

Abstract
Prostaglandin (PG)I2 has important regulatory functions on the innate and adaptive immune systems. Recent experimental evidence reveals that PGI2 modulates the development and function of CD4+ T cells subsets, including Th1, Th2, and Th17 cell responses. In vitro and in vivo studies support that PGI2 generally has an inhibitory effect on Th1 and Th2 activation, differentiation, and cytokine production. In contrast, PGI2 seems to enhance Th17-favoring polarization conditions, resulting in Th17 cytokine production. Therefore, PGI2 may either promote or inhibit individual CD4+ cell subsets and impact adaptive immune responses.
Keywords: PGI2, T cell, Interferon-γ, Dendritic cell
Introduction
In 1976, a research group from the Royal College of Surgeons in the UK led by Sir John Vane, was the first to describe a novel eicosanoid extracted from the aortas of sheep and pigs [1]. The molecule was initially termed PGX by the research team. This investigative team found that a synthetic form of this molecule, which they termed epoprostenol, was over 30 times more potent than PGE2 in inhibiting human platelet aggregation. The molecule was also found to cause relaxation of certain isolated blood vessels and smooth muscle constriction of gastrointestinal muscles, and was later renamed PGI2 [2].
PGI2 is formed from diacylglycerols or cell membrane phospholipids that enter the arachidonic acid metabolic pathway [3]. Phospholipase A2 hydorlyzes fatty acids at the sn-2 position of membrane phospholipids, forming free fatty acids including arachidonic acid. Arachidonic acid is then converted to PGH2, an unstable intermediate, by the cyclooxygenase (COX) enzymes, COX-1 or COX-2. PGI synthase (PGIS) is the terminal enzyme that converts PGH2 into PGI2 and is most abundantly expressed in vascular tissues. PGI2 is a transient molecule, with a half life of only a few seconds within the body and signals through a seven transmembrane G Protein-coupled prostanoid receptor called IP [4]. Upon binding IP expressed on endothelial cells, PGI2 modulates relaxation of vascular smooth muscle and vasodilation of vascular beds. Due to these effects on the vascular system, in the early 1980s PGI2 was first used successfully in the treatment of pulmonary hypertension [5].
Over the last decade, many groups have reported that IP is expressed on immune cells present in the lung. The presence of IP on these immune cells suggests an importance for PGI2 in host defense. IP is known to be expressed on alveolar macrophages [6], conventional dendritic cells [7], and T lymphocytes [8]. In addition, PGIS has been recently discovered in certain immune cells, specifically follicular dendritic cells [9]. Follicular dendritic cells (FDCs) are stromal cells derived from nonhematopoetic cells that present unprocessed antigen to B lymphocytes [10]. These cells create a microenvironmental niche for B and T cells in the primary and secondary lymphoid organ follicles. FDCs are necessary for a fully robust humoral immune response, and PGI synthase expression in FDCs suggests an important effect of PGI2 on humoral immunity [10].
CD4+ T cell activation and subset differentiation
CD4+ T cells are critical for fully functional immune response as these cells provide help to B lymphocytes as well as innate immune cells such as macrophages [11]. CD4+ T cell activation occurs when “professional” antigen presenting cells (APCs) such as dendritic cells, B lymphocytes, and macrophages, display antigen to the CD4+ cells in the context of major histocompatibility complex (MHC) II molecules. The most important APC in the activation of naïve CD4+ T cells is the conventional dendritic cell (cDC). cDCs pinocytose exogenous environmental antigens and then process the antigen internally so that it can fit in the MHC class II binding groove. Upon activation, cDCs migrate to the draining lymph nodes where they contact naïve CD4+ T cells in the T cell zone via interaction of the APCs MHC class II molecule with the CD4+ T cell receptor (TCR). A fully function T cell response also requires the interaction of APC costimulatory molecules, such as CD80 and CD86, with CD28 expressed on the surface of the T cell. In the absence of co-stimulation, the CD4+ T cell becomes anergic. The newly activated CD4+ T cell then may differentiate down one of a number of T helper (Th) defined lineages. The precise lineage is determined by the cytokine milieu present in the microenvironment at the time of activation [12]. The three most common lineages, and the ones addressed in this review, are Th1, Th2, and Th17 cells (Figure 1). These cell lineages are defined by their cytokine secretion profile.
Figure 1.

Cytokines responsible for and produced by CD4+ Th1, Th2, and Th17 cells
Th1 cells are defined by their production of interferon (IFN)-γ and differentiate under the control of the master transcriptional regulator T-bet [12]. Th1 cells are differentiated in vitro when T cell activation occurs in the presence of interleukin (IL)-12, produced by APCs, and IL-4 antagonism, as IL-4 negatively regulates Th1 development. Th1 cells are particularly important in immune responses against intracellular pathogens, such as viruses, and are also involved in tumor immunity. Th2 cells are defined by their production of IL-4, IL-5, and IL-13, and these cells differentiate under the control of the master transcriptional regulator GATA-3 [12]. Th2 cells are differentiated in vitro when T cell activation occurs in the presence of IL-4 and IFN-γ antagonism, as IFN-γ negatively regulates Th2 development. Th2 cells are particularly important in immune responses against parasitic infections and are also pathogenic in allergic inflammation. Th17 cells are defined by their production of IL-17A, IL-17F, IL-21, and IL-22, and these cells differentiate under the control of the master transcriptional regulator RORC [13]. Th17 cells are differentiated in vitro when T cell activation occurs in the presence of IL-1β, IL-6, and TGF-β, and antagonism of IFN-γ, IL-4, and IL-13, as these cytokines negatively regulate Th17 development [14, 15]. Th17 cells are particularly important in anti-bacterial immunity and may also have a role in the pathogenesis of autoimmune diseases such as inflammatory bowel disease, rheumatoid arthritis, and multiple sclerosis [16].
In specific instances as noted above in the conditions of CD4+ subset differentiation, the cytokines produced by one lineage of Th cells inhibits the differentiation of the others. This evolutionary adaptation allows for the T helper response to be specific and self-reinforcing. In each of the sections below, we will first focus on how PGI2 modulates the function and cytokine secretion of dendritic cells since these are key cells in regulating CD4+ T cell differentiation. We will then focus on the ability of PGI2 to directly modulate CD4+ cell subset differentiation in antigen-independent protocols. Finally, we will review mouse models of disease that inform us on how PGI2 regulates cytokine production and inflammation in vivo.
Role of PGI2 in regulating Th1 responses
As noted above, naïve T helper cells activated in the presence of the cytokine IL-12 differentiate into Th1 cells and cDCs are the primary producers of IL-12 in vivo. Our lab explored the ability of PGI2 to modulate cDC production of IL-12 [7]. Such modulation could possibly alter Th1 differentiation and thus affect the CD4+ immune response. In these studies, bone marrow-derived dendritic cells (BMDCs) were cultured for 8 days and then activated with lipopolysaccharide (LPS) in the presence of either of two PGI2 analogs, iloprost or cicaprost, or the analogs’ respective vehicles. Iloprost and cicaprost both have biologic half-lives that are in the range of 20–30 minutes, each substantially longer than the 1–2 minute half-life of PGI2, and therefore of greater utility in the tissue culture setting. Both iloprost and cicaprost inhibited BMDC production of IL-12 in a dose-dependent manner, compared to their vehicle controls (Figure 2) [7]. The effect of both cicaprost and iloprost was fully ablated in BMDCs from mice genetically deficient in the PGI2 receptor IP, revealing that the effects of the two PGI2 analogs on BMDC production was specific for IP signaling [7].
Figure 2.
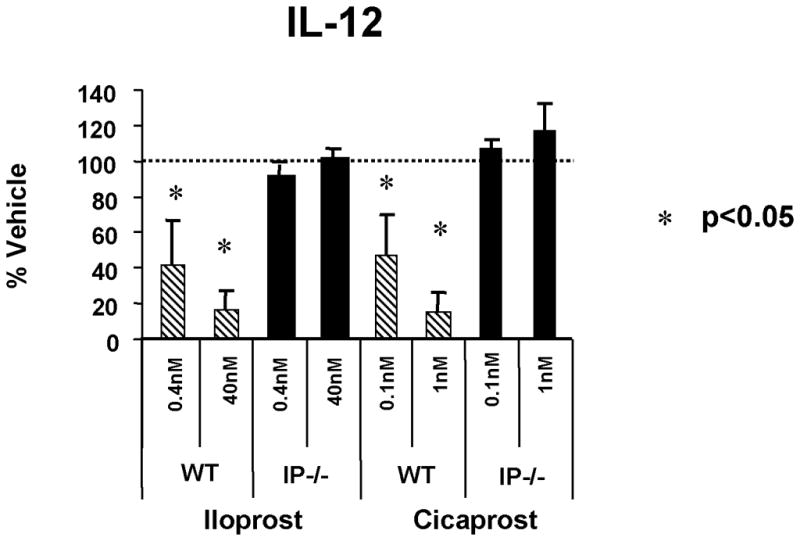
PGI2 analogs inhibited IL-12 production by BMDCs. BMDC cells were cultured in GM-CSF for 8 days. The cell population was treated with PGI2 analogs (iloprost and cicaprost) at increasing concentrations in the presence of LPS (1 μg/ml) for 24 h. The levels of IL-12 in the culture supernatants were measured by ELISA. Data represent mean ± SD of three experiments. *, p < 0.05 compared with vehicle-treated cells.
In addition to its effects on BMDCs, PGI2 directly inhibited Th1 effector cytokine production, suggesting a dampening effect on Th1 differentiation [8]. In this experiment, naïve CD4+ T cells from C57BL/6 mice were activated by anti-CD3 and anti-CD28 in the presence of IL-12. After 4 days, these cells were restimulated using anti-CD3 in the presence of cicaprost, iloprost, or the respective vehicle controls. In each case, treatment with the PGI2 analogs significantly reduced IFN-γ production by the activated T cells. This reduction was only modestly ablated in IP−/− mice, suggesting that the effect of PGI2 analogs on CD4+ Th1 cell IFN- γ production was only partially IP-specific [8]. However, in another report, PGI2 promoted Th1 differentiation in a mouse model of contact hypersensitivity [17]. Naïve T helper cells from C57BL/6 mice were activated using plate-bound anti-CD3 and increasing concentrations of anti-CD28 (1–100 μg/ml range) under Th1-skewing conditions. After 2 days, cells were washed and re-plated in Th1-skewing conditions. Iloprost increased IFN-γ production in a dose-dependent manner based on the amount of anti-CD28 present. Iloprost did not increase IFN-γ production in CD4+ cells from IP−/− mice, showing the effect of this analog in these conditions was IP-specific [17]. The reason for this seemingly conflicting finding is not fully understood as both groups used cells from the same strain of mice, but may involve the inclusion of anti-CD28 during cell restimulation, which possibly could lead to different effector cytokine production.
The effect on PGI2 on human mononuclear cell expression of the Th1 related chemokine expression of CXCL10 has also been examined [18]. Human monocytes were grown in vitro for 24 hours and were pre-treated with iloprost or treprostinil 2 hours before stimulation with LPS [18]. Treprostinil is another PGI2 analog, but is not as IP-specific as either iloprost or cicaprost as it signals through the PGE2 receptor EP2 at lower concentrations [6]. Monocytes treated with PGI2 analogs had decreased CXCL10 expression compared to vehicle-treated cells, but the effect was only partially IP-dependent in studies using an IP antagonist, CAY10449 [18]. These findings reinforce the notion that PGI2 restrains the Th1 immune response in a partially IP-specific manner.
The restraining effect of PGI2 on the Th1 response has been confirmed in an in vivo model of respiratory syncytial virus (RSV) infection [19]. RSV is the leading cause of respiratory failure of infants worldwide and the mouse model of RSV has many features similar to human disease, including abundant infiltration of IFN-γ-producing cells [20]. To test the effect of PGI2 in regulating IFN-γ production, mice which overexpress PGIS (PGIS OE) selectively in the respiratory epithelium and their wild type (WT) FVB strain littermates were infected with RSV laboratory strain A2 [19]. Six days after infection, the peak of IFN-γ expression in the mouse model of RSV, the RSV infected PGIS OE mice had significantly decreased lung IFN-γ protein expression compared to WT littermate controls. Interestingly, the PGIS OE mice also had significantly reduced weight loss [19], which reflects the notion that weight loss in this model is a result of inflammation, and that illness as defined by weight loss is a consequence of the ability to clear the infection [20]. The ability of PGI2 to negatively regulate IFN-γ production in vivo was confirmed with RSV infection of IP−/− mice and their WT controls. In this experiment, there was a significant increase in lung IFN-γ expression six days after infection in the IP−/− mice and these mice also had greater RSV-induced weight loss and delayed recovery compared to WT controls. Therefore, the inability to signal through IP significantly increased the expression of this Th1 cytokine, suggesting that PGI2 inhibits Th1 responses.
In summary, PGI2 analogs decreased BMDC expression of IL-12, the principal cytokine that drives Th1 responses, in an IP-specific fashion [7], while PGI2 directly decreased CD4 Th1 cytokine production, in a partially IP-specific manner [8]. In vivo airway epithelial PGIS OE inhibited lung IFN-γ expression following RSV infection, while the reverse was true in IP−/− mice. Taken together, these results certainly suggest that PGI2 signaling limits Th1 immune responses.
Role of PGI2 in regulating Th2 responses
In addition to its generally inhibitory effect on Th1 responses, PGI2 also restrains the ability of dendritic cells to optimally present antigen to CD4+ T cells and reduces Th2 immune responses both in vitro and in vivo. Our lab explored the ability of PGI2 to regulate dendritic cell-mediated Th2 differentiation in vitro [7]. First, BMDCs were cultured for 8 days and then pulsed overnight with chicken egg ovalbumin (OVA) protein so that the protein could be taken up by the cells, processed, and then presented on the cell surface in the context of MHC class II. PGI2 analogs, or their respective vehicles, were added at the time of the addition of OVA to the culture to determine the effect of these analogs on BMDC cell surface molecule expression. The PGI2 analogs significantly decreased BMDC MHC class II, CD86, and CD40 surface expression, thus reducing expression of molecules critical for antigen presentation [7].
In other experiments to assess the ability of PGI2 to regulate the capacity of BMDCs to present antigen and stimulate T cell proliferation, the BMDCs were again cultured for 8 days and prior to overnight addition of OVA protein in the presence or absence of the PGI2 analogs [7]. These BMDCs that now presented OVA peptides in the context of MHC class II were then cultured with CD4+ cells obtained from D011.10 mice for 4 days. For this experiment D011.10 transgenic mice, which have an OVA-specific T cell receptor, were used as a source of CD4+ T lymphocytes. Therefore the precursor frequency of OVA-specific naïve CD4+ T cells in D011.10 mice is much greater than in WT mice and can be used to assess the dendritic cell’s ability to activate T cells in an antigen-specific manner. Prior to culture, the D011.10 CD4+ cells were incubated with carboxyfluorescein succinimidyl ester (CFSE), so that CD4+ cell proliferation could be assessed. Briefly, CFSE is a fluorescent dye which is passed equally to the two daughter cells during mitosis. Each daughter cell will contain half as much CFSE as the parent cell, therefore cell division is tracked by flow cytometry. Cell division, in turn, is directly associated with the level of activation, as more activation leads to more rounds of division. Both iloprost and cicaprost treatment of dendritic cells at the time of exposure to OVA significantly inhibited subsequent T cell proliferation and secretion of the Th2 cytokines IL-5 and IL-13 [7]. These in vitro results indicate that PGI2 negatively regulates the ability of BMDC to stimulate T cell proliferation and cytokine production in an antigen-specific fashion.
PGI2 also directly down-regulates Th2 polarized CD4+ cell expression of IL-4, IL-5, and IL-13 in vitro (Figure 3) [8]. Purified splenic CD4+ cells from WT and IP−/− mice were activated with anti-CD3 and anti-CD28 in the presence of Th2-skewing conditions. After 4 days of culture, the cells were re-stimulated with anti-CD3 with or without PGI2 analogs, mimicking restimulation by antigen in the presence or absence of PGI2. Both PGI2 analogs suppressed Th2 effector cytokine production at concentrations in a dose-dependent manner [8]. This suppression was partially ablated in cells taken from IP−/− mice, suggesting that PGI2 reduction of Th2 cytokine production is only partially IP-specific [8].
Figure 3.

PGI2 analogs inhibited effector cytokine production by differentiated Th2 cells. CD4 T cells from WT and IP−/− mice were stimulated under Th2 conditions for 4 days followed by TCR restimulation for 2 days. The PGI2 analogs cicaprost and iloprost were added at a concentration of 20 nM at the time of restimulation. Cytokines were measured by ELISA. Data shown are the mean ± SEM of n = 4 experiments. *, P < 0.05, versus the respective vehicles for each of the analogs.
Several groups have reported results that strongly suggest that PGI2 analogs restrain Th2 responses in vivo. The first used the OVA model of allergic lung inflammation which incorporates a sensitization phase where mice are intraperitoneally injected with OVA formulated with aluminum hydroxide on days 0 and 12, followed by a challenge phase 10 days later where the mice are exposed to aerosolized OVA [21]. Takahashi and colleagues reported that IP−/− mice had a significant increase in serum antigen specific IgE, lung leukocyte accumulation, and Th2 cytokine protein expression [21]. In addition, there was an approximate three-fold greater production of IL-4 by spleen cells from sensitized and challenged IP−/− mice compared to WT mice that had undergone the same protocol. This same group extended the OVA challenge period so that mice were exposed to antigen daily for three weeks [22]. In this setting, IP−/− mice had increased goblet cell hyperplasia and subepithelial fibrosis compared to WT mice. These results strongly suggest that the inability to signal through IP augments allergen-induced lung inflammation.
Jaffar and colleagues have also investigated the role of PGI2 in Th2-mediated allergic responses [23, 24]. In the first report, they adoptively transferred Th2-polarized CD4+ cells from D011.10 mice into naïve recipients and then challenged the mice while they were being treated with a selective COX-2 inhibitor or vehicle [23]. The COX-2 inhibitor treated mice had decreased levels of the stable PGI2 metabolite 6-keto-PGF1α, yet enhanced airway responsiveness and lung production of Th2 cytokines compared to the vehicle treated mice. Additionally, CD4+ D011.10 cells polarized to Th2 cells in the presence of the PGI2 analog carbaprostacyclin had increased production of IL-10, a cytokine that suppresses Th2 immune responses [23].
A more recent report by this same team of investigators proposed a mechanism for the inhibition of Th2 immune responses by PGI2, suggesting that reduced cell recruitment is the driving force of the inhibition, rather than alteration of Th2 cell function [24]. In this study, CD4+ cells from DO11.10 mice were activated through exposure to OVA peptide under Th2-skewing conditions. After 4 days, cells were restimulated with OVA peptide and IL-2 and simultaneously treated with either PGI2, which was continuously replenished due to its short half life, or vehicle. After 4 more days of restimulation, Th2 cells were adoptively transferred into naïve BALB/c recipients. Mice were exposed to aerosolized OVA peptide for 20 minutes a day over 7 days. Th2 cytokine levels mirrored previous findings, with PGI2 having an inhibitory effect on the Th2 response [24]. The investigators concluded that PGI2 was responsible for decreased Th2 immune responses due to inhibition of cell recruitment to the lung [24]. If mice were not treated immediately with aerosolized OVA and were allowed to rest for 2 weeks, the reduction in Th2 immune responses was lost, suggesting that the inhibitory effects of PGI2 are transitory [24]. The reduction of Th2 response was independent of cAMP levels, as mice who received cells pre-treated with cAMP did not differ from control [24]. Further experiments proved the effect of PGI2 in downregulating allergic inflammation in this model to be IP-specific.
Another report implicates PGI2 in altering cDC function, offering an alternate explanation for the reduction of OVA-induced Th2 immune response. Idzko and colleagues sensitized mice to OVA and then treated the mice intranasally with either iloprost or vehicle at the time of OVA challenge [25]. Mice treated with iloprost were found to have decreased airway responsiveness to OVA, as well as an overall decreased Th2 response. This group found that iloprost treatment reduced the ability of cDCs to migrate to the lymph nodes, as well as decreased expression of co-stimulatory molecules on the surface of the cDCs [25]. Such co-stimulatory molecules are necessary for proper T cell activation, and the lack of correct cDC function offers a strong theory for how Th2 immune responses are dampened.
While several reports suggest that PGI2 is a critical negative regulator of allergic inflammation, it is important to recognize that other prostanoids may also be involved in modulating allergic responses. Carey and colleagues identified a differential role for COX-1 and COX-2 in regulating Th2 responses [26]. They found that the airways of COX-1−/− mice had exaggerated bronchoalveolar lavage (BAL) fluid levels of Th2 cytokines, augmented BAL concentrations of chemokines such as eotaxin and thymus- and activation regulated chemokine (TARC), and increased airway CD4+ and CD8+ cells compared to either WT or COX-2−/− mice [26]. These results suggest that PGI2 may not be the predominant COX product negatively regulating allergic inflammation, in that COX-2−/− mice have reduced production of PGI2, and one might have expected augmented allergic inflammation in the COX-2−/− mice if PGI2 restrained allergic inflammation. However, our lab reported that both COX-1 and COX-2 inhibitors each augmented allergic inflammation compared to vehicle-treated animals [27]. The discordance of these results suggest that there may be compensatory changes in the COX-2−/− mice that negate the inhibitory effect of PGI2 on allergic inflammation, and that these compensatory changes are no longer present when acute pharmacologic inhibition of COX-2 is effected.
In summary, PGI2 analogs decreased the ability BMDC to stimulate T cell proliferation and Th2 cytokine production in an antigen-specific fashion [7], while PGI2 directly decreased CD4 Th2 cytokine production, in a partially IP-specific manner [8]. In vivo studies from several groups strongly suggest that PGI2 restrains allergen-induced lung inflammation [21–25]. Taken together, these results certainly suggest that PGI2 signaling limits Th2 immune responses.
Role of PGI2 in regulating Th17 responses
The overwhelming evidence is that PGI2 signaling decreases both Th1 and Th2 responses. Th1 and Th2 cytokines such as IL-12, IFN-γ, IL-4, and IL-13 hinder Th17 differentiation [28, 29]. Through these actions, one might hypothesize that PGI2 enhances Th17 immune responses by dampening Th17-suppressing factors. Recent published data, coupled with unpublished data from our lab, suggest that PGI2 does in fact enhance Th17 immune responses. Mice which lack the ability to synthesize PGI2 have blunted Th17 immune responses as shown in an in vivo mouse model of ovalbumin sensitization and challenge [30]. WT mice, COX-1−/−, and COX-2−/− mice were sensitized to OVA, and 2 weeks later mice were exposed to aerosolized OVA for 30 minutes each day for 4 consecutive days, and the next day lungs were harvested and Th17 cytokine levels were measured. Th17 cytokines were significantly lowered in the lungs, lymph nodes, and BAL fluid of COX-2−/− mice compared to WT, while no significant differences were detected between WT and COX-1−/− mice [30]. This reduction of Th17 differentiation could be rescued in vitro through the addition of PGI2 analogs to cultured CD4+ cells, returning Th17 cytokine levels to that of WT mice [30].
One possible mechanism by which PGI2 skewed Th17 differentiation could involve the balance of the Th1-driving cytokine IL-12, to the Th17-priming cytokine, IL- 23 at the time of CD4+ T cell differentiation. IL-23 is secreted by DCs, and promotes maturation of the Th17 cell phenotype [31]. As mentioned earlier, PGI2 significantly decreased expression of IL-12 by BMDCs in a dose-dependent manner following LPS stimulation; however, PGI2 had no effect on the expression of IL-23 by these cells [32]. Therefore, the ratio of IL-12 to IL-23 in the microenvironment heavily favors Th17 cell differentiation. This effect of PGI2 on the ratio of IL-23 to IL-12 expression is IP-specific as prostacyclin analogs such as iloprost and cicaprost had no effect on either IL-12 or IL-23 production from cultured BMDCs from IP−/− mice (Figure 4) [32].
Figure 4.

PGI2 analogs increased the ratio of IL-23/IL-12 produced by BMDCs from WT and IP−/− mice. BMDC cells were cultured in GM-CSF for 8 days. The cell population was treated with PGI2 analogs (iloprost and cicaprost) at increasing concentrations in the presence of LPS (1 μg/ml) for 24 h. The levels of IL-23 and IL-12 in the culture supernatants were measured by ELISA. Data represent the ratio of IL- 23/IL-12 at each concentration of the PGI2 analogs. *, p < 0.05 compared with vehicle- treated cells.
This upregulation of Th17 differentiation and cytokine expression by PGI2 suggests that mice lacking the ability to signal through PGI2-IP would be protected against Th17-mediated diseases. Our lab tested this supposition in a mouse model of experimental autoimmune encephalitis (EAE) [32]. EAE has features similar to the human disorder Multiple Sclerosis and the early features of this disease have been shown to be largely mediated by Th17 inflammation. In this model, mice are infected with myelin basic protein (MOG) emulsified in Complete Freund’s Adjuvant. This mixture induces the recruitment of autoimmune T Cells that specifically react to myelin antigen found in nerve sheaths. As the disease progresses, mice undergo ascending paralysis from the tail to the forelimbs. This paralysis may be remitting. IP deficient mice had statistically significantly delayed onset and decreased severity of EAE [32]. The inability to signal through IP therefore led to a decreased Th17 response and protection against EAE [32].
In summary, we have reviewed that PGI2 suppresses Th1 and Th2 differentiation and function in most cases. This inhibition may be fully or partially IP-specific, depending on the cell type involved in the immune response. In the limited reports published so far, PGI2 seems to enhance Th17 immune responses [30, 32]. PGI2 therefore plays a critical role not only in vascular tissues, but also as a broad central mediator of immune responses in the lung and beyond.
Highlights.
PGI2 has important regulatory functions on the innate and adaptive immune systems.
PGI2 inhibits CD4+ Th1 differentiation and function by decreasing dendritic cell expression of IL-12 and by directly suppressing IFN-γ production by polarized Th1 cells
PGI2 inhibits CD4+ Th2 differentiation and function by decreasing the ability of dendritic cells to stimulate T cell proliferation and cytokine secretion in an antigen-specific manner.
PGI2 promotes CD4+ Th17 differentiation and function by increasing the ratio of IL-23 to IL-12 produced by dendritic cells and by inhibiting CD4+ T cell production of cytokines that suppress Th17 development.
Acknowledgments
This work was supported by R01-HL-090664, R01-AI070672, R21-HL 106446, and 1I01BX000624
Abbreviations
- APCs
antigen presenting cells
- BMDCs
bone marrow-derived dendritic cells
- BAL
bronchoalveolar lavage
- CFSE
carboxyfluorescein succinimidyl ester
- cDC
conventional dendritic cell
- COX
cyclooxygenase
- FDCs
follicular dendritic cells
- IFN
interferon
- IL
interleukin
- MHC
major histocompatibility complex
- OVA
ovalbumin
- PGIS
PGI synthase
- PG
prostaglandin
- RSV
respiratory syncytial virus
- Th
T helper
- TARC
thymus- and activation regulated chemokine
- WT
Wild type
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Moncada S, Gryglewski R, Bunting S, Vane JR. An enzyme isolated from arteries transforms prostaglandin endoperoxides to an unstable substance that inhibits platelet aggregation. Nature. 1976;263:663–665. doi: 10.1038/263663a0. [DOI] [PubMed] [Google Scholar]
- 2.Whittaker N, Bunting S, Salmon J, et al. The chemical structure of prostaglandin X (prostacyclin) Prostaglandins. 1976;12:915–928. doi: 10.1016/0090-6980(76)90126-x. [DOI] [PubMed] [Google Scholar]
- 3.Helliwell RJ, Adams LF, Mitchell MD. Prostaglandin synthases: recent developments and a novel hypothesis. Prostaglandins Leukot Essent Fatty Acids. 2004;70:101–113. doi: 10.1016/j.plefa.2003.04.002. [DOI] [PubMed] [Google Scholar]
- 4.Smyth EM, Fitzgerald GA. Human prostacyclin receptor. Vitam Horm. 2002;65:149–165. doi: 10.1016/s0083-6729(02)65063-0. [DOI] [PubMed] [Google Scholar]
- 5.Szczeklik J, Szczeklik A, Nizankowski R. Prostacyclin for pulmonary hypertension. Lancet. 1980;2:1076. doi: 10.1016/s0140-6736(80)92291-6. [DOI] [PubMed] [Google Scholar]
- 6.Aronoff DM, Peres CM, Serezani CH, et al. Synthetic Prostacyclin Analogs Differentially Regulate Macrophage Function via Distinct Analog-Receptor Binding Specificities. J Immunol. 2007;178:1628–1634. doi: 10.4049/jimmunol.178.3.1628. [DOI] [PubMed] [Google Scholar]
- 7.Zhou W, Hashimoto K, Goleniewska K, et al. Prostaglandin I2 analogs inhibit proinflammatory cytokine production and T cell stimulatory function of dendritic cells. J Immunol. 2007;178:702–710. doi: 10.4049/jimmunol.178.2.702. [DOI] [PubMed] [Google Scholar]
- 8.Zhou W, Blackwell TS, Goleniewska K, et al. Prostaglandin I2 analogs inhibit Th1 and Th2 effector cytokine production by CD4 T cells. J Leukoc Biol. 2007;81:809–817. doi: 10.1189/jlb.0606375. [DOI] [PubMed] [Google Scholar]
- 9.Lee IY, Ko EM, Kim SH, Jeoung DI, Choe J. Human follicular dendritic cells express prostacyclin synthase: a novel mechanism to control T cell numbers in the germinal center. J Immunol. 2005;175:1658–1664. doi: 10.4049/jimmunol.175.3.1658. [DOI] [PubMed] [Google Scholar]
- 10.Gatto D, Brink R. The germinal center reaction. J Allergy Clin Immunol. 2010;126:898–907. doi: 10.1016/j.jaci.2010.09.007. [DOI] [PubMed] [Google Scholar]
- 11.Abbas AK. The control of T cell activation vs. tolerance. Autoimmun Rev. 2003;2:115–118. doi: 10.1016/s1568-9972(03)00028-4. [DOI] [PubMed] [Google Scholar]
- 12.Amsen D, Spilianakis CG, Flavell RA. How are T(H)1 and T(H)2 effector cells made? Curr Opin Immunol. 2009;21:153–160. doi: 10.1016/j.coi.2009.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang XP, Ghoreschi K, Steward-Tharp SM, et al. Opposing regulation of the locus encoding IL-17 through direct, reciprocal actions of STAT3 and STAT5. Nat Immunol. 2011;12:247–254. doi: 10.1038/ni.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Manel N, Unutmaz D, Littman DR. The differentiation of human T(H)-17 cells requires transforming growth factor-beta and induction of the nuclear receptor RORgammat. Nat Immunol. 2008;9:641–649. doi: 10.1038/ni.1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Newcomb DC, Boswell MG, Zhou W, et al. Human Th17 cells express a functional IL-13 receptor and IL-13 attenuates IL-17A production. J Allergy Clin Immunol. 2011 doi: 10.1016/j.jaci.2010.11.043. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jager A, Kuchroo VK. Effector and regulatory T-cell subsets in autoimmunity and tissue inflammation. Scand J Immunol. 2010;72:173–184. doi: 10.1111/j.1365-3083.2010.02432.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nakajima S, Honda T, Sakata D, et al. Prostaglandin I2-IP signaling promotes Th1 differentiation in a mouse model of contact hypersensitivity. J Immunol. 2010;184:5595–5603. doi: 10.4049/jimmunol.0903260. [DOI] [PubMed] [Google Scholar]
- 18.Kuo CH, Ko YC, Yang SN, et al. Effects of PGI2 analogues on Th1- and Th2-related chemokines in monocytes via epigenetic regulation. J Mol Med. 2011;89:29–41. doi: 10.1007/s00109-010-0694-2. [DOI] [PubMed] [Google Scholar]
- 19.Hashimoto K, Graham BS, Geraci MW, et al. Signaling through the prostaglandin I2 receptor IP protects against respiratory syncytial virus-induced illness. J Virol. 2004;78:10303–10309. doi: 10.1128/JVI.78.19.10303-10309.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Graham BS, Johnson TR, Peebles RS. Immune-mediated disease pathogenesis in respiratory syncytial virus infection. Immunopharmacology. 2000;48:237–247. doi: 10.1016/s0162-3109(00)00233-2. [DOI] [PubMed] [Google Scholar]
- 21.Takahashi Y, Tokuoka S, Masuda T, et al. Augmentation of allergic inflammation in prostanoid IP receptor deficient mice. Br J Pharmacol. 2002;137:315–322. doi: 10.1038/sj.bjp.0704872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nagao K, Tanaka H, Komai M, Masuda T, Narumiya S, Nagai H. Role of prostaglandin I2 in airway remodeling induced by repeated allergen challenge in mice. Am J Respir Cell Mol Biol. 2003 doi: 10.1165/rcmb.2003-0035OC. [DOI] [PubMed] [Google Scholar]
- 23.Jaffar Z, Wan KS, Roberts K. A key role for prostaglandin I2 in limiting lung mucosal Th2, but not Th1, responses to inhaled allergen. J Immunol. 2002;169:5997–6004. doi: 10.4049/jimmunol.169.10.5997. [DOI] [PubMed] [Google Scholar]
- 24.Jaffar Z, Ferrini ME, Buford MC, Fitzgerald GA, Roberts K. Prostaglandin I2-IP signaling blocks allergic pulmonary inflammation by preventing recruitment of CD4+ Th2 cells into the airways in a mouse model of asthma. J Immunol. 2007;179:6193–6203. doi: 10.4049/jimmunol.179.9.6193. [DOI] [PubMed] [Google Scholar]
- 25.Idzko M, Hammad H, van Nimwegen M, et al. Inhaled iloprost suppresses the cardinal features of asthma via inhibition of airway dendritic cell function. J Clin Invest. 2007;117:464–472. doi: 10.1172/JCI28949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Carey MA, Germolec DR, Bradbury JA, et al. Accentuated T helper type 2 airway response after allergen challenge in cyclooxygenase-1-/- but not cyclooxygenase-2-/- mice. Am J Respir Crit Care Med. 2003;167:1509–1515. doi: 10.1164/rccm.200211-1383OC. [DOI] [PubMed] [Google Scholar]
- 27.Peebles RS, Jr, Hashimoto K, Morrow JD, et al. Selective cyclooxygenase-1 and -2 inhibitors each increase allergic inflammation and airway hyperresponsiveness in mice. Am J Respir Crit Care Med. 2002;165:1154–1160. doi: 10.1164/ajrccm.165.8.2106025. [DOI] [PubMed] [Google Scholar]
- 28.Harrington LE, Hatton RD, Mangan PR, et al. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. 2005;6:1123–1132. doi: 10.1038/ni1254. [DOI] [PubMed] [Google Scholar]
- 29.Newcomb DC, Zhou W, Moore ML, et al. A functional IL-13 receptor is expressed on polarized murine CD4+ Th17 cells and IL-13 signaling attenuates Th17 cytokine production. J Immunol. 2009;182:5317–5321. doi: 10.4049/jimmunol.0803868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li H, Bradbury JA, Dackor RT, et al. Cyclooxygenase-2 (COX-2) Regulates Th17 Cell Differentiation During Allergic Lung Inflammation. Am J Respir Crit Care Med. 2011 doi: 10.1164/rccm.201010-1637OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Iwakura Y, Ishigame H. The IL-23/IL-17 axis in inflammation. J Clin Invest. 2006;116:1218–1222. doi: 10.1172/JCI28508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhou W, Huckabee MM, Dowell DR, et al. Prostagalndin I2 signaling drives Th17 differentiation and exacerbates experimental autoimmune encephalitis. PLoS One. doi: 10.1371/journal.pone.0033518. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]