

Abstract
Background
The A341V mutation in the pore-forming KCNQ1 subunit of the slowly activating delayed-rectifier potassium current (IKs) underlies a common form of the long QT syndrome, and is associated with an unusually severe phenotype. However, there is controversy regarding the underlying mechanism responsible for the clinically observed phenotype. We investigated the biophysical characteristics of A341V in a cardiac environment by utilizing a cardiac cell line, and in particular the impact of the KCNE1 β -subunit.
Methods
Whole-cell current were recorded from transiently transfected HL-1 cells, a cardiac cell line. Mutant KCNQ1 and KCNE1 were constructed by site-directed mutagenesis.
Results
The A341V mutant resulted in a non-functional channel when expressed alone. When co-expressed with wild type KCNE1, A341V produced a slowly activating current, with a smaller current density, slower rates of activation, and a depolarized shift in its activation curve compared to the wild type KCNQ1+KCNE1. Confocal microscopy confirmed the surface expression of GFP-tagged A341V, suggesting a functionally defective protein. A T58A mutation in KCNE1 abolished functional restoration of A341V. Under heterozygous conditions, the expression of A341V+KCNQ1+KCNE1 reduced but did not abolish the electrophysiological changes observed in A341V+KCNE1. A dominant negative effect of A341V was also observed. Action potential simulations revealed that the A341V mutation is arrhythmogenic.
Conclusions
The KCNE1 β-subunit partially rescued the non-functional A341V mutant, with electrophysiological properties distinct from the wild type IKs.
General Significance
The severity of the A341V phenotype may be due to a combination of a significant suppression of the IKs with altered biophysical characteristics.
Keywords: long QT syndrome, IKs, A341V, KCNE1, arrhythmia, action potentials
1. Introduction
The long QT syndrome (LQTS) is an inherited or acquired form of cardiac arrhythmia with abnormal prolongation of the QT interval in the electrocardiogram, which can lead to syncope and sudden death, usually due to ventricular fibrillation [1]. The slowly activating delayed-rectifier potassium current (IKs) mediates repolarization of the cardiac action potential, and its dysfunction is one of the leading causes of inherited LQTS [2]. The IKs channel consists of at least two subunits: the pore-forming α-subunit and accessory β-subunit encoded by KCNQ1 and KCNE1, respectively [3]. The KCNQ1 subunit is composed of six transmembrane regions (S1-S6) with the voltage-sensor at S4 and KCNE1 is a single transmembrane protein [4]. KCNQ1 alone forms a functional channel with fast activation kinetics [5], but it is the co-assembly of KCNQ1 with KCNE1 that produces the salient biophysical features of the native IKs channel with the characteristic slow rate of activation [6-8].
LQTS caused by mutations in KCNQ1 is categorized as long QT syndrome type 1 or LQT1. LQT1 is the most common variant of the inherited form of LQTS [9]. A number of LQT1 associated mutations have been identified that result in the prolongation of the QT interval due to reduction of the IKs [10, 11]. One of the most common mutations described to date is the substitution of alanine at position 341 to valine in the S6 transmembrane domain of KCNQ1 [12-16]. This A341V substitution is associated with an unusually severe phenotype [17]. Compared to the overall LQT1 population, the mutation carriers of A341V have been found to be symptomatic earlier, with a 79% incidence of a first cardiac event by 40 years of age [18]. Furthermore, compared with the event-free survival rate of 80% by age 15 within carriers of LQT1 mutations, carriers of the specific A341V mutation have a significantly lower rate of 20% [18].
Previous studies have reported on the electrophysiological characteristics of A341V expressed in Chinese hamster ovary (CHO) cells [18], Xenopus oocytes [19] or COS7 cells [20]. Depending on the expression system, the biophysical properties of the expressed mutant current were varied, ranging from a dominant negative suppression to loss of function in the absence of a dominant negative effect. Thus, the mechanism underlying the electrophysiological changes triggered by A341V is unclear. As a result, there is a discrepancy correlating the clinical phenotype to the physiological outcome of the A341V mutant, whereby a loss of function may not account for the observed clinical severity related to this specific mutation. This is likely due to the various inherent differences in the expression system. Consequently, the aim of this study was to characterize the electrophysiological properties of A341V in a cardiac environment by utilizing HL-1 cells. These cells, derived from mouse atrial cardiomyocytes, maintain the ability to contract and retain the differentiated cardiac morphological, biochemical and electrophysiological properties [21, 22]. In particular, the role of the KCNE1 subunit in modulating A341V was determined.
2. Materials and Methods
2.1. Cell Culture and Transfection
Experiments were conducted using the HL-1 cardiac cell line, kindly provided by Dr. William Claycomb (Louisiana State University Health Science Center, New Orleans, LA). Cells were cultured in Claycomb Medium (SAFC Biosciences, Lenexa, KS) supplemented with 10% fetal bovine serum, 4 mM L-glutamine, 10 μM noradrenaline and Penicillin/Streptomycin as previously described [21].
Cells were transiently transfected with the cDNAs using Lipofectamine 2000 CD (Invitrogen, Carlsbad, CA). Human KCNQ1 and KCNE1 cDNAs cloned into pcDNA3.1 vector (Invitrogen) under the CMV promoter were generous gifts from Dr. Michael Sanguinetti (University of Utah, Salt Lake City, UT). Briefly, for a 35 mm dish of HL-1 cardiomyocytes, 2.4 μg of human KCNQ1 cDNA alone or with 2.4 μg of human KCNE1 cDNA were premixed with 0.8 μg of green fluorescent protein (GFP) cDNA and Lipofectamine 2000 CD (ratio of cDNAs : Lipofectamine 2000 CD was 1 μg : 3 μl) in 0.4 ml serum-free medium and incubated 20 min at room temperature. The solution was then added to the dish and cells were grown at 37 °C for 6 h, when medium was changed. After 2-3 days, the cells were trypsinized and used for recording. Only those cells showing GFP fluorescence were used for the electrophysiological studies.
2.2. Site-directed Mutagenesis
The mutant KCNQ1 A341V was constructed using the QuikChange Site-Directed Mutagenesis Kit (Agilent, Palo Alto, CA) and confirmed by DNA sequencing. To assess the interaction between A341V and KCNE1, a mutant KCNE1 T58A (substitution of a threonine residue to an alanine at position 58) was also constructed and confirmed by DNA sequencing. To determine the cellular localization of A341V, a GFP-tagged A341V was constructed for visualization by confocal microscopy. The A341V mutant was cloned in-frame into the pcDNA3.1-CT-GFP-topo vector (Invitrogen), thereby creating a GFP fusion with the C-terminus of mutant KCNQ1 A341V.
2.3. Electrophysiology
A drop of cells suspended in a modified Tyrode solution was placed in a flow-through chamber mounted on the stage of an inverted microscope. Patch pipettes were pulled from borosilicate glass capillaries (Garner Glass, Claremont, CA) with a micropipette puller (PC-10; Narishige, Tokyo, Japan) and heat-polished by using a microforge (MF-830; Narishige). The resistance of the recording pipettes, when filled with pipette solution and immersed in the modified Tyrode solution, ranged from 3 to 5 MΩ. Standard whole-cell configuration of the patch clamp technique was used to monitor currents at room temperature using an Axopatch 200B amplifier and Digidata 1322A interface (Axon Instruments, Foster City, CA). The pClamp10 software (Axon Instruments) was used for data acquisition and analysis. Current signal was sampled at 10 kHz and low-pass filtered at a cutoff frequency of 3 kHz.
The voltage-dependence of current activation was determined by constructing a normalized isochronal activation curve. The activation curves were fitted to a Boltzmann equation of the following form: I/Imax=(A-B)/[1+exp((V0.5-Vt)/k)]+B, where I/Imax is the normalized current amplitude, V0.5 is the voltage of half-maximal activation, Vt is the test pulse potential, k is the slope factor, and A and B are constants. Activation time to half-maximal amplitude (T0.5) was measured at +60 mV. The deactivating tail current was monitored on return to -50 mV from a +60 mV test pulse and fitted with a standard double-exponential function: I(t)=A1·exp(-t/τf)+A2·exp(-t/τs)+C, where I is the current amplitude; τf and τs are the fast and slow time constants, respectively, and A1, A2 and C are constants.
2.4. Confocal Microscopy
A confocal microscope (Nikon C1, Tokyo, Japan) was used to visualize the cellular localization of the GFP-tagged A341V. GFP fluorescence was excited using an argon laser at 488 nm and emission was recorded using a B-2E/C filter.
2.5. Solutions and Drugs
All chemicals were purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise noted. Modified Tyrode solution contained 132 mM NaCl, 4.8 mM KCl, 1.2 mM MgCl2, 1.0 mM CaCl2, 5 mM dextrose, and 10 mM HEPES (pH adjusted to 7.4 with NaOH). The external solution contained 132 mM N-methyl-D-glucamine, 1 mM CaCl2, 2 mM MgCl2, 10 mM HEPES, 5 mM dextrose, 2 mM 4-aminopyridine (to block the transient outward potassium current), 0.2 mM CdCl2 (to block the L-type calcium current), 5 μM E-4031 (Wako, Osaka, Japan; to block the rapidly activating delayed-rectifier potassium current) and 0.1 mM KCl (pH adjusted to 7.4 with HCl). The pipette solution contained 60 mM K-glutamate, 50 mM KCl, 10 mM HEPES, 1 mM MgCl2, 11 mM EGTA, 1 mM CaCl2, and 5 mM K2-adenosine triphosphate (pH adjusted to 7.4 with KOH).
2.6. Simulation of the Action Potential
Simulations of the cardiac action potential were conducted using Cellular Open Resource, a modeling environment developed by the Oxford Cardiac Electrophysiology Group in the Department of Physiology at the University of Oxford (Great Britain) [23, 24]. Specifically, action potential simulations were run using a computational model of the human left ventricular epicardial myocyte developed by Iyer, Mazhari and Winslow with modifications [25]. This computational model utilized Markov models for the gating of cardiac ion channel currents.
2.7. Data Analysis and Statistical Analysis
Data are presented as mean ± SE. The number of experiments is denoted by n. Unpaired Student's t-tests and one-way ANOVA with the Scheffe's test for multiple pairwise comparisons were used for statistical analysis. Statistical analyses were performed using Origin 7 (OriginLab, Northampton, MA) and Prism 5 (GraphPad Software, Lo Jolla, CA). A value of P < 0.05 was considered statistically significant.
3. Results
3.1. Electrophysiological Properties of A341V
Fig. 1 shows the representative current traces of untransfected and transfected HL-1 cardiomyocytes. Untransfected cells and those transfected with the wild type KCNE1 alone did not exhibit any time-dependent outward current, demonstrating the absence of endogenous IKs and KCNQ1 in these cells (Fig. 1, Panels A and B). Cells transfected with the wild type KCNQ1 gene exhibited current with fast activation kinetics as previously described (Fig. 1C) [6, 7]. Co-expression of KCNQ1 with KCNE1 (KCNQ1+KCNE1) produced a slowly activating current that recapitulated the biophysical characteristics of IKs (Fig. 1D). In contrast, when A341V was transfected alone into the HL-1 cells, no time-dependent outward current was evident (Fig. 1E). Thus, this single point mutation rendered the channel non-functional. However, co-expression of A341V with KCNE1 (A341V+KCNE1) produced a slowly activating time dependent current, suggesting that KCNE1 restored, at least partially, function to A341V (Fig. 1F).
Fig. 1.

Representative whole cell current traces recorded from untransfected and transiently transfected HL-1 cardiomyocytes. A. Current recording from an untransfected HL-1 cardiomyocyte is shown. A small, time-independent background current was detectable. The inset shows the 4-s test pulse voltage protocol. B. Current recording from an HL-1 cell transiently expressing KCNE1 β-subunit is presented. No difference was observed compared to the untransfected HL-1 cardiomyocyte. C. HL-1 cell expressing the KCNQ1 α-subunit resulted in functional channels with fast activation kinetics. D. The co-expression of KCNQ1 with KCNE1 resulted in functional channels with slow activation kinetics, characteristic of native wild type IKs. E. The KCNQ1 A341V mutant did not result in functional channels. No differences were observed compared to the untransfected cells. F. The co-expression of A341V with KCNE1 resulted in functional channels with markedly slow activation kinetics.
Although KCNE1 appeared to functionally rescue A341V, the electrophysiological features were distinct from those of the wild type KCNQ1+KCNE1. As shown in Fig. 2A, the current density of A341V+KCNE1 was significantly smaller than that of KCNQ1+KCNE1. For comparisons, current densities obtained from non-transfected HL-1 cells and those transfected with KCNE1 alone are also depicted. The suppressed current density of A341V+KCNE1 was accompanied by a significant depolarizing shift with a steeper slope in the voltage dependence of activation compared to KCNQ1+KCNE1 (Fig. 2B and Table 1). The resultant mutant also exhibited markedly slower current activation kinetics but had a faster deactivating tail current as demonstrated in Fig. 2C. To quantify the current activation kinetics, the activation time to half-maximal amplitude was determined during a test pulse to +60 mV. The deactivation kinetics were quantified by fitting a standard double-exponential function to the tail current. The results are summarized in Table 1 and indicate significantly slower activation and faster deactivation kinetics for the A341V+KCNE1 current.
Fig. 2.

Electrophysiological comparison between wild type KCNQ1+KCNE1 and A341V+KCNE1. A. Current-voltage relationships of KCNQ1+KCNE1 and A341V+KCNE1. Current amplitudes were normalized to cell capacitance to yield current density (n=8-11/group). The current densities obtained from untransfected HL-1 cells and cells transfected with KCNE1 alone are also depicted (n=7-8/group). Significant differences were observed between the A341V+KCNE1 and KCNQ1+KCNE1 groups (P < 0.01 versus KCNQ1+KCNE1). B. Voltage dependence of activation of KCNQ1+KCNE1 and A341V+KCNE1. The isochronal activation curves were fitted with the Boltzmann equation as described in Materials and methods (n=8-11/group; Table 1). C. Comparisons of current activation and deactivation kinetics between A341V+KCNE1 and KCNQ1+KCNE1. Currents were monitored during a test pulse to +60 mV from a -50 mV holding potential, peak-adjusted, and superimposed. The activation and deactivation parameters are summarized in Table 1.
Table 1. Biophysical Variables of Wild Type IKs (KCNQ1+KCNE1) and A341V Constructs.
| KCNQ1+KCNE1 | A341V+KCNE1 | A341V[GFP]+KCNE1 | A341V+2KCNE1 | |
|---|---|---|---|---|
| V0.5, mV | 35.6 ± 0.9 | 46.2 ± 0.9* | 40.4 ± 1.3 | 41.4 ± 0.7 |
| k | 16.3 ± 0.8 | 9.0± 0.8* | 7.2 ± 0.9 | 5.9 ± 0.7 |
| T0.5, ms | 447 ± 76 | 1172 ± 72* | 1231 ± 69 | 980 ± 90 |
| τf, ms | 215 ± 15 | 142 ± 21 † | 163 ± 9 | 117 ± 5 |
| τs, ms | 2357 ± 465 | 564 ± 74 * | 760 ± 90 | 628 ± 67 |
| n | 8 | 11 | 7 | 11 |
V0.5 = voltage of half-maximal activation; k = slope factor; T0.5 = activation time to half-maximal amplitude at +60 mV; τf = fast time constant; τs = slow time constant; n = number of experiments
V0.5, k, T0.5, τf and τs were determined as described in Methods. Data are presented as mean ± SE.
P < 0.01 versus KCNQ1+KCNE1,
P < 0.05 versus KCNQ1+KCNE1
3.2. Cellular Localization of the A341V Mutant
The co-expression of A341V with KCNE1 culminated in a functional channel, suggesting the ability of KCNE1 to recover, at least partially, the non-functional mutant. That KCNE1 can partially restore channel activity to A341V in a cardiac environment may contribute to some of the observed discrepancies regarding the dominant negative suppression of the channel. To determine whether this partial “rescue” is due to restoration of trafficking of the A341V to the sarcolemma, or a functional rescue due to the interaction of A341V with KCNE1, a GFP-tagged A341V (A341V[GFP]) was constructed for visualization by confocal microscopy. We first established that A341V[GFP] acted in a manner analogous to the original A341V construct to confirm that the GFP did not alter any channel properties. As shown in Fig. 3A, A341V[GFP] alone did not express a functional channel, while the co-expression with KCNE1 restored the channel activity as expected (Fig. 3B). There were no significant differences in the biophysical characteristics between A341V[GFP]+KCNE1 and A341V+KCNE1 (Fig. 3C and 3D; Table 1). Confocal microscopy revealed that both A341V[GFP] and A341V[GFP]+KCNE1 were similarly expressed on the HL-1 sarcolemmal membrane (Fig. 3, E and F). This suggested that the A341V was a non-functional protein and its rescue by KCNE1 was due to a functional interaction between the two subunits, and likely not due to restoration of trafficking of A341V to the sarcolemmal membrane.
Fig. 3.
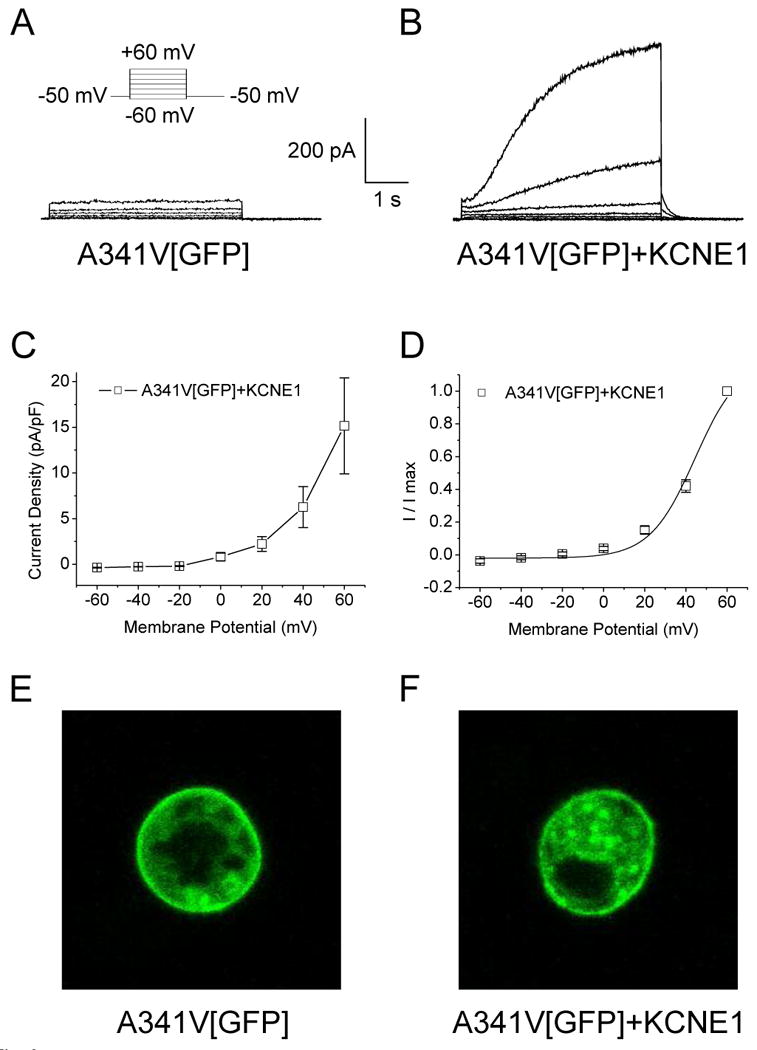
Cellular localization of A341V expressed in HL-1 cells. A GFP-tagged A341V (A341V[GFP]) construct was transiently expressed in HL-1 cells. Representative current traces of A341V[GFP] in the absence (A) or co-expressed with KCNE1 (B) are shown. The current-voltage relationship, normalized to cell capacitance (C) and the isochronal activation curve (D) for A341V[GFP]+KCNE1 (n=7) revealed no significant changes compared to those of A341V+KCNE1 (in the absence of GFP; Table 1). Cellular localization of A341V[GFP] in the absence (E) or co-expressed with KCNE1 (F) were visualized by confocal microscopy. In both cases, surface expression of the mutant protein was evident.
3.3. Dependence on KCNE1 in Restoring A341V Function
The results obtained above suggested that the interaction of A341V with KCNE1 was critical for functional restoration of the mutant protein. To test this hypothesis, a mutant KCNE1, T58A, was co-expressed with A341V. The T58 residue on KCNE1 has been reported to be essential for its interaction with KCNQ1, and mutation of this residue interferes with this interaction [26]. As shown in Fig. 4A, the T58A mutant was unable to functionally rescue A341V. This observation supported our hypothesis. Since previous reports have suggested that the stoichiometry of KCNQ1 and KCNE1 can be variable, whereby increasing KCNE1 have been demonstrated to slow activation kinetics [27], we subsequently tested whether increasing the expression of KCNE1 can further restore the A341V function closer to that of the wild type KCNQ1+KCNE1. HL-1 cells were co-transfected with a 1:2 cDNA ratio of A341V to KCNE1 (A341V+2KCNE1; 2.4 μg of A341V cDNA and 4.8 μg of KCNE1). Fig. 4B shows the representative current traces of A341V+2KCNE1, with a slowly activating time-dependent current with activation kinetics similar to those of A341V+KCNE1. There were no significant differences in the current-voltage relationships and the isochronal activation curves between A341V+2KCNE1 and A341V+KCNE1 (Table 1 and Fig. 4, C and D). Thus, further restoration of the A341V mutant was not achieved by increasing KCNE1 twofold.
Fig. 4.

Dependence on KCNE1 in restoring function to A341V. A. Representative current traces recorded from an HL-1 cardiomyocyte co-expressing the KCNQ1 A341V mutant with a KCNE1 T58A mutant. The T58A mutant was unable to restore function to A341V. B. Current recordings were obtained from an HL-1 cardiomyocyte transiently transfected with 2.4 μg of A341V and 4.8 μg of KCNE1 cDNA (A341V+2KCNE1; 1:2 ratio of A341V to KCNE1). The corresponding current-voltage relationship (C) and isochronal activation curve (D) for A341V+2KCNE1 are shown (n=11). No significant changes were observed compared to the A341V+KCNE1 group (Table 1).
3.4. Impact of A341V under Heterozygous Conditions
The results above revealed that when the cDNAs for A341V and KCNE1 were transfected into HL-1 cells in a 1:1 ratio, a functional channel was formed with significantly altered biophysical characteristics compared to the wild type IKs. This could potentially address the severe clinical phenotype associated with the specific mutation of the KCNQ1 protein. However, since the heterozygous expression of the wild type KCNQ1 and KCNE1 with A341V better mimics the clinical setting, experiments were conducted on HL-1 cells transfected with the cDNAs of each in a 1:1:1 (2.4 μg of cDNA/subunit) ratio. Fig. 5A shows a representative set of current traces of the expressed A341V+KCNQ1+KCNE1. When compared to the corresponding wild type IKs recorded from an HL-1 cell transfected with 4.8 μg of KCNQ1 and 2.4 μg of KCNE1 cDNAs (2KCNQ1+KCNE1; Fig. 5A, right panel), the A341V+KCNQ1+KCNE1 current was markedly smaller. As summarized in Fig. 5B, the current density in cells expressing A341V+KCNQ1+KCNE1 was significantly smaller than in those expressing 2KCNQ1+KCNE1. Comparison of the isochronal activation curves also showed a significant depolarizing shift in A341V+KCNQ1+KCNE1 compared to the 2KCNQ1+KCNE1 wild type (Fig. 5C and Table 2). The A341V+KCNQ1+KCNE1 channel also exhibited slower current activation kinetics than the wild type, as shown in Fig. 5D. The activation time to half-maximal current amplitude of A341V+KCNQ1+KCNE1 was significantly longer than that of 2KCNQ1+KCNE1 (Table 2). In contrast, the deactivating tail current of A341V+KCNQ1+KCNE1 had faster kinetics where the slow time constant was significantly smaller than that of 2KCNQ1+KCNE1 (Table 2). Based on these results, significant changes in the biophysical characteristics of A341V+KCNQ1+KCNE1 were still apparent compared to the wild type in a heterozygous setting.
Fig. 5.

The impact of A341V under heterozygous conditions. A. To mimic the heterozygous clinical conditions, current recordings were obtained in HL-1 cardiomyocytes expressing A341V+KCNQ1+KCNE1 and compared to those with 2xKCNQ1+KCNE1 (2KCNQ1+KCNE1). Representative recordings from each group are shown. The corresponding current-voltage relationships (B) and isochronal activation curves (C) are shown for each group (n=11-12/group). P < 0.01 versus 2KCNQ1+KCNE1, †P < 0.05 versus 2KCNQ1+KCNE1. The activation curves were fitted with the Boltzmann equation and the parameters are summarized in Table 2. D. Comparisons of the current activation and deactivation kinetics between A341V+KCNQ1+KCNE1 and 2KCNQ1+KCNE1 are demonstrated. Currents were monitored during a depolarizing test pulse to +60 mV from a -50 mV holding potential, peak-adjusted, and superimposed. The activation and deactivation parameters are summarized in Table 2. E. The current density (obtained from a +60 mV test pulse) of A341V+KCNQ1+KCNE1 was compared to that of KCNQ1+KCNE1 to determine whether A341V triggered a dominant negative effect. A significant suppression of current density in the presence of A341V was observed (n=8-12/group), indicative of a dominant negative effect.
Table 2. Biophysical Variables of A341V Constructs: Heterozygous Clinical Situation (A341V+KCNQ1+KCNE1) and Corresponding Wild Type IKs (2KCNQ1+KCNE1).
| A341V+KCNQ1+KCNE1 | 2KCNQ1+KCNE1 | |
|---|---|---|
| V0.5, mV | 30.2 ± 1.2 * | 23.3 ± 1.7 |
| k | 12.2 ± 0.5 † | 14.6 ± 0.7 |
| T0.5, ms | 514 ± 55 * | 263 ± 30 |
| τf, ms | 197 ± 9 | 219 ± 19 |
| τs, ms | 729 ± 74 * | 2095 ± 239 |
| n | 12 | 11 |
V0.5 = voltage of half-maximal activation; k = slope factor; T0.5 = activation time to half-maximal amplitude at +60 mV; τf = fast time constant; τs = slow time constant; n = number of experiments
V0.5, k, T0.5, τf and τs were determined as described in Methods. Data are presented as mean ± SE.
P < 0.01 versus 2KCNQ1+KCNE1,
P < 0.05 versus 2KCNQ1+KCNE1
To address whether the impact of A341V on KCNQ1+KCNE1 was due to a dominant negative effect, we compared the current amplitudes recorded at a test pulse of +60 mV of two transfected groups: KCNQ1+KCNE1 and A341V+KCNQ1+KCNE1. The results are summarized in Fig. 5E. A significant difference between the A341V+KCNQ1+KCNE1 and KCNQ1+KCNE1 groups was observed. This result suggested that the suppression of current by A341V was due to a dominant negative effect.
3.5. Simulation of the Impact of A341V on the Action Potential
To determine the impact of the significant changes in the biophysical characteristics incurred by A341V under heterozygous conditions on the action potential, simulations were conducted using a modified computational model of the human left ventricular epicardial myocytes by Iyer, Mazhari and Winslow [25]. The model was modified according to the changes in whole-cell current obtained under the heterozygous condition. Because the computational model by Iyer, Mazhari and Winslow was empirically derived and based on the time-rate of change of channel-state occupancy probabilities, our ability to modify the parameters was limited to relative changes. Specifically, activation kinetics and deactivation kinetics were slowed and accelerated, respectively, by a factor of 2 in the mutant (A341V+KCNQ1+KCNE1) compared to the wild type (2KCNQ1+KCNE1). Furthermore, initial current density was halved in the mutant relative to the wild type. The resultant simulations are shown in Fig. 6. In the wild type (control) case, stimulation frequencies of 2 and 2.8 Hz did not result in the deformation of the action potential profile (Panel A). In contrast, computer simulations in the A341V mutant case yielded a different outcome. While a stimulation frequency of 2 Hz yielded a normal action potential profile, stimulations at 2.8 Hz triggered arrhythmias as evidenced by the altered, complex action potential waveforms (Panel B). In particular, key features in this simulation of the arrhythmogenic response to the A341V mutation were a progressive reduction in the amplitude of the action potential upstroke, a gradual depolarization of the baseline resting membrane potential, the failure to elicit a full action potential near the 5.5-second mark, and subsequent prolongation of the action potential duration. The small but gradually increasing depolarization of the resting membrane potential could account for the accompanying decrease in the upstroke amplitude via decrease in the sodium (Na) current. This is shown in the corresponding simulated Na current amplitude (Figure 7, Panel A). There was a notable time-dependent decrease in the Na current amplitude likely due to accumulation of channel inactivation as the result of the gradual depolarization of the resting membrane potential. This ultimately led to the failure to elicit an action potential near the 5.5-second mark. Subsequently, as sufficient sodium channels recovered from inactivation, an action potential was triggered near the 6-second mark. However, the resulting prolonged action potential again prevented the subsequent elicitation of an action potential until the sodium channel has recovered sufficiently from inactivation. The A341V mutation also resulted in a marked loss of IKs with slower activation kinetics compared to the wild type control, as expected (Figure 7, Panel B).
Fig. 6.

Impact of A341V: simulation of the action potential. A computational model of the human left ventricular epicardial myocytes developed by Iyer, Mazhari and Winslow was utilized. Results were obtained from two stimulation frequencies, 2 and 2.8 Hz, are depicted. A. Under control conditions (wild type IKs), normal action potential profiles were obtained at both stimulation frequencies. No evidence of arrhythmogenicity was evident. B. The computational model was modified to account for changes in the current density and in the activation and deactivation kinetics observed in the mutant IKs (A341V+KCNQ1+KCNE1). At the lower stimulation frequency of 2 Hz, a normal action potential profile was obtained. In contrast, at the higher stimulation frequency of 2.8 Hz, the action potential profile exhibited characteristics indicative of arrhythmias.
Fig. 7.

Simulation of INa and IKs. Simulations of the sodium (INa, Panel A) and potassium (IKs, Panel B) currents were conducted using the Iyer, Mazhari and Winslow model. Resultant currents were obtained at a stimulation frequency of 2.8 Hz in control (wild type) and in the A341V mutant (A341V+KCNQ1+KCNE1). A progressive decrease in INa amplitude accompanied by a depressed IKs current with slower activation kinetics was observed in the A341V mutant compared to the control.
4. Discussion
The results from this study utilizing a cardiac cell line revealed a previously unreported effect of the KCNE1 β-subunit to “restore” function to the A341V mutant protein. A341V alone was non-functional, consistent with a previous finding [28]. However, studies have also reported that A341V co-expressed with KCNE1 did not produce measurable current in Xenopus oocytes [28] and COS7 cells [20]. The discrepancy is likely due to the different expression systems used in the studies. Our choice of using the HL-1 cardiac cell line better recapitulated the physiological environment of the IKs channel. Our results show that the mechanism underlying the functional recovery of A341V was via its interaction with the KCNE1 β-subunit at the level of the sarcolemmal membrane. Defective trafficking of KCNQ1 has been reported for several LQTS associated mutations [29, 30]. For A341V, this was likely not the case as surface expression of the mutant protein was evident. However, we cannot entirely exclude the possibility of diminished trafficking as a contributing factor. Nevertheless, the surface expression of A341V and the absence of current confirmed that the mutant was a non-functional protein.
The use of the KCNE1 T58A mutant confirmed that the functional interaction with KCNE1 was necessary to restore function to A341V. The amino acids 338-340 of KCNQ1 and 57-59 of KCNE1 are critical for the functional interaction between the KCNQ1 and KCNE subunits [31, 32], and a key interaction between T58 of KCNE1 and F340 of KCNQ1 has been identified [26]. The T58A mutation on KCNE1 interferes with the β-subunit's interaction with F340 on KCNQ1. In the absence of this interaction, the T58A mutant was unable to impart function to the A341V mutant. Interestingly, the electrophysiological properties of the functionally rescued A341V construct (A341V+KCNE1) were significantly different from those of the wild type IKs (KCNQ1+KCNE1). Specifically, a decrease in the mutant current density was accompanied by a depolarizing shift and a greater slope factor in the isochronal activation curve, indicative of a very steep voltage-dependence of activation. The A341V+KCNE1 also exhibited a significantly slower rate of current activation, but had a faster rate of deactivation. One caveat to the decreased current density of the mutant is the possibility that KCNE1 did not co-assemble equally well with A341V as compared to its assembly with KCNQ1. The “reluctance” of KCNE1 to co-assemble with A341V could, at least in part, account for the decreased density. This may also play a role in the dominant negative effect observed in the heterozygous condition. However, the “reluctance” to co-assemble likely would not account for the changes in the biophysical characteristics exhibited by the mutant channel.
Although the molecular mechanism underlying the non-functional outcome of the A341V mutation has not been determined, this alanine residue is predicted to form part of the wall of the KCNQ1 central cavity [33]. Consequently, the abrogation of function of KCNQ1 by this mutation suggested a structural collapse or instability of the pore region. How an interaction with KCNE1 would lead to restoration of function to the mutant is unknown, but a “stabilization” of the pore region by the β-subunit is implicated based on our results. Since the restored functional characteristics were significantly different from those of the wild type IKs, the “stabilization” was likely to have been partial.
The precise molecular nature of the interaction between KCNE1 and KCNQ1 has not been established. Recent studies have shown that KCNE1 stabilizes the voltage-sensing S4 segment on KCNQ1 and slows activation [34], but others have demonstrated that it does not affect the equilibration rate of the KCNQ1 voltage sensor [35]. Furthermore, cytoplasmic and extracellular domains have also been identified in the interactions between the two subunits that underlie IKs [36-38]. Our results implicate a modification of the coupling between A341V and KCNE1 compared to the wild type situation as evidenced by the significant alteration in the voltage-dependence of gating in the mutant current. Whether the potential instability triggered by this mutation is specific to a valine substitution has yet to be established. Previous studies using the oocyte expression system reported a functional KCNQ1 mutant channel with an A341C mutation (substitution of alanine at position 341 to cysteine) [33]. However, the A341C mutant construct expressed in HL-1 cells resulted in a non-functional channel (data not shown), similar to the A341V. Therefore, in a cardiac environment, alanine at position 341 in the S6 domain of KCNQ1 may play a key role in stabilizing the channel pore region.
Previous studies have reported that the A341V mutation had little or no dominant negative effect in Xenopus oocytes [39] or COS7 cells [20]. These results implied that under heterozygous conditions, KCNQ1 did not co-assemble with A341V, and consequently, loss of channel function was attributed to the mutant phenotype. In contrast, in CHO cells, A341V was found to exert a dominant negative effect on IKs [18]. In our present study using the HL-1 cardiac cells, the current density of A341V+KCNQ1+KCNE1 was significantly smaller than that of KCNQ1+KCNE1, implicative of a dominant negative effect and indicative of a channel assembly that includes KCNQ1, A341V and KCNE1. In this specific regard, the results are similar to those reported in CHO cells. However, in addition to the suppression of current, our results also showed altered biophysical characteristics in the resultant current. The combination of suppression and altered biophysical characteristics of the current is likely a significant contributor to the clinically observed A341V phenotype.
The electrophysiological impact of the altered channel characteristic was demonstrated in a computational model of the human left ventricular epicardial myocytes. Compared to the wild-type control, a high stimulation frequency triggered arrhythmias in the A341V mutant. At the lower frequency, arrhythmias were not present. Thus, the A341V mutation was arrhythmogenic. Interestingly, based on the model, this was due a progressive reduction in the Na current amplitude as the result of a gradual depolarization of the resting membrane potential. This would suggest that the A341V mutation can trigger alterations in cardiac excitability and potentially lead to conduction block. A limitation in our simulation is that the relative changes in current kinetics were based on our recordings conducted at room temperature while the computational model by Iyer, Mazhari and Winslow used gating kinetics at physiological temperature. The results from our simulations could potentially extrapolate to a more severe breakdown in the action potential profile at physiological temperature due to temperature-dependent changes in the current kinetics.
The basis of the discrepancy in the results obtained in our study using HL-1 cells compared to those from Xenopus oocytes or COS7 cells is not entirely clear. HL-1 cells are a commonly used cardiac cell line that retains the gene expression pattern of normal adult myocytes [21]. In contrast to other cardiac cell lines, HL-1 cells can be repeatedly passaged without reverting to an embryonic phenotype. Thus, this is a distinct advantage over other cardiac cell lines. Since HL-1 cells are derived from atrial myocytes, some of the distinct cellular factors pertinent to a ventricular environment may be lacking. On the other hand, in the absence of a comparable cell line derived from ventricular myocytes, these cells have been used quite extensively as a cell culture model in the investigation of cardiac signal transduction pathways, oxidative stress, and transcriptional regulation [22]. An argument can also be made that differences in the cellular environment in the atrial and ventricular cardiomyocytes are likely smaller than those between HL-1 cells and Xenopus oocytes. However, the precise properties among the various expression systems that would account for the discrepant results of A341V are not known. A recent study has shown that ion channel proteins diffuse at different rates when expressed in HEK293 cells compared to HL-1 cells [40]. This was likely due to differences in the lipid environment and cytoskeleton between the two cell lines. Thus, variations in the microenvironment of ion channels in the various expression systems can potentially contribute to some of the observed functional differences.
Conclusions
In a cardiac environment, the KCNE1 β-subunit restored functionality to a relatively common LQT1 associated mutant, A341V. The resultant biophysical characteristics were significantly altered from the wild type IKs. These changes were diminished under clinically relevant heterozygous conditions, but remained significantly different from the wild type. Based on a computational model, particularly under conditions where the heart rate is high, the observed electrophysiological changes triggered by A341V may account for some of the severe clinical phenotype associated with this mutation.
Research Highlights.
KCNE1 functionally restored a long QT syndrome-associated A341V mutant.
The resultant A341V+KCNE1 current had distinct biophysical characteristics.
A341V had a dominant negative effect on channel current.
Heterozygous expression of A341V+KCNQ1+KCNE1 altered cardiac excitability in a computational model of ventricular action potentials.
These changes contribute to the severe clinical phenotype of carriers of A341V.
Acknowledgments
We thank Dr. Sang H. Lee and his graduate student, Eric Danielson, Department of Pharmacology & Toxicology, Medical College of Wisconsin, Milwaukee, Wisconsin, USA, for their expert help in acquiring the confocal images. We also thank Dr. J. Bruce McCallum, Department of Anesthesiology, Medical College of Wisconsin, for his expert advice and input on statistical analysis.
Disclosures: This study was supported by National Institutes of Health grant RO1-GM-067675 (to WMK) and the Medical College of Wisconsin Diversity Summer Health-related Research Education Program (to CT).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Ikuomi Mikuni, Department of Anesthesiology, Medical College of Wisconsin.
Carlos G. Torres, Department of Anesthesiology, Medical College of Wisconsin.
Martin W. Bienengraeber, Departments of Anesthesiology and Pharmacology & Toxicology, Medical College of Wisconsin.
Wai-Meng Kwok, Departments of Anesthesiology and Pharmacology & Toxicology, Medical College of Wisconsin.
References
- 1.Roden DM. Clinical practice. Long-QT syndrome. The New England Journal of Medicine. 2008;358:169–176. doi: 10.1056/NEJMcp0706513. [DOI] [PubMed] [Google Scholar]
- 2.Keating MT, Sanguinetti MC. Molecular and cellular mechanisms of cardiac arrhythmias. Cell. 2001;104:569–580. doi: 10.1016/s0092-8674(01)00243-4. [DOI] [PubMed] [Google Scholar]
- 3.Jespersen T, Grunnet M, Olesen SP. The KCNQ1 potassium channel: from gene to physiological function. Physiology (Bethesda, Md. 2005;20:408–416. doi: 10.1152/physiol.00031.2005. [DOI] [PubMed] [Google Scholar]
- 4.Melman YF, Krummerman A, McDonald TV. KCNE regulation of KvLQT1 channels: structure-function correlates. Trends in Cardiovascular Medicine. 2002;12:182–187. doi: 10.1016/s1050-1738(02)00158-5. [DOI] [PubMed] [Google Scholar]
- 5.Sesti F, Goldstein SA. Single-channel characteristics of wild-type IKs channels and channels formed with two minK mutants that cause long QT syndrome. The Journal of General Physiology. 1998;112:651–663. doi: 10.1085/jgp.112.6.651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barhanin J, Lesage F, Guillemare E, Fink M, Lazdunski M, Romey G. K(V)LQT1 and lsK (minK) proteins associate to form the I(Ks) cardiac potassium current. Nature. 1996;384:78–80. doi: 10.1038/384078a0. [DOI] [PubMed] [Google Scholar]
- 7.Sanguinetti MC, Curran ME, Zou A, Shen J, Spector PS, Atkinson DL, Keating MT. Coassembly of K(V)LQT1 and minK (IsK) proteins to form cardiac I(Ks) potassium channel. Nature. 1996;384:80–83. doi: 10.1038/384080a0. [DOI] [PubMed] [Google Scholar]
- 8.Yang Y, Sigworth FJ. Single-channel properties of IKs potassium channels. The Journal of General Physiology. 1998;112:665–678. doi: 10.1085/jgp.112.6.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morita H, Wu J, Zipes DP. The QT syndromes: long and short. Lancet. 2008;372:750–763. doi: 10.1016/S0140-6736(08)61307-0. [DOI] [PubMed] [Google Scholar]
- 10.Splawski I, Shen J, Timothy KW, Lehmann MH, Priori S, Robinson JL, Moss AJ, Schwartz PJ, Towbin JA, Vincent GM, Keating MT. Spectrum of mutations in long-QT syndrome genes. KVLQT1, HERG, SCN5A, KCNE1, and KCNE2. Circulation. 2000;102:1178–1185. doi: 10.1161/01.cir.102.10.1178. [DOI] [PubMed] [Google Scholar]
- 11.Moss AJ, Shimizu W, Wilde AA, Towbin JA, Zareba W, Robinson JL, Qi M, Vincent GM, Ackerman MJ, Kaufman ES, Hofman N, Seth R, Kamakura S, Miyamoto Y, Goldenberg I, Andrews ML, McNitt S. Clinical aspects of type-1 long-QT syndrome by location, coding type, and biophysical function of mutations involving the KCNQ1 gene. Circulation. 2007;115:2481–2489. doi: 10.1161/CIRCULATIONAHA.106.665406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Donger C, Denjoy I, Berthet M, Neyroud N, Cruaud C, Bennaceur M, Chivoret G, Schwartz K, Coumel P, Guicheney P. KVLQT1 C-terminal missense mutation causes a forme fruste long-QT syndrome. Circulation. 1997;96:2778–2781. doi: 10.1161/01.cir.96.9.2778. [DOI] [PubMed] [Google Scholar]
- 13.Anastasakis A, Kotta CM, Kyriakogonas S, Wollnik B, Theopistou A, Stefanadis C. Phenotype reveals genotype in a Greek long QT syndrome family. Europace. 2006;8:241–244. doi: 10.1093/europace/eul012. [DOI] [PubMed] [Google Scholar]
- 14.Crotti L, Monti MC, Insolia R, Peljto A, Goosen A, Brink PA, Greenberg DA, Schwartz PJ, George AL., Jr NOS1AP is a genetic modifier of the long-QT syndrome. Circulation. 2009;120:1657–1663. doi: 10.1161/CIRCULATIONAHA.109.879643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heradien MJ, Goosen A, Crotti L, Durrheim G, Corfield V, Brink PA, Schwartz PJ. Does pregnancy increase cardiac risk for LQT1 patients with the KCNQ1-A341V mutation? Journal of the American College of Cardiology. 2006;48:1410–1415. doi: 10.1016/j.jacc.2006.05.060. [DOI] [PubMed] [Google Scholar]
- 16.Schwartz PJ, Vanoli E, Crotti L, Spazzolini C, Ferrandi C, Goosen A, Hedley P, Heradien M, Bacchini S, Turco A, La Rovere MT, Bartoli A, George AL, Jr, Brink PA. Neural control of heart rate is an arrhythmia risk modifier in long QT syndrome. Journal of the American College of Cardiology. 2008;51:920–929. doi: 10.1016/j.jacc.2007.09.069. [DOI] [PubMed] [Google Scholar]
- 17.Crotti L, Spazzolini C, Schwartz PJ, Shimizu W, Denjoy I, Schulze-Bahr E, Zaklyazminskaya EV, Swan H, Ackerman MJ, Moss AJ, Wilde AA, Horie M, Brink PA, Insolia R, De Ferrari GM, Crimi G. The common long-QT syndrome mutation KCNQ1/A341V causes unusually severe clinical manifestations in patients with different ethnic backgrounds: toward a mutation-specific risk stratification. Circulation. 2007;116:2366–2375. doi: 10.1161/CIRCULATIONAHA.107.726950. [DOI] [PubMed] [Google Scholar]
- 18.Brink PA, Crotti L, Corfield V, Goosen A, Durrheim G, Hedley P, Heradien M, Geldenhuys G, Vanoli E, Bacchini S, Spazzolini C, Lundquist AL, Roden DM, George AL, Jr, Schwartz PJ. Phenotypic variability and unusual clinical severity of congenital long-QT syndrome in a founder population. Circulation. 2005;112:2602–2610. doi: 10.1161/CIRCULATIONAHA.105.572453. [DOI] [PubMed] [Google Scholar]
- 19.Seebohm G, Pusch M, Chen J, Sanguinetti MC. Pharmacological activation of normal and arrhythmia-associated mutant KCNQ1 potassium channels. Circulation Research. 2003;93:941–947. doi: 10.1161/01.RES.0000102866.67863.2B. [DOI] [PubMed] [Google Scholar]
- 20.Kobori A, Sarai N, Shimizu W, Nakamura Y, Murakami Y, Makiyama T, Ohno S, Takenaka K, Ninomiya T, Fujiwara Y, Matsuoka S, Takano M, Noma A, Kita T, Horie M. Additional gene variants reduce effectiveness of beta-blockers in the LQT1 form of long QT syndrome. Journal of Cardiovascular Electrophysiology. 2004;15:190–199. doi: 10.1046/j.1540-8167.2004.03212.x. [DOI] [PubMed] [Google Scholar]
- 21.Claycomb WC, Lanson NA, Jr, Stallworth BS, Egeland DB, Delcarpio JB, Bahinski A, Izzo NJ., Jr HL-1 cells: a cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocyte. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:2979–2984. doi: 10.1073/pnas.95.6.2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.White SM, Constantin PE, Claycomb WC. Cardiac physiology at the cellular level: use of cultured HL-1 cardiomyocytes for studies of cardiac muscle cell structure and function. American Journal of Physiology. 2004;286:H823–829. doi: 10.1152/ajpheart.00986.2003. [DOI] [PubMed] [Google Scholar]
- 23.Garny A, Noble D, Hunter PJ, Kohl P. CELLULAR OPEN RESOURCE (COR): current status and future directions. Philosophical Transactions. 2009;367:1885–1905. doi: 10.1098/rsta.2008.0289. [DOI] [PubMed] [Google Scholar]
- 24.Tampo A, Hogan CS, Sedlic F, Bosnjak ZJ, Kwok WM. Accelerated inactivation of cardiac L-type calcium channels triggered by anaesthetic-induced preconditioning. British Journal of Pharmacology. 2009;156:432–443. doi: 10.1111/j.1476-5381.2008.00026.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Iyer V, Mazhari R, Winslow RL. A computational model of the human left-ventricular epicardial myocyte. Biophysical Journal. 2004;87:1507–1525. doi: 10.1529/biophysj.104.043299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Panaghie G, Tai KK, Abbott GW. Interaction of KCNE subunits with the KCNQ1 K+ channel pore. The Journal of Physiology. 2006;570:455–467. doi: 10.1113/jphysiol.2005.100644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang W, Xia J, Kass RS. MinK-KvLQT1 fusion proteins, evidence for multiple stoichiometries of the assembled IsK channel. The Journal of Biological Chemistry. 1998;273:34069–34074. doi: 10.1074/jbc.273.51.34069. [DOI] [PubMed] [Google Scholar]
- 28.Westenskow P, Splawski I, Timothy KW, Keating MT, Sanguinetti MC. Compound mutations: a common cause of severe long-QT syndrome. Circulation. 2004;109:1834–1841. doi: 10.1161/01.CIR.0000125524.34234.13. [DOI] [PubMed] [Google Scholar]
- 29.Boulet IR, Raes AL, Ottschytsch N, Snyders DJ. Functional effects of a KCNQ1 mutation associated with the long QT syndrome. Cardiovascular Research. 2006;70:466–474. doi: 10.1016/j.cardiores.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 30.Delisle BP, Anson BD, Rajamani S, January CT. Biology of cardiac arrhythmias: ion channel protein trafficking. Circulation Research. 2004;94:1418–1428. doi: 10.1161/01.RES.0000128561.28701.ea. [DOI] [PubMed] [Google Scholar]
- 31.Melman YF, Domenech A, de la Luna S, McDonald TV. Structural determinants of KvLQT1 control by the KCNE family of proteins. The Journal of Biological Chemistry. 2001;276:6439–6444. doi: 10.1074/jbc.M010713200. [DOI] [PubMed] [Google Scholar]
- 32.Melman YF, Um SY, Krumerman A, Kagan A, McDonald TV. KCNE1 binds to the KCNQ1 pore to regulate potassium channel activity. Neuron. 2004;42:927–937. doi: 10.1016/j.neuron.2004.06.001. [DOI] [PubMed] [Google Scholar]
- 33.Seebohm G, Chen J, Strutz N, Culberson C, Lerche C, Sanguinetti MC. Molecular determinants of KCNQ1 channel block by a benzodiazepine. Molecular Pharmacology. 2003;64:70–77. doi: 10.1124/mol.64.1.70. [DOI] [PubMed] [Google Scholar]
- 34.Nakajo K, Kubo Y. KCNE1 and KCNE3 stabilize and/or slow voltage sensing S4 segment of KCNQ1 channel. The Journal of General Physiology. 2007;130:269–281. doi: 10.1085/jgp.200709805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rocheleau JM, Kobertz WR. KCNE peptides differently affect voltage sensor equilibrium and equilibration rates in KCNQ1 K+ channels. The Journal of General Physiology. 2008;131:59–68. doi: 10.1085/jgp.200709816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu X, Jiang M, Hsu KL, Zhang M, Tseng GN. KCNQ1 and KCNE1 in the IKs channel complex make state-dependent contacts in their extracellular domains. The Journal of General Physiology. 2008;131:589–603. doi: 10.1085/jgp.200809976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chung DY, Chan PJ, Bankston JR, Yang L, Liu G, Marx SO, Karlin A, Kass RS. Location of KCNE1 relative to KCNQ1 in the I(KS) potassium channel by disulfide cross-linking of substituted cysteines. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:743–748. doi: 10.1073/pnas.0811897106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lvov A, Gage SD, Berrios VM, Kobertz WR. Identification of a protein-protein interaction between KCNE1 and the activation gate machinery of KCNQ1. The Journal of General Physiology. 135:607–618. doi: 10.1085/jgp.200910386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang Z, Tristani-Firouzi M, Xu Q, Lin M, Keating MT, Sanguinetti MC. Functional effects of mutations in KvLQT1 that cause long QT syndrome. Journal of Cardiovascular Electrophysiology. 1999;10:817–826. doi: 10.1111/j.1540-8167.1999.tb00262.x. [DOI] [PubMed] [Google Scholar]
- 40.Mashanov GI, Nobles M, Harmer SC, Molloy JE, Tinker A. Direct observation of individual KCNQ1 potassium channels reveals their distinctive diffusive behavior. The Journal of Biological Chemistry. 2010;285:3664–3675. doi: 10.1074/jbc.M109.039974. [DOI] [PMC free article] [PubMed] [Google Scholar]