte layers, coinciding with the peak expression of biologically active TGFß1 (Haapasalmi et al., 1996; Yang et al., 1999; Häkkinen et al., 2000; Fig. 6). However, whether αvß6 integrin is involved in TGFß activation during wound healing, or whether it serves other functions, remains to be shown. Interestingly, αvß6 integrin seems to be dispensable during normal wound healing, but may play a significant role in chronic wounds and in wounds compromised by corticosteroids (Häkkinen et al., 2004; Xie et al., 2009).
Concluding Remarks
Cell adhesion and integrins regulate many crucial functions of oral epithelial cells. Activation of ß1 integrins together with α6ß4 integrin-mediated laminin 332 binding regulates adhesion of JE cells to enamel. These mechanisms appear to be poorly developed in PIE. In addition, αvß6 integrin in JE may control periodontal inflammation via TGFß1 activation. Many integrins and matrix molecules collectively regulate re-epithelialization during wound healing. Overall, epithelial cells have been proven to function far beyond their traditional role in providing a protective barrier for connective tissues. Future research should be focused on identifying in more detail the cell adhesion molecules that are expressed in tooth and implant interfaces.
Footnotes
A supplemental appendix to this article is published electronically only at http://jdr.sagepub.com/supplemental.
This study is supported by the following NIH grants: U01- DE018903, U01-HG004438, U01-HG004423, U01-HG004446, R01-DE014899, R01-DE0 9551, R01-DE12101, R03-DE021425, and P60-DE-13076, and by NIH contract HHSN268200782-096C. Other support was provided by the Danish NRF, Danish Pharmacists’ Fund, Egmont Foundation, March of Dimes, Augustinus Foundation, and Health Fund of the Danish Health Insurance Societies.
References
- Abiko Y, Nishimura M, Rahemtulla F, Mizoguchi I, Kaku T. (2001). Immunohistochemical localization of large chondroitin sulphate proteoglycan in porcine gingival epithelia. Eur J Morphol 39:99-104 [DOI] [PubMed] [Google Scholar]
- Abrahamsson I, Berglundh T, Wennstrom J, Lindhe J. (1996). The peri-implant hard and soft tissue characteristics at different implant systems. A comparative study in dogs. Clin Oral Implants Res 7:212-219 [DOI] [PubMed] [Google Scholar]
- Abrahamsson I, Berglundh T, Moon IS, Lindhe J. (1999). Peri-implant tissues at submerged and non-submerged titanium implants. J Clin Periodontol 26:600-607 [DOI] [PubMed] [Google Scholar]
- Amano S, Akutsu N, Ogura Y, Nishiyama T. (2004). Increase of laminin 5 synthesis in human keratinocytes by acute wound fluid, inflammatory cytokines and growth factors, and lysophospholipids. Br J Dermatol 151:961-970 [DOI] [PubMed] [Google Scholar]
- Arnaout MA, Mahalingam B, Xiong JP. (2005). Integrin structure, allostery, and bidirectional signaling. Annu Rev Cell Dev Biol 21:381-410 [DOI] [PubMed] [Google Scholar]
- Atsuta I, Yamaza T, Yoshinari M, Mino S, Goto T, Kido MA, et al. (2005a). Changes in the distribution of laminin-5 during peri-implant epithelium formation after immediate titanium implantation in rats. Biomaterials 26:1751-1760 [DOI] [PubMed] [Google Scholar]
- Atsuta I, Yamaza T, Yoshinari M, Goto T, Kido MA, Kagiya T, et al. (2005b). Ultrastructural localization of laminin-5 (γ2 chain) in the rat peri-implant oral mucosa around a titanium-dental implant by immuno-electron microscopy. Biomaterials 26:6280-6287 [DOI] [PubMed] [Google Scholar]
- Aumailley M, El Khal A, Knöss N, Tunggal L. (2003). Laminin 5 processing and its integration into the ECM. Matrix Biol 22:49-54 [DOI] [PubMed] [Google Scholar]
- Berglundh T, Lindhe J. (1996). Dimension of the periimplant mucosa. Biological width revisited. J Clin Periodontol 23:971-973 [DOI] [PubMed] [Google Scholar]
- Borradori L, Sonnenberg A. (1999). Structure and function of hemidesmosomes: more than simple adhesion complexes. J Invest Dermatol 112:411-418 [DOI] [PubMed] [Google Scholar]
- Bosshardt DD, Lang NP. (2005). The junctional epithelium: from health to disease. J Dent Res 84:9-20 [DOI] [PubMed] [Google Scholar]
- Breuss JM, Gillett N, Lu L, Sheppard D, Pytela R. (1993). Restricted distribution of integrin ß6 mRNA in primate epithelial tissues. J Histochem Cytochem 41:1521-1527 [DOI] [PubMed] [Google Scholar]
- Buser D, Weber HP, Donath K, Fiorellini JP, Paquette DW, Williams RC. (1992). Soft tissue reactions to non-submerged unloaded titanium implants in beagle dogs. J Periodontol 63:225-235 [DOI] [PubMed] [Google Scholar]
- Carter WG, Ryan MC, Gahr PJ. (1991). Epiligrin, a new cell adhesion ligand for integrin α3β1 in epithelial basement membranes. Cell 65:599-610 [DOI] [PubMed] [Google Scholar]
- Cavani A, Zambruno G, Marconi A, Manca V, Marchetti M, Giannetti A. (1993). Distinctive integrin expression in the newly forming epidermis during wound healing in humans. J Invest Dermatol 101:600-604 [DOI] [PubMed] [Google Scholar]
- Chang H, Brown CW, Matzuk MM. (2002). Genetic analysis of the mammalian transforming growth factor-ß superfamily. Endocr Rev 23:787-823 [DOI] [PubMed] [Google Scholar]
- Clark RA, Ashcroft GS, Spencer MJ, Larjava H, Ferguson MW. (1996). Re-epithelialization of normal human excisional wounds is associated with a switch from αvß5 to αvß6 integrins. Br J Dermatol 135:46-51 [PubMed] [Google Scholar]
- Cochran DL, Hermann JS, Schenk RK, Higginbottom FL, Buser D. (1997). Biologic width around titanium implants. A histometric analysis of the implanto-gingival junction around unloaded and loaded nonsubmerged implants in the canine mandible. J Periodontol 68:186-198 [DOI] [PubMed] [Google Scholar]
- Csiszar A, Wiebe C, Larjava H, Häkkinen L. (2007). Distinct molecular composition of human interdental papilla. J Periodontol 78:304-314 [DOI] [PubMed] [Google Scholar]
- Davis GE, Bayless KJ, Davis MJ, Meininger GA. (2000). Regulation of tissue injury responses by the exposure of matricryptic sites within extracellular matrix molecules. Am J Pathol 156:1489-1498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Décline F, Rousselle P. (2001). Keratinocyte migration requires α2ß1 integrin-mediated interaction with the laminin 5γ2 chain. J Cell Sci 114(Pt 4):811-823 [DOI] [PubMed] [Google Scholar]
- deHart GW, Healy KE, Jones JC. (2003). The role of α3ß1 integrin in determining the supramolecular organization of laminin-5 in the extracellular matrix of keratinocytes. Exp Cell Res 283:67-79 [DOI] [PubMed] [Google Scholar]
- Dutzan N, Gamonal J, Silva A, Sanz M, Vernal R. (2009). Over-expression of forkhead box P3 and its association with receptor activator of nuclear factor-κ B ligand, interleukin (IL) -17, IL-10 and transforming growth factor-ß during the progression of chronic periodontitis. J Clin Periodontol 36:396-403 [DOI] [PubMed] [Google Scholar]
- Ffrench-Constant C, van de Water L, Dvorak HF, Hynes RO. (1989). Re-appearance of an embryonic pattern of fibronectin splicing during wound healing in the adult rat. J Cell Biol 109:903-914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiseki M, Matsuzaka K, Yoshinari M, Shimono M, Inoue T. (2003). An experimental study on the features of peri-implant epithelium: immunohistochemical and electron-microscopic observations. Bull Tokyo Dent Coll 44:185-199 [DOI] [PubMed] [Google Scholar]
- Gailit J, Clark RA. (1994). Wound repair in the context of extracellular matrix. Curr Opin Cell Biol 6:717-725 [DOI] [PubMed] [Google Scholar]
- Gargiulo AW, Wentz FM, Orban B. (1961). Dimensions and relations of the dentogingival junction in humans. J Periodontol 32:261-267 [Google Scholar]
- Ghannad F, Nica D, Fulle MI, Grenier D, Putnins EE, Johnston S, et al. (2008). Absence of αvß6 integrin is linked to initiation and progression of periodontal disease. Am J Pathol 172:1271-1286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillan L, Matei D, Fishman DA, Gerbin CS, Karlan BY, Chang DD. (2002). Periostin secreted by epithelial ovarian carcinoma is a ligand for alpha(V)beta(3) and alpha(V)beta(5) integrins and promotes cell motility. Cancer Res 62:5358-5364 [PubMed] [Google Scholar]
- Glick AB, Kulkarni AB, Tennenbaum T, Hennings H, Flanders KC, O’Reilly M, et al. (1993). Loss of expression of transforming growth factor ß in skin and skin tumors is associated with hyperproliferation and a high risk for malignant conversion. Proc Natl Acad Sci USA 90:6076-6080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldfinger LE, Hopkinson SB, deHart GW, Collawn S, Couchman JR, Jones JC. (1999). The α3 laminin subunit, α6ß4 and α3ß1 integrin coordinately regulate wound healing in cultured epithelial cells and in the skin. J Cell Sci 112(Pt 16):2615-2629 [DOI] [PubMed] [Google Scholar]
- Gould TR, Westbury L, Brunette DM. (1984). Ultrastructural study of the attachment of human gingiva to titanium in vivo. J Prosthet Dent 52:418-420 [DOI] [PubMed] [Google Scholar]
- Grose R, Hutter C, Bloch W, Thorey I, Watt FM, Fassler R, et al. (2002). A crucial role of ß1 integrins for keratinocyte migration in vitro and during cutaneous wound repair. Development 129:2303-2315 [DOI] [PubMed] [Google Scholar]
- Haapasalmi K, Makela M, Oksala O, Heino J, Yamada KM, Uitto VJ, et al. (1995). Expression of epithelial adhesion proteins and integrins in chronic inflammation. Am J Pathol 147:193-206 [PMC free article] [PubMed] [Google Scholar]
- Haapasalmi K, Zhang K, Tonnesen M, Olerud J, Sheppard D, Salo T, et al. (1996). Keratinocytes in human wounds express αvß6 integrin. J Invest Dermatol 106:42-48 [DOI] [PubMed] [Google Scholar]
- Hahm K, Lukashev ME, Luo Y, Yang WJ, Dolinski BM, Weinreb PH, et al. (2007). αvß6 integrin regulates renal fibrosis and inflammation in Alport mouse. Am J Pathol 170:110-125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Häkkinen L, Hildebrand HC, Berndt A, Kosmehl H, Larjava H. (2000). Immunolocalization of tenascin-C, α9 integrin subunit, and αvß6 integrin during wound healing in human oral mucosa. J Histochem Cytochem 48:985-998 [DOI] [PubMed] [Google Scholar]
- Häkkinen L, Koivisto L, Gardner H, Saarialho-Kere U, Carroll JM, Lakso M, et al. (2004). Increased expression of ß6 integrin in skin leads to spontaneous development of chronic wounds. Am J Pathol 164:229-242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamidi S, Salo T, Kainulainen T, Epstein J, Lerner K, Larjava H. (2000). Expression of αvß6 integrin in oral leukoplakia. Br J Cancer 82:1433-1440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartwig B, Borm B, Schneider H, Arin MJ, Kirfel G, Herzog V. (2007). Laminin-5-deficient human keratinocytes: defective adhesion results in a saltatory and inefficient mode of migration. Exp Cell Res 313:1575-1587 [DOI] [PubMed] [Google Scholar]
- Has C, Herz C, Zimina E, Qu HY, He Y, Zhang ZG, et al. (2009). Kindlin-1 is required for RhoGTPase-mediated lamellipodia formation in keratinocytes. Am J Pathol 175:1442-1452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasturk H, Kantarci A, Ohira T, Arita M, Ebrahimi N, Chiang N, et al. (2006). RvE1 protects from local inflammation and osteoclast-mediated bone destruction in periodontitis. FASEB J 20:401-403 [DOI] [PubMed] [Google Scholar]
- Hauzenberger D, Olivier P, Gundersen D, Ruegg C. (1999). Tenascin-C inhibits ß1 integrin-dependent T lymphocyte adhesion to fibronectin through the binding of its fnIII 1-5 repeats to fibronectin. Eur J Immunol 29:1435-1447 [DOI] [PubMed] [Google Scholar]
- Hermann JS, Buser D, Schenk RK, Schoolfield JD, Cochran DL. (2001). Biological width around one- and two-piece titanium implants. Clin Oral Implants Res 12:559-571 [DOI] [PubMed] [Google Scholar]
- Herz C, Aumailley M, Schulte C, Schlötzer-Schrehardt U, Bruckner-Tuderman L, Has C. (2006). Kindlin-1 is a phosphoprotein involved in regulation of polarity, proliferation, and motility of epidermal keratinocytes. J Biol Chem 281:36082-36090 [DOI] [PubMed] [Google Scholar]
- Hodivala-Dilke KM, DiPersio CM, Kreidberg JA, Hynes RO. (1998). Novel roles for α3ß1 integrin as a regulator of cytoskeletal assembly and as a trans-dominant inhibitor of integrin receptor function in mouse keratinocytes. J Cell Biol 142:1357-1369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hormia M, Virtanen I, Quaranta V. (1992). Immunolocalization of integrin α6ß4 in mouse junctional epithelium suggests an anchoring function to both the internal and the external basal lamina. J Dent Res 71:1503-1508 [DOI] [PubMed] [Google Scholar]
- Hormia M, Sahlberg C, Thesleff I, Airenne T. (1998). The epithelium-tooth interface—a basal lamina rich in laminin-5 and lacking other known laminin isoforms. J Dent Res 77:1479-1485 [DOI] [PubMed] [Google Scholar]
- Hormia M, Owaribe K, Virtanen I. (2001). The dento-epithelial junction: cell adhesion by type I hemidesmosomes in the absence of a true basal lamina. J Periodontol 72:788-797 [DOI] [PubMed] [Google Scholar]
- Huang XZ, Wu JF, Cass D, Erle DJ, Corry D, Young SG, et al. (1996). Inactivation of the integrin ß6 subunit gene reveals a role of epithelial integrins in regulating inflammation in the lung and skin. J Cell Biol 133:921-928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hynes RO. (2002). Integrins: bidirectional, allosteric signaling machines. Cell 110:673-687 [DOI] [PubMed] [Google Scholar]
- Ikeda H, Yamaza T, Yoshinari M, Ohsaki Y, Ayukawa Y, Kido MA, et al. (2000). Ultrastructural and immunoelectron microscopic studies of the peri-implant epithelium-implant (Ti-6Al-4V) interface of rat maxilla. J Periodontol 71:961-973 [DOI] [PubMed] [Google Scholar]
- Ikeda H, Shiraiwa M, Yamaza T, Yoshinari M, Kido MA, Ayukawa Y, et al. (2002). Difference in penetration of horseradish peroxidase tracer as a foreign substance into the peri-implant or junctional epithelium of rat gingivae. Clin Oral Implants Res 13:243-251 [DOI] [PubMed] [Google Scholar]
- Impola U, Uitto VJ, Hietanen J, Häkkinen L, Zhang L, Larjava H, et al. (2004). Differential expression of matrilysin-1 (MMP-7), 92 kD gelatinase (MMP-9), and metalloelastase (MMP-12) in oral verrucous and squamous cell cancer. J Pathol 202:14-22 [DOI] [PubMed] [Google Scholar]
- Ingham KC, Brew SA, Erickson HP. (2004). Localization of a cryptic binding site for tenascin on fibronectin. J Biol Chem 279:28132-28135 [DOI] [PubMed] [Google Scholar]
- Inoue T, Takeda T, Lee CY, Abiko Y, Ayukawa Y, Tanaka T, et al. (1997). Immunolocalization of proliferating cell nuclear antigen in the peri-implant epithelium. Bull Tokyo Dent Coll 38:187-193 [PubMed] [Google Scholar]
- Johansson S, Svineng G, Wennerberg K, Armulik A, Lohikangas L. (1997). Fibronectin-integrin interactions. Front Biosci 2:d126-d146 [DOI] [PubMed] [Google Scholar]
- Jones JC, Asmuth J, Baker SE, Langhofer M, Roth SI, Hopkinson SB. (1994). Hemidesmosomes: extracellular matrix/intermediate filament connectors. Exp Cell Res 213:1-11 [DOI] [PubMed] [Google Scholar]
- Juhasz I, Murphy GF, Yan HC, Herlyn M, Albelda SM. (1993). Regulation of extracellular matrix proteins and integrin cell substratum adhesion receptors on epithelium during cutaneous human wound healing in vivo. Am J Pathol 143:1458-1469 [PMC free article] [PubMed] [Google Scholar]
- Kainulainen T, Häkkinen L, Hamidi S, Larjava K, Kallioinen M, Peltonen J, et al. (1998). Laminin-5 expression is independent of the injury and the microenvironment during reepithelialization of wounds. J Histochem Cytochem 46:353-360 [DOI] [PubMed] [Google Scholar]
- Kane CJ, Knapp AM, Mansbridge JN, Hanawalt PC. (1990). Transforming growth factor-ß1 localization in normal and psoriatic epidermal keratinocytes in situ. J Cell Physiol 144:144-150 [DOI] [PubMed] [Google Scholar]
- Kim JP, Zhang K, Chen JD, Wynn KC, Kramer RH, Woodley DT. (1992a). Mechanism of human keratinocyte migration on fibronectin, unique roles of RGD site and integrins. J Cell Physiol 151:443-450 [DOI] [PubMed] [Google Scholar]
- Kim JP, Zhang K, Kramer RH, Schall TJ, Woodley DT. (1992b). Integrin receptors and RGD sequences in human keratinocyte migration: unique anti-migratory function of α3ß1 epiligrin receptor. J Invest Dermatol 98:764-770 [DOI] [PubMed] [Google Scholar]
- Kinumatsu T, Hashimoto S, Muramatsu T, Sasaki H, Jung HS, Yamada S, et al. (2009). Involvement of laminin and integrins in adhesion and migration of junctional epithelium cells. J Periodontal Res 44:13-20 [DOI] [PubMed] [Google Scholar]
- Koivisto L, Larjava K, Häkkinen L, Uitto V-J, Heino J, Larjava H. (1999). Different integrins mediate cell spreading, haptotaxis and lateral migration of HaCaT keratinocytes on fibronectin. Cell Adhes Commun 7:245-257 [DOI] [PubMed] [Google Scholar]
- Koldsland OC, Scheie AA, Aass AM. (2010). Prevalence of peri-implantitis related to severity of the disease with different degrees of bone loss. J Periodontol 81:231-238 [DOI] [PubMed] [Google Scholar]
- Kulkarni AB, Huh CG, Becker D, Geiser A, Lyght M, Flanders KC, et al. (1993). Transforming growth factor ß1 null mutation in mice causes excessive inflammatory response and early death. Proc Natl Acad Sci USA 90:770-774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai-Cheong JE, Ussar S, Arita K, Hart IR, McGrath JA. (2008). Colocalization of kindlin-1, kindlin-2, and migfilin at keratinocyte focal adhesion and relevance to the pathophysiology of Kindler syndrome. J Invest Dermatol 128:2156-2165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larjava H, Salo T, Haapasalmi K, Kramer RH, Heino J. (1993). Expression of integrins and basement membrane components by wound keratinocytes. J Clin Invest 92:1425-1435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larjava H, Plow EF, Wu C. (2008). Kindlins: essential regulators of integrin signaling and cell-matrix adhesion. EMBO Rep 9:1203-1208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Firth JD, Putnins EE. (2005). Keratinocyte growth factor-1 expression in healthy and diseased human periodontal tissues. J Periodontal Res 40:118-128 [DOI] [PubMed] [Google Scholar]
- Li MO, Sanjabi S, Flavell RA. (2006). Transforming growth factor-ß controls development, homeostasis, and tolerance of T cells by regulatory T cell dependent and independent mechanisms. Immunity 25:455-471 [DOI] [PubMed] [Google Scholar]
- Liao YF, Gotwals PJ, Koteliansky VE, Sheppard D, Van De Water L. (2002). The EIIIA segment of fibronectin is a ligand for integrins α9ß1 and α4ß1 providing a novel mechanism for regulating cell adhesion by alternative splicing. J Biol Chem 277:14467-14474 [DOI] [PubMed] [Google Scholar]
- Litjens SH, de Pereda JM, Sonnenberg A. (2006). Current insights into the formation and breakdown of hemidesmosomes. Trends Cell Biol 16:376-383 [DOI] [PubMed] [Google Scholar]
- Ludlow A, Yee KO, Lipman R, Bronson R, Weinreb P, Huang X, et al. (2005). Characterization of integrin ß6 and thrombospondin-1 double-null mice. J Cell Mol Med 9:421-437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackie EJ, Tucker RP. (1999). The tenascin-C knockout revisited. J Cell Sci 112(Pt 22):3847-3853 [DOI] [PubMed] [Google Scholar]
- Margadant C, Raymond K, Kreft M, Sachs N, Janssen H, Sonnenberg A. (2009). Integrin α3ß1 inhibits directional migration and wound re-epithelialization in the skin. J Cell Sci 122(Pt 2):278-288 [DOI] [PubMed] [Google Scholar]
- Máximo MB, de Mendonça AC, Alves JF, Cortelli SC, Peruzzo DC, Duarte PM. (2008). Peri-implant diseases may be associated with increased time loading and generalized periodontal bone loss: preliminary results. J Oral Implantol 34:268-273 [DOI] [PubMed] [Google Scholar]
- Meves A, Stremmel C, Gottschalk K, Fässler R. (2009). The Kindlin protein family: new members to the club of focal adhesion proteins. Trends Cell Biol 19:504-513 [DOI] [PubMed] [Google Scholar]
- Mitchell K, Szekeres C, Milano V, Svenson KB, Nilsen-Hamilton M, Kreidberg JA, et al. (2009). α3ß1 integrin in epidermis promotes wound angiogenesis and keratinocyte-to-endothelial-cell crosstalk through the induction of MRP3. J Cell Sci 122(Pt 11):1778-1787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munger JS, Huang X, Kawakatsu H, Griffiths MJ, Dalton SL, Wu J, et al. (1999). The integrin αvß6 binds and activates latent TGF-ß1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell 96:319-328 [DOI] [PubMed] [Google Scholar]
- Murgia C, Blaikie P, Kim N, Dans M, Petrie HT, Giancotti FG. (1998). Cell cycle and adhesion defects in mice carrying a targeted deletion of the integrin ß4 cytoplasmic domain. EMBO J 17:3940-3951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muro AF, Chauhan AK, Gajovic S, Iaconcig A, Porro F, Stanta G, et al. (2003). Regulated splicing of the fibronectin EDA exon is essential for proper skin wound healing and normal lifespan. J Cell Biol 162:149-160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myshin HL, Wiens JP. (2005). Factors affecting soft tissue around dental implants: a review of the literature. J Prosthet Dent 94:440-444 [DOI] [PubMed] [Google Scholar]
- Nikolopoulos SN, Blaikie P, Yoshioka T, Guo W, Puri C, Tacchetti C, et al. (2005). Targeted deletion of the integrin ß4 signaling domain suppresses laminin-5-dependent nuclear entry of mitogen-activated protein kinases and NF-κB, causing defects in epidermal growth and migration. Mol Cell Biol 25:6090-6102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oksonen J, Sorokin LM, Virtanen, Hormia M. (2001). The junctional epithelium around murine teeth differs from gingival epithelium in its basement membrane composition. J Dent Res 80:2093-2097 [DOI] [PubMed] [Google Scholar]
- Pankov R, Yamada KM. (2002). Fibronectin at a glance. J Cell Sci 115(Pt 20):3861-3863 [DOI] [PubMed] [Google Scholar]
- Petricca G, Leppilampi M, Jiang G, Owen GR, Wiebe C, Tu Y, et al. (2009). Localization and potential function of kindlin-1 in periodontal tissues. Eur J Oral Sci 117:518-527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilcher BK, Dumin JA, Sudbeck BD, Krane SM, Welgus HG, Parks WC. (1997). The activity of collagenase-1 is required for keratinocyte migration on a type I collagen matrix. J Cell Biol 137:1445-1457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prieto AL, Edelman GM, Crossin KL. (1993). Multiple integrins mediate cell attachment to cytotactin/tenascin. Proc Natl Acad Sci USA 90:10154-10158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghavan S, Bauer C, Mundschau G, Li Q, Fuchs E. (2000). Conditional ablation of ß1 integrin in skin. Severe defects in epidermal proliferation, basement membrane formation, and hair follicle invagination. J Cell Biol 150:1149-1160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds LE, Conti FJ, Silva R, Robinson SD, Iyer V, Rudling R, et al. (2008). α3ß1 integrin-controlled Smad7 regulates reepithelialization during wound healing in mice. J Clin Invest 118:965-974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rios H, Koushik SV, Wang H, Wang J, Zhou HM, Lindsley A, et al. (2005). Periostin null mice exhibit dwarfism, incisor enamel defects, and an early-onset periodontal disease-like phenotype. Mol Cell Biol 25:11131-11144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robson CN, Gnanapragasam V, Byrne RL, Collins AT, Neal DE. (1999). Transforming growth factor-ß1 up-regulates p15, p21 and p27 and blocks cell cycling in G1 in human prostate epithelium. J Endocrinol 160:257-266 [DOI] [PubMed] [Google Scholar]
- Rompen E, Domken O, Degidi M, Pontes AE, Piattelli A. (2006). The effect of material characteristics, of surface topography and of implant components and connections on soft tissue integration: a literature review. Clin Oral Implants Res 17(Suppl 2):55-67 [DOI] [PubMed] [Google Scholar]
- Roos-Jansåker AM, Lindahl C, Renvert H, Renvert S. (2006). Nine- to fourteen-year follow-up of implant treatment. Part II: presence of peri-implant lesions. J Clin Periodontol 33:290-295 [DOI] [PubMed] [Google Scholar]
- Russell AJ, Fincher EF, Millman L, Smith R, Vela V, Waterman EA, et al. (2003). α6ß4 integrin regulates keratinocyte chemotaxis through differential GTPase activation and antagonism of α3ß1 integrin. J Cell Sci 116(Pt 17):3543-3556 [DOI] [PubMed] [Google Scholar]
- Saarialho-Kere UK, Kovacs SO, Pentland AP, Olerud JE, Welgus HG, Parks WC. (1993). Cell-matrix interactions modulate interstitial collagenase expression by human keratinocytes actively involved in wound healing. J Clin Invest 92:2858-2866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salonen J, Oda D, Funk SE, Sage H. (1991). Synthesis of type VIII collagen by epithelial cells of human gingiva. J Periodontal Res 26: 355-360 [DOI] [PubMed] [Google Scholar]
- Shull MM, Ormsby I, Kier AB, Pawlowski S, Diebold RJ, Yin M, et al. (1992). Targeted disruption of the mouse transforming growth factor-ß1 gene results in multifocal inflammatory disease. Nature 359: 693-699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidhu SS, Yuan S, Innes AL, Kerr S, Woodruff PG, Hou L, et al. (2010). Roles of epithelial cell-derived periostin in TGF-beta activation, collagen production, and collagen gel elasticity in asthma. Proc Natl Acad Sci U S A. 107:14170-14175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh P, Reimer CL, Peters JH, Stepp MA, Hynes RO, Van De Water L. (2004). The spatial and temporal expression patterns of integrin α9ß1 and one of its ligands, the EIIIA segment of fibronectin, in cutaneous wound healing. J Invest Dermatol 123:1176-1181 [DOI] [PubMed] [Google Scholar]
- Singh P, Chen C, Pal-Ghosh S, Stepp MA, Sheppard D, Van De Water L. (2009). Loss of integrin α9ß1 results in defects in proliferation, causing poor re-epithelialization during cutaneous wound healing. J Invest Dermatol 129:217-228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smola H, Stark HJ, Thiekötter G, Mirancea N, Krieg T, Fusenig NE. (1998). Dynamics of basement membrane formation by keratinocyte-fibroblast interactions in organotypic skin culture. Exp Cell Res 239:399-410 [DOI] [PubMed] [Google Scholar]
- Taipale J, Miyazono K, Heldin CH, Keski-Oja J. (1994). Latent transforming growth factor-ß1 associates to fibroblast extracellular matrix via latent TGF-ß binding protein. J Cell Biol 12:171-181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas GJ, Nyström ML, Marshall JF. (2006). αvß6 integrin in wound healing and cancer of the oral cavity. J Oral Pathol Med 35:1-10 [DOI] [PubMed] [Google Scholar]
- Vacek JS, Gher ME, Assad DA, Richardson AC, Giambarresi LI. (1994). The dimensions of human dentogingival junction. Int Periodontics Restorative Dent 14:154-165 [PubMed] [Google Scholar]
- Van Dyke TE, Serhan CN. (2003). Resolution of inflammation: a new paradigm for the pathogenesis of periodontal diseases. J Dent Res 82:82-90 [DOI] [PubMed] [Google Scholar]
- Verrecchia F, Mauviel A. (2002). Control of connective tissue gene expression by TGFß: role of Smad proteins in fibrosis. Curr Rheumatol Rep 4:143-149 [DOI] [PubMed] [Google Scholar]
- Wahl SM, Swisher J, McCartney-Francis N, Chen W. (2004). TGF-ß: the perpetrator of immune suppression by regulatory T cells and suicidal T cells. J Leukoc Biol 76:15-24 [DOI] [PubMed] [Google Scholar]
- Watanabe K, Petro BJ, Sevandal M, Anshuman S, Jovanovic A, Tyner AL. (2004). Histochemical examination of periodontal junctional epithelium in p21/p27 double knockout mice. Eur J Oral Sci 112: 253-258 [DOI] [PubMed] [Google Scholar]
- Wiebe CB, Petricca G, Häkkinen L, Jiang G, Wu C, Larjava H. (2008). Kindler syndrome and periodontal disease: review of the literature and 12-year follow-up of treatment outcome. J Periodontol 79: 961-966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelmsen K, Litjens SH, Kuikman I, Margadant C, van Rheenen J, Sonnenberg A. (2007). Serine phosphorylation of the integrin ß4 subunit is necessary for epidermal growth factor receptor induced hemidesmosome disruption. Mol Biol Cell 18:3512-3522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong JW, Gallant-Behm C, Wiebe C, Mak K, Hart DA, Larjava H, et al. (2009). Wound healing in oral mucosa results in reduced scar formation as compared with skin: evidence from the red Duroc pig model and humans. Wound Repair Regen 17:717-729 [DOI] [PubMed] [Google Scholar]
- Xie Y, Gao K, Häkkinen L, Larjava H. (2009). Mice lacking ß6 integrin in skin show accelerated wound repair in dexamethasone impaired wound healing model. Wound Repair Regen 17:326-339 [DOI] [PubMed] [Google Scholar]
- Yang L, Qiu CX, Ludlow A, Ferguson MW, Brunner G. (1999). Active transforming growth factor-ß in wound repair: determination using a new assay. Am J Pathol 154:105-111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Mu Z, Dabovic B, Jurukovski V, Yu D, Sung J, et al. (2007). Absence of integrin-mediated TGFß1 activation in vivo recapitulates the phenotype of TGFß1-null mice. J Cell Biol 176:787-793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokosaki Y, Palmer EL, Prieto AL, Crossin KL, Bourdon MA, Pytela R, et al. (1994). The integrin α9ß1 mediates cell attachment to a non-RGD site in the third fibronectin type III repeat of tenascin. J Biol Chem 269:26691-26696 [PubMed] [Google Scholar]
- Zhang K, Kramer RH. (1996). Laminin 5 deposition promotes keratinocyte motility. Exp Cell Res 227:309-322 [DOI] [PubMed] [Google Scholar]
- Zhang Z, Morla AO, Vuori K, Bauer JS, Juliano RL, Ruoslahti E. (1993). The αvß1 integrin functions as a fibronectin receptor but does not support fibronectin matrix assembly and cell migration on fibronectin. J Cell Biol 122:235-242 [DOI] [PMC free article] [PubMed] [Google Scholar]