

Abstract
TIMAP is a regulatory subunit of protein phosphatase 1, whose role remains largely unknown. Our recent data suggested that TIMAP is involved in the regulation of barrier function in cultured pulmonary endothelial monolayers (Csortos et al., Am J Physiol Lung Cell Mol Physiol 295: L440-450, 2008). Here we showed that TIMAP depletion exacerbates lipopolysaccharide (LPS)-induced vascular leakage in murine lung, suggesting that TIMAP has a barrier-protective role in vivo. Real-Time RT PCR analysis revealed that treatment with LPS significantly suppressed Timap mRNA level. This suppression was not achieved via the down-regulation of Timap promoter activity, suggesting that LPS decreased Timap mRNA stability. Pretreatment with protein kinase A (PKA) inhibitor H-89 reduced TIMAP mRNA level, whereas pretreatment with PKA activator, bnz-cAMP, increased this level and attenuated LPS-induced decrease in TIMAP mRNA. Altogether, these data confirmed the barrier-protective role of TIMAP and suggested that barrier-disruptive and barrier-protective agents may employ modulation of TIMAP expression as a mechanism affecting barrier permeability.
Keywords: TIMAP, PPP1R16B, LPS, PKA, barrier dysfunction
1. Inroduction
TIMAP is a regulatory protein, functioning as a targeting subunit for the catalytic subunit of protein phosphatase 1 (PP1C). TIMAP is a member of MYPT family, which also includes MYPT1, MYPT2, MBS85, and MYPT3 (Grassie et al., 2011). While a great deal is known about myosin targeting subunit MYPT1, TIMAP function is studied to a lesser extent. MYPT1 is a key protein regulating smooth muscle contraction and the motility of many other types of cells (Grassie et al., 2011); therefore, it is not surprising that Mypt1 knockouts manifest early embryonic lethality (Okamoto et al., 2005). On the contrary, Timap knockouts do not display any obvious anatomical abnormalities (Heinzel and Bleul, 2007). Some information about TIMAP putative function can be deducted from the patterns of TIMAP expression and localization. Analysis of data from Gene Atlas database (www.biogps.org) reveals that TIMAP is highly expressed in murine brain and vascularized tissues such as lung and kidney. In endothelial cells, where TIMAP was originally characterized as a protein down-regulated by TGFβ treatment (Cao et al., 2002), TIMAP is localized to the plasma membrane. Attempts to characterize TIMAP substrates led to the identification of non-integrin laminin receptor 1 (LAMR1) (Kim et al., 2005; Li et al., 2007) and members of Ezrin-Radixin-Moesin (ERM) family of actin-binding proteins (Csortos et al., 2008). Although the ability of TIMAP to regulate PP1C activity toward phosphorylated MLC was documented in vitro (Czikora et al., 2011), there is no evidence that TIMAP-PP1C complex can act as a myosin phosphatase in vivo.
The information about cellular processes involving TIMAP is also very limited. TIMAP was suggested to participate in filopodia formation in glomerular endothelial cells (Li et al., 2007). TIMAP involvement in the regulation of endothelial barrier function was implied from the experiments showing that TIMAP depletion exacerbated the effect of barrier disruptors (thrombin and nocodazole) and attenuated the effect of barrier protectors (sphingosine-1-phosphate and ATP). Based on these results, TIMAP was viewed as a barrier-protective protein, whose action is exerted via dephosphorylation of membrane-associated ERM (Csortos et al., 2008), and, since TIMAP expression is suppressed in the presence of barrier disruptor TGFβ (Cao et al., 2002), these data let us hypothesize that TIMAP down-regulation can serve as one of the mechanisms contributing to the endothelial barrier dysfunction.
In this manuscript, we first assessed whether TIMAP indeed plays a barrier protective role in vivo. Use of Timap-deficient mice in the established model of lipopolysaccharide (LPS) -induced Acute Lung Injury let us approach this question. We next assessed whether LPS is able to suppress TIMAP expression, and whether this suppression is achieved via the down-regulation of promoter activity. Finally, we studied the effect of barrier-protective enzyme, PKA, on TIMAP expression in endothelium. Our data confirmed that TIMAP is involved in the regulation of endothelial barrier and pinpointed a major pathway regulating TIMAP expression.
2. Materials and Methods
2.1. Reagents
LPS from E. coli (0127:B8, with activity of 900, 000 u/mg) was purchased from Sigma (St. Louis, MO). Protein kinase A (PKA) inhibitor H-89 was from Calbiochem (La Jolla, CA). PKA activator N6-benzoyl-cAMP (bnz-cAMP) was from Biolog Life Science Institute (Bremen, Germany).
2.2. Animals
Mice carrying Timap knockout mutation (Heinzel and Bleul, 2007) were generously donated by Prof. Thomas Boehm from Max Plank Institute (Freiburg, Germany). Timap+/− mice were repeatedly backcrossed to C57BL/6N (Charles River, Wilmington, MA) to transfer the mutation into a homogenous genetic background. The genotyping was performed by PCR as described in (Heinzel and Bleul, 2007).
Mice carrying Luciferase under the control of Timap promoter (TimapGetwise) were generated via ES-based genetic engineering and described in (Poirier et al., unpublished data). Luciferase was inserted in the 5′ UTR of the 3rd exon of Timap. Timap promoter-driven luciferase expression in brain and lung is demonstrated on the Suppl. Fig. S1.
2.3. Measurement of lung permeability and luciferase activity
All animal studies conformed to NIH guidelines. The experimental procedure was approved by the Georgia Health Sciences University Institutional Animal Care and Use Committee. For intratracheal instillation of LPS, we used 1) 20–25 g male C57BL/6N mice (Charles River Laboratories, Wilmington, MA); 2) wild-type, heterozygous, or homozygous Timap−/− littermates; 3) heterozygous TimapGetwise mice. LPS dissolved in saline was administered intratracheally to the animals anesthetized with ketamine (150 mg/kg of body weight)/acetopromazine (15 mg/kg); animals were allowed to recover from the anesthesia. Anesthesia was readministered, and Evans Blue Dye (EBD)-albumin conjugate was injected in the tail vein (20 mg/kg) 1 hour prior to animal sacrifice.
In each anesthetized animals, the chest cavities were opened, and lungs were washed from blood by injecting saline/EDTA via the right ventricle. For EBD extraction, lungs were homogenized in 3 ml of formamide (18h, 60°C). The homogenate was spun for 30 min at 5000g; the optical density of supernatant was determined at 620 nm and 750 nm. The extravasated EBD concentration was calculated using a standard curve and normalized to body weight. For luciferase activity analysis, lungs were snap-frozen in liquid nitrogen, homogenized using Besson tissue pulverizer, and extracted with 0.5 ml of 1× luciferase extraction buffer (Promega, Madison, WI). Measurement of activity was carried out with Steady-Glow luciferase substrate (Promega) using Paradigm modular detection platform (Hewlett-Packard, Palo Alto, CA). Protein level was assessed using BCA kit and used to normalize luminescence data.
2.4. Cell culture
Human Pulmonary Artery Endothelial Cells (HPAEC) were purchased from Lonza (Walkerville, MD) and used at passages 5–7. Bovine Pulmonary Artery Endothelial Cells (BPAEC), received as a generous gift from Prof. J. Catravas (Georgia Health Sciences University), were used at passages 2–4. Cells were cultured in media containing 5% (HPAEC) or 10% (BPAEC) FBS and maintained at 37°C in a humidified atmosphere of 5% CO2-95% air.
2.5. Real-Time RT PCR
Comparative CT method was used to assess the level of TIMAP mRNA. For the mouse mRNA, Timap and Hprt Taqman assays from Applied Biosystems (Foster City, CA) were used. For human and bovine mRNA, TIMAP expression with HPRT expression endogenous control was monitored by incorporation of SYBR Green dye (Applied Biosystems) into the double strand cDNA. The analysis was done using StepOne Real-Time PCR system from Applied Biosystems. The primers used for Real-Time PCR of human and bovine cDNA are summarized in Suppl. Table S1.
2.6. Statistical Analysis
Data were analyzed using T-test; results with p<0.05 were considered significantly different.
3. Results
3.1. Effect of TIMAP deficiency on LPS-induced pulmonary vascular permeability
To assess the effect of TIMAP depletion on LPS-induced vascular leakage, we used an established model of intratracheal LPS instillation. Based on our earlier data (Csortos et al., 2008), TIMAP depletion was expected to exacerbate LPS-induced barrier dysfunction. Therefore, it was paramount first to establish the sub-maximal concentrations of LPS and to characterize the time course of response to LPS in mice from the same genetic background. Using EBD extravasation from vascular lumen to lung tissue as a criterion of vascular permeability, we established that the most significant accumulation of EBD occurred within the first 6h of exposure to LPS, followed by a further increase in EBD accumulation during the subsequent 18h (Fig 1A). We also observed that LPS dose-dependently increased vascular leak in the range of 0.45–2 mg/kg (Fig 1B). Based on these data, the dose of 0.45 mg/kg was chosen as a sub-maximal dose, which will allow investigators to observe the exacerbation of lung permeability.
Fig. 1.
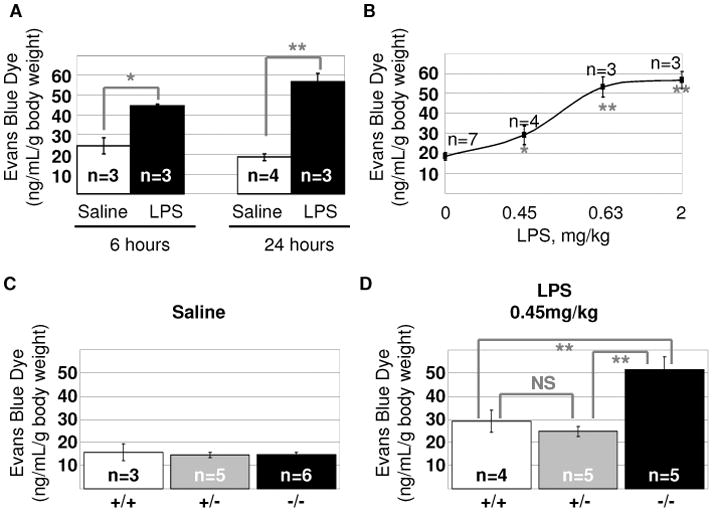
Timap deficiency exacerbates vascular leakage in response to LPS. A–B. 22–25g male C57BL/6N mice were subjected to intratracheal instillation of saline or LPS for the time indicated (A, 2mg/kg LPS) or 24h (B). C–D. 22–25g wild-type, heterozygous, and homozygous for Timap mutation male littermates were subjected to intratracheal instillation of saline (C) or LPS (D) for 24h. EBD extravasation into lung tissue was expressed as mean±SEM. *p<0.05, **p<0.01 (t-test).
We next assessed whether pulmonary vascular barrier is affected by the Timap knockout. Analysis of the vascular permeability in wild-type, heterozygous Timap+/−, and homozygous Timap−/−mice revealed that Timap deficiency does not affect basal permeability (Fig 1C). However, instillation of 0.45mg/kg LPS resulted in a higher rate of vascular leakage in Timap-deficient mice (Fig 1D), evident of the exacerbation of LPS-induced barrier dysfunction by Timap knockout.
3.2. Effect of LPS on TIMAP expression
Using Real Time RT PCR, we next studied how LPS treatment affects TIMAP expression in vitro and in vivo. Analysis of mRNA from LPS-treated HPAEC (Fig 2A) and BPAEC (Fig 2B) monolayers confirmed that TIMAP expression is significantly down-regulated by LPS treatment. Maximal decrease occurred 6h post-LPS exposure, with a partial recovery of TIMAP mRNA level seen at 24h. Analysis of mRNA from the lungs of LPS-treated mice revealed that 6h exposure to LPS induced a marked decrease in Timap mRNA level. 24h post-LPS exposure, the Timap mRNA level recovered to the level observed in naïve lungs (Fig 2C). We have developed earlier a transgenic line, TimapGetwise, in which the endogenous Timap promoter controls the expression of the reporter gene Luciferase (Poirier et al., unpublished data). Here, we used this line to elucidate whether LPS-induced down-regulation of Timap mRNA is achieved via the suppression of the Timap promoter activity or the destabilization of mRNA itself. Our data show that exposure to LPS does not suppress luciferase activity in TimapGetwise mice. To the contrary, 6h exposure to LPS induced a ~50% increase in murine lung Timap promoter activity. This increase was sustained 24h post-LPS exposure.
Fig. 2.
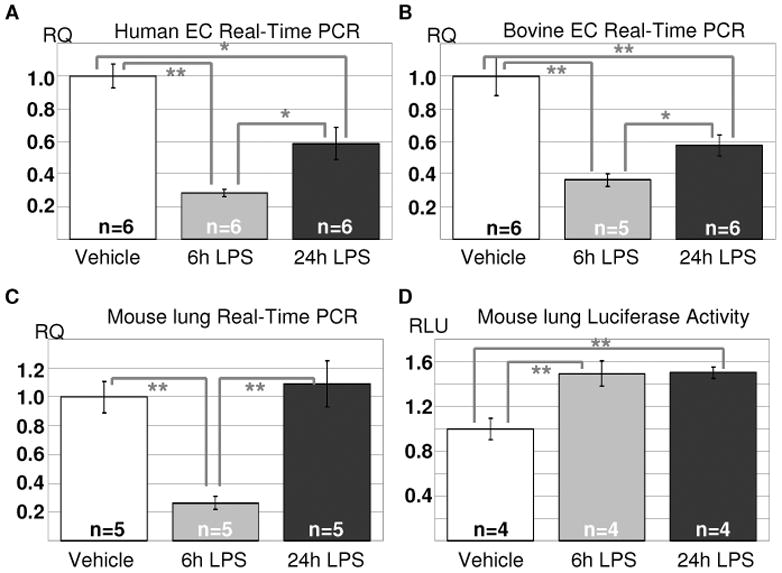
LPS induces decrease in TIMAP mRNA expression. A–B. HPAEC (A) and BPAEC (B) grown to confluence on 6-well plates were stimulated with 100ng/ml LPS for the time indicated. RNA was extracted, and TIMAP mRNA level was assessed by Real-Time PCR. C–D. 22–25g male C57BL/6N mice (C) or heterozygous TimapGetwise mice (D) were subjected to intratracheal instillation of saline or LPS for the time indicated. RNA (C) or protein (D) were extracted from the lungs. Timap mRNA level was assessed by Real-Time RT PCR and normalized to the level of the house-keeping gene Hprt (C). Luciferase activity was normalized to the protein concentration and expressed as fold of control. Relative mRNA quantitation and luciferase activity are shown as mean±SEM. *p<0.05, **p<0.01 (t-test).
We have shown previously that an increase in intracellular cAMP, and, in particular, PKA activation attenuates LPS-induced vascular leakage (Bogatcheva et al., 2009). Here, we assessed how PKA inhibition and activation affect LPS-induced alteration in TIMAP mRNA expression. We observed that pretreatment of HPAEC with PKA inhibitor H-89 significantly decreased TIMAP mRNA level (Table 1). On the contrary, pretreatment of HPAEC with PKA activator bnz-cAMP increased this level. Importantly, bnz-cAMP pretreatment attenuated LPS-induced drop in TIMAP mRNA level.
Table 1.
The effect of PKA inhibition and activation on Timap mRNA level in LPS-treated HPAEC. Relative quantitation is expressed as mean±SEM.
| no LPS | 100ng/ml LPS, 6h | |
|---|---|---|
| vehicle | 1±0.07 | 0.16±0.02 |
| 10 μM H-89 | 0.44±0.05** | 0.12±0.01 |
| 100 μM bnz-cAMP | 2.56±0.32** | 0.61±0.14* |
p<0.05,
p<0.01 (t-test)
4. Discussion
In this manuscript, we studied the role of phosphatase1 targeting subunit, TIMAP, in the regulation of vascular function. The use of bacterial endotoxin/LPS let us model vascular leakage occurring in gram-negative sepsis and assess the involvement of TIMAP in the signaling events mediating sepsis-induced pulmonary edema. We have shown earlier that TIMAP acts as a barrier-protective protein in cultured human pulmonary endothelial monolayers (Csortos et al., 2008). Here we established for the first time that TIMAP has a barrier-protective role in vivo. Mice deficient for Timap expression exhibited exaggerated vascular leakage in response to LPS treatment. These data confirmed the protective role of TIMAP in the regulation of vascular barrier and prompted us to examine the involvement of TIMAP more carefully.
We next studied how TIMAP mRNA expression is affected by LPS. We have shown that TIMAP mRNA level undergoes a marked down-regulation 6h post-LPS treatment. Depending on the species, complete or partial recovery from this down-regulation occurs within the following 18h. Analysis of the luciferase activity in TimapGetwise mice revealed that LPS-induced down-regulation of Timap mRNA level is not achieved via the suppression of Timap promoter activity. To the contrary, Timap promoter activity is up-regulated by LPS exposure. These data suggest that LPS-induced decrease in Timap mRNA occurs due to the destabilization of Timap mRNA. The initial drop in Timap mRNA level is followed by the recovery, which is likely to be mediated by the significant increase in Timap promoter activity.
The time course of Timap mRNA alteration correlated with the time course of LPS-induced barrier dysfunction in cultured cell monolayers (Bogatcheva et al., 2011), where maximal drop in transendothelial resistance (6–8h post-LPS) was followed by the partial recovery. Our in vivo data also confirmed that the majority of EBD extravasated from the vascular lumen within the first 6h of exposure to LPS. Based on these data, we hypothesized that LPS-induced down-regulation of barrier-protective protein TIMAP can be one of the mechanisms, by which LPS elicits its barrier-disruptive action. In Timap-deficient mice, the protection rendered by the declining TIMAP levels is void, hence the advanced susceptibility of these mice to LPS-induced leakage.
We next assessed how an activation or inactivation of a barrier-protective signaling enzyme PKA would affect TIMAP mRNA expression. The ability of PKA to counteract LPS effects on barrier permeability was shown by us before (Bogatcheva et al., 2009). Here we questioned whether this ability relies on the modulation of TIMAP expression as a barrier-protective mechanism. Our data showed that in HPAEC, PKA inactivation causes a significant decrease in TIMAP mRNA expression level; vice versa, PKA activation with a PKA-specific cAMP analogue causes a significant increase in TIMAP mRNA expression level. Moreover, PKA activation is able to attenuate LPS-induced decrease in TIMAP mRNA expression. Altogether, these data suggest that the modulation of TIMAP expression level can be a part of the PKA-dependent barrier-protective mechanisms. Earlier, TIMAP phosphorylation by PKA was shown to affect dephosphorylation of LAMR1 (Li et al., 2007) and moesin (Csortos et al., 2008), and, via these substrates, to modulate filopodia formation and barrier function. Taken together, these data indicate that TIMAP can be regulated by PKA both on mRNA level and by posttranslational modifications.
In conclusion, our data supported the idea that TIMAP plays a barrier-protective role in vascular endothelium. We showed that TIMAP down-regulation can be one of the barrier-disruptive mechanisms exerted by LPS, whereas TIMAP up-regulation is one of the mechanism induced by barrier-protective enzyme PKA. As information about the role of TIMAP in pathology and physiology is rather scarce, our results significantly contribute to the understanding of the part played by TIMAP in the regulation of vascular function. Our data also add to an understanding of the mechanisms of barrier disruption occurring during sepsis-induced pulmonary edema.
Supplementary Material
Highlights.
Timap deficiency exacerbates LPS-induced pulmonary vascular leak
TIMAP mRNA level is down-regulated by LPS in different species
Timap promoter activity is not down-regulated by LPS
TIMAP mRNA level is up-regulated by PKA
Acknowledgments
This research was supported by extramural grants from the American Heart Association to N.V.B. (SDG #0930038N), and the National Institutes of Health to A.D.V. (HL080675, HL083327, HL067307). The sponsors played no role in the study design, data collection, analysis, and interpretation, and the decision to submit the manuscript. The authors would like to thank Prof. Thomas Boehm from Max Plank Institute (Freiburg, Germany) for Timap knockout mice and Prof. John Catravas and Mrs. Connie Snead from Georgia Health Sciences University (Augusta, Georgia) for the isolation of BPAEC.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Christophe Poirier, Email: cpoirier@georgiahealth.edu.
Boris A. Gorshkov, Email: bgorshkov@georgiahealth.edu.
Marina A. Zemskova, Email: mzemskova@georgiahealth.edu.
Natalia V. Bogatcheva, Email: nbogatcheva@georgiahealth.edu.
Alexander D. Verin, Email: averin@georgiahealth.edu.
References
- Bogatcheva NV, Zemskova MA, Kovalenkov Y, Poirier C, Verin AD. Molecular mechanisms mediating protective effect of cAMP on lipopolysaccharide (LPS)-induced human lung microvascular endothelial cells (HLMVEC) hyperpermeability. J Cell Physiol. 2009;221:750–759. doi: 10.1002/jcp.21913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogatcheva NV, Zemskova MA, Poirier C, Mirzapoiazova T, Kolosova I, Bresnick AR, Verin AD. The suppression of myosin light chain (MLC) phosphorylation during the response to lipopolysaccharide (LPS): beneficial or detrimental to endothelial barrier? J Cell Physiol. 2011 doi: 10.1002/jcp.22669. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao W, Mattagajasingh SN, Xu H, Kim K, Fierlbeck W, Deng J, Lowenstein CJ, Ballermann BJ. TIMAP, a novel CAAX box protein regulated by TGF-beta1 and expressed in endothelial cells. Am J Physiol Cell Physiol. 2002;283:C327–337. doi: 10.1152/ajpcell.00442.2001. [DOI] [PubMed] [Google Scholar]
- Csortos C, Czikora I, Bogatcheva NV, Adyshev DM, Poirier C, Olah G, Verin AD. TIMAP is a positive regulator of pulmonary endothelial barrier function. Am J Physiol Lung Cell Mol Physiol. 2008;295:L440–450. doi: 10.1152/ajplung.00325.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czikora I, Kim KM, Kasa A, Becsi B, Verin AD, Gergely P, Erdodi F, Csortos C. Characterization of the effect of TIMAP phosphorylation on its interaction with protein phosphatase 1. Biochimie. 2011;93:1139–1145. doi: 10.1016/j.biochi.2011.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grassie ME, Moffat LD, Walsh MP, Macdonald JA. The myosin phosphatase targeting protein (MYPT) family: A regulated mechanism for achieving substrate specificity of the catalytic subunit of protein phosphatase type 1delta. Arch Biochem Biophys. 2011;510:147–159. doi: 10.1016/j.abb.2011.01.018. [DOI] [PubMed] [Google Scholar]
- Heinzel K, Bleul CC. The Foxn1-dependent transcripts PCOLCE2 and mPPP1R16B are not required for normal thymopoiesis. Eur J Immunol. 2007;37:2562–2571. doi: 10.1002/eji.200637474. [DOI] [PubMed] [Google Scholar]
- Kim K, Li L, Kozlowski K, Suh HS, Cao W, Ballermann BJ. The protein phosphatase-1 targeting subunit TIMAP regulates LAMR1 phosphorylation. Biochem Biophys Res Commun. 2005;338:1327–1334. doi: 10.1016/j.bbrc.2005.10.089. [DOI] [PubMed] [Google Scholar]
- Li L, Kozlowski K, Wegner B, Rashid T, Yeung T, Holmes C, Ballermann BJ. Phosphorylation of TIMAP by glycogen synthase kinase-3beta activates its associated protein phosphatase 1. J Biol Chem. 2007;282:25960–25969. doi: 10.1074/jbc.M703532200. [DOI] [PubMed] [Google Scholar]
- Okamoto R, Ito M, Suzuki N, Kongo M, Moriki N, Saito H, Tsumura H, Imanaka-Yoshida K, Kimura K, Mizoguchi A, Hartshorne DJ, Nakano T. The targeted disruption of the MYPT1 gene results in embryonic lethality. Transgenic Res. 2005;14:337–340. doi: 10.1007/s11248-005-3453-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.