

Abstract
Hereditary Spastic Paraplegia (HSP) is a clinically and genetically heterogeneous group of neurological disorders that are characterized by progressive spasticity of the lower extremities. We describe an extended consanguineous Saudi family in which HSP is linked to SPG18, a previously reported autosomal recessive locus, and show that it is associated with a nullimorphic deletion of ERLIN2, a component of endoplasmic reticulum associated degradation. This finding adds to the growing diversity of cellular functions that are now known to be involved in the maintenance of the corticospinal tract neurons.
Keywords: ERAD, Aphasia, Intellectual disability, ERLIN2
Brief report
Hereditary Spastic Paraplegia (HSP) is a clinically and genetically heterogeneous group of neurological disorders that are characterized by progressive spasticity that typically affects the lower extremities but can extend cranially as the disease advances [1]. When manifestations other than motor dysfunction are present, the designation complicated or complex (as opposed to “pure”) HSP is often used [2]. Autosomal recessive and dominant as well as X-linked forms have been described clinically and later corroborated by the delineation of the underlying loci (designated SPG), many of which have been solved at the gene level [3]. Interestingly, the genes identified to date serve myriad functions including membrane trafficking, myelination, neurotrophy, cytoskeleton organization, chaperone activity and mitochondrial functions [3]. Identifying new HSP genes will likely, therefore, shed light on other aspects of the cellular process required for the maintenance of motor neurons.
We have identified an extended consanguineous Saudi family with five affected members with HSP (Fig. 1a). The two siblings (1 and 2) were referred to us for evaluation because of progressively worsening spasticity and intellectual disability. Patient 1 is an 8 year old boy whose pregnancy and neonatal history was unremarkable. He sat at 1 year and walked holding onto furniture at 24 months. At 30 months parents noticed progressive tightening of his lower extremities which progressed to the upper extremities at 4 years of age at which point he was completely wheel chair-bound. His expressive language never progressed beyond mama and dada but he regressed to babbling. Seizures were first noted at 7 years and EEG was severely abnormal with generalized slowing of background, and presence of generalized slow spike and wave activities compatible with the diagnosis of atypical absence epilepsy. MRI/MRS brain was normal as was a metabolic screen, blood indices, CPK level, thyroid function test and EM of lymphocytes. Patient 2 is the 3 year old sister of Patient 1. Her delivery was via C/S because of breech presentation and she was diagnosed with congenital hip dislocation on newborn exam. Her subsequent course was remarkably similar to her brother’s except that she did not have seizures. A similar panel of investigations revealed normal results although EEG was not performed. Unfortunately, the three maternal uncles could not come for evaluation but reports available indicate severe intellectual disability, aphasia and marked hypertonia but no seizures.
Given the consanguinity loop that connects all affected members of this family, we hypothesized that a common autozygous interval will be shared by the affected members consistent with autosomal recessive inheritance caused by a homoallelic mutation [4]. Indeed, autozygosity mapping revealed only a single locus shared across all the patients we analyzed, an 18.2 Mb interval on 8p12–8q11.22. This was further confirmed by linkage analysis which revealed a genome-wide maximal LOD score of 4.205 to this interval (Fig. 1b). No other peak crossed the 3.0 linkage threshold. We assessed all peaks of >2.5 LOD score for overlap with any SPG locus, whether autosomal dominant or recessive. The only overlap we noted was with our maximal linkage peak, which overlapped with the previously reported SPG18 locus [5]. The remarkable similarity in phenotype with the two Omani families who defined that locus led us to hypothesize that a similar locus underlies the disease in our family, which allowed us to further narrow the interval to 7.6 Mb flanked by the heterozygous SNP rs10954899 at the proximal end and D8S532 at the distal end (Fig. 1c). This reduced the number of genes in the candidate interval from 118 to 57 genes. Of these, UNC5D, RAB11FIP1 and ERLIN2 were selected for capillary sequencing based on cell biology function and mouse phenotype if known. No pathogenic mutations were identified in the first two genes. However, the two alternative initiation exons of ERLIN2 consistently failed to amplify in patients compared to controls and unaffected siblings, whereas the remaining exons amplified normally (primer sequences are provided in Table 1). This pattern indicated the possibility of a large-scale genomic deletion and segregated with the disease state in all five patients and in individuals IV:9–IV:16 (Fig. 1a).
Fig. 1.
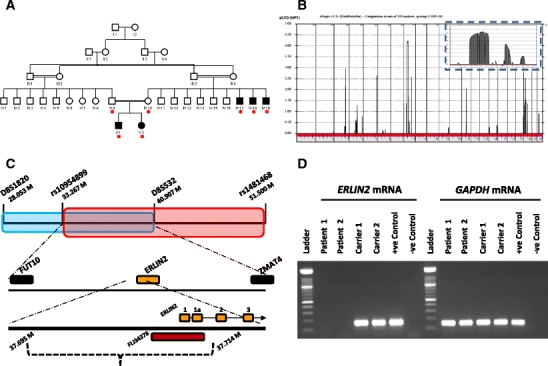
a Pedigree of the family described in this study. Clinical details for patients V:1 and V:2 are provided in the text. Individuals with red asterisks underneath were utilized for linkage analysis. b Genome-wide linkage analysis using the Affymetrix Axiom microarray platform and EasyLinkage v5.08 software revealed a single maximal peak on chromosome 8 with a LOD score of 4.2. Inset: chromosome-specific display showing close-up of linkage region on chromosome 8. c Overlap of the linkage interval in this study (in red) with the one previously published by Al-Yahyaee et al. [5] (in blue) refined the critical interval to a region flanked by the FUT10 and ZMAT4 genes. Physical position of markers is based on the 2006 assembly (hg18). The dashed curly bracket indicates the 20 kb deleted region, along with the approximate breakpoint positions and the genes affected. d Electrophoresis gel image showing results of RT-PCR conducted on two patients (V:1, V:2 from A), two obligate carriers (IV:9, IV:10), one normal control and one negative control containing no template. The GAPDH data confirms presence of cDNA in patient samples
Table 1.
Sequences of primers used for genomic DNA and cDNA amplification in this study
| Primer name | Sequence |
|---|---|
| ERLIN2_Ex1_F | 5′ CCAGCTACGGCCATTGAC 3′ |
| ERLIN2_Ex1_R | 5′ GAAAGTGACGGGTCAGCC 3′ |
| ERLIN2_Ex1b_F | 5′ AGGAAGTCGCGTGCTGAG 3′ |
| ERLIN2_Ex1b_R | 5′ GGTGACTGACTGCAAAGTTCA 3′ |
| ERLIN2_Ex2_F | 5′ GAGGTCCTCTCGCTGTTGTG 3′ |
| ERLIN2_Ex2_R | 5′ AGGAAAGCAATGACCAGGAA 3′ |
| ERLIN2_Ex3_F | 5′ GAAGGGAAAGTTGACCCTCA 3′ |
| ERLIN2_Ex3_R | 5′ TCTCTGGCTTTATCTTGAAGGA 3′ |
| ERLIN2_Ex4_F | 5′ CCAGGACAAAGGCATTTAGG 3′ |
| ERLIN2_Ex4_R | 5′ TCACTGTCTCTTCCTGCAACC 3′ |
| ERLIN2_Ex5+6_F | 5′ ACATCTTACGCCATCACCCT 3′ |
| ERLIN2_Ex5+6_R | 5′ TTCTAGAATGGACCTTGTCTTCA 3′ |
| ERLIN2_Ex6b_F | 5′ TCTGTTTAAACTCACTGCCAGAA 3′ |
| ERLIN2_Ex6b_R | 5′ CCTCTTCAGGGACATTAGGG 3′ |
| ERLIN2_Ex7+8_F | 5′ GCACTTTACCTCATCCTGCC 3′ |
| ERLIN2_Ex7+8_R | 5′ GCAGCTTTGGTTACCTCTGG 3′ |
| ERLIN2_Ex9_F | 5′ GGAGTTTGCCTCTCTTCAGC 3′ |
| ERLIN2_Ex9_R | 5′ CTCCTTGCATCTTCCCACC 3′ |
| ERLIN2_Ex10_F | 5′ ACACTCAGCTCGGGTGAAAG 3′ |
| ERLIN2_Ex10_R | 5′ CAGTTAAAGGGCAAAGGCAA 3′ |
| ERLIN2_Ex11_F | 5′ TTCCCATAGCCTCTGCACTT 3′ |
| ERLIN2_Ex11_R | 5′ CAACACTGGAAACTCTCCTGC 3′ |
| ERLIN2_Ex12_F | 5′ TGCTCCAAGATCTGAGATGACT 3′ |
| ERLIN2_Ex12_R | 5′ CAAAGGTCAAGCCCATGATT 3′ |
| ERLIN2_RT-PCR_F | 5′ TGCTCCCTTTCATCACATCA 3′ |
| ERLIN2_RT-PCR_R | 5′ CCAGGAAGTTCACCACTTCAA 3′ |
| GAPDH_RT-PCR_F | 5′ GGTGAAGGTCGGAGTCAAC 3′ |
| GAPDH_RT-PCR_R | 5′ ATGGGTGGAATCATATTGGA 3′ |
To delineate the deletion breakpoints we designed primers specific for the genomic DNA upstream of ERLIN2, at 1 kb intervals. Amplification of patient DNA alongside normal controls revealed an approximately 20 kb deletion, with the distal breakpoint near physical position 37,694,857 (Build NCBI36) and the proximal breakpoint near 37,714,575 immediately upstream of ERLIN2 exon 2 (Fig. 1c).This deletion is not described in the Toronto database of genomic variants (http://projects.tcag.ca/variation). Further experiments indicated that exon 2, while not deleted, had been genomically repositioned (data not shown). This 20 kb interval spans two protein-coding genes, ERLIN2 and FLJ34378 (RP11-863 K10.7). The latter gene is of unknown function and is totally deleted in our patients. To assay the effects of this genomic deletion on ERLIN2 transcription we performed reverse-transcriptase PCR (RT-PCR) on lymphoblast RNA. Intriguingly there was no transcription evident in the patient samples, unlike obligate carriers and normal controls who displayed robust RT-PCR products (Fig. 1d).Our data indicate that the loss of the ERLIN2 initiation exons along with mislocalization of exon 2 is sufficient to cause a nullimorphic allele.
While there is also loss of genomic DNA (and thus transcription) from the neighboring FLJ34378 gene, a recent article by Yildirim et al. describes a familial case of motor dysfunction and intellectual disability that was linked to a 2 base pair frameshift insertion in ERLIN2. Although not classified as spastic paraplegia, their patient phenotype displays certain similarities with ours [6]. In addition, a survey of the 1,000 Genomes Project (http://browser.1000genomes.org) does not reveal any truncating variants within the protein-coding sequences of ERLIN2, while FLJ34378 includes rs112819064, a deletion variant within the putative coding region that is predicted to cause premature truncation. Hence, while we cannot exclude the possibility of FLJ34378 contributing a modifier effect to our family, a review of the literature strongly implicates ERLIN2 depletion as the likely cause of our patients’ phenotype. We have therefore identified a novel ERLIN2 mutation that defines the SPG18 locus.
ERLIN2 (also known as SPFH2) is an SPFH domain-containing protein. The domain, named based on similarities to the proteins stomatin, prohibitin, flotillin, and HflC/K, defines a family of around 100 mammalian proteins that have in common the ability to assemble into large oligomeric structures and localize to cholesterol-rich, detergent-resistant membranes [7]. This protein has been identified as a mediator of the endoplasmic reticulum degradation (ERAD) pathway, a multistep process that involves the proper ubiquitin–proteasome mediated degradation of proteins [8]. ERAD does not only target aberrant proteins but also certain normal proteins that are needed to be maintained in a tightly regulated level of activity to maintain cellular homeostasis [9]. Inositol 1,4,5-trisphosphate(IP3) receptors (IP3Rs) are a good example of the latter since ERAD of IP3Rs is coupled with their activation by IP3 so the resulting channel opening is kept transient [10]. ERLIN2 was found, among other factors, to mediate ERAD of IP3R. Specifically, ERLIN2 was found to form a heterodimer with SPFH1 and deficiency of this complex induced by RNAi led to failure of ERAD of IP3R. In fact ERLIN2 deficiency, while still permitting the formation of stable SPFH1 homodimers, led to failure of SPFH1 to effect activation of IP3R ERAD [11]. Our mutation, in the event it permits transcription, leads to loss of a domain that is essential for the formation of the heterodimer with SPFH1 [11].
An attractive model, therefore, arises for the pathogenesis of HSP in our patients. Deficiency of ERLIN2 leads to impaired ERAD of IP3R which leads to persistent activation of IP3 signaling and channel opening thus keeping neurons in a state of hyperactivity. This is consistent with the recently demonstrated role of IP3R in the neuronalcalcium signaling repertoire [12]. However, we caution that experimental evidence is needed to support this model and that ERLIN2 may exert its pathogenesis through the impaired degradation of other proteins.
In summary, we have defined the locus for SPG18 using a Saudi family that bears a null mutation in ERLIN2. The biology behind ERLIN2 function opens intriguing possibilities as to the mechanism underlying HSP in our patients and others who link to this gene.
Acknowledgments
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
References
- 1.Salinas S, Proukakis C, Crosby A, Warner TT. Hereditary spastic paraplegia: clinical features and pathogenetic mechanisms. Lancet Neurol. 2008;7(12):1127–1138. doi: 10.1016/S1474-4422(08)70258-8. [DOI] [PubMed] [Google Scholar]
- 2.Fink JK (1993) Hereditary spastic paraplegia overview [PubMed]
- 3.Blackstone C, O’Kane CJ, Reid E. Hereditary pastic paraplegias: membrane traffic and the motor pathway. Nat Rev Neurosci. 2011;12(1):31–42. doi: 10.1038/nrn2946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alkuraya FS. Autozygome decoded. Genet Med. 2010;12(12):765–771. doi: 10.1097/GIM.0b013e3181fbfcc4. [DOI] [PubMed] [Google Scholar]
- 5.Al-Yahyaee S, Al-Gazali LI, De Jonghe P, Al-Barwany H, Al-Kindi M, De Vriendt E, Chand P, Koul R, Jacob PC, Gururaj A, Sztriha L, Parrado A, Van Broeckhoven C, Bayoumi RA. A novel locus for hereditary spastic paraplegia with thin corpus callosum and epilepsy. Neurology. 2006;66(8):1230–1234. doi: 10.1212/01.wnl.0000208501.52849.dd. [DOI] [PubMed] [Google Scholar]
- 6.Yildirim Y, Orhan EK, Iseri SA, Serdaroglu-Oflazer P, Kara B, Solakoglu S, Tolun A. A frameshift mutation of ERLIN2 in recessive intellectual disability, motor dysfunction and multiple joint contractures. Hum Mol Genet. 2011;20(10):1886–1892. doi: 10.1093/hmg/ddr070. [DOI] [PubMed] [Google Scholar]
- 7.Browman DT, Hoegg MB, Robbins SM. The SPFH domain-containing proteins: more than lipid raft markers. Trends Cell Biol. 2007;17(8):394–402. doi: 10.1016/j.tcb.2007.06.005. [DOI] [PubMed] [Google Scholar]
- 8.Pearce MM, Wang Y, Kelley GG, Wojcikiewicz RJ. SPFH2 mediates the endoplasmic reticulum-associated degradation of inositol 1,4,5-trisphosphate receptors and other substrates in mammalian cells. J Biol Chem. 2007;282(28):20104–20115. doi: 10.1074/jbc.M701862200. [DOI] [PubMed] [Google Scholar]
- 9.Vembar SS, Brodsky JL. One step at a time: endoplasmic reticulum-associated degradation. Nat Rev Mol Cell Biol. 2008;9(12):944–957. doi: 10.1038/nrm2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wojcikiewicz RJ. Regulated ubiquitination of proteins in GPCR-initiated signaling pathways. Trends Pharmacol Sci. 2004;25(1):35–41. doi: 10.1016/j.tips.2003.11.008. [DOI] [PubMed] [Google Scholar]
- 11.Pearce MM, Wormer DB, Wilkens S, Wojcikiewicz RJ. An endoplasmic reticulum (ER) membrane complex composed of SPFH1 and SPFH2 mediates the ER-associated degradation of inositol 1,4,5-trisphosphate receptors. J Biol Chem. 2009;284(16):10433–10445. doi: 10.1074/jbc.M809801200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ryglewski S, Pflueger HJ, Duch C. Expanding the neuron’s calcium signaling repertoire: intracellular calcium release via voltage-induced PLC and IP3R activation. PLoS Biol. 2007;5(4):e66. doi: 10.1371/journal.pbio.0050066. [DOI] [PMC free article] [PubMed] [Google Scholar]