

Abstract
Neurons of the ventrolateral preoptic nucleus (VLPO) promote sleep and VLPO loss produces insomnia. Previous studies show that general anesthetics including isoflurane activate VLPO neurons, and may contribute to their sedative effects. However, it is not clear to what extent the activation of VLPO neurons contributes to general anesthesia. We tested whether destruction of the VLPO neurons would affect the onset, depth, or recovery from isoflurane's general anesthetic effects. The VLPO was ablated in 25 rats by bilateral local injection of orexin-saporin, and polysomnography was performed to measure baseline sleep loss and responses to isoflurane anesthesia at 1% and 2%. Eight rats received sham (saline) injections. We measured isoflurane effects on time to loss of righting reflex, onset of continuous slow wave activity, and burst suppression; burst-suppression ratio; and time to recovery of righting reflex and desynchronized EEG. VLPO neuron cell loss was quantified by post hoc histology. Loss of VLPO neurons as well as lesion size were associated with cumulative sleep loss (r=0.77 and r=0.62, respectively), and cumulative sleep loss was the strongest predictor of high sensitivity to anesthesia, expressed as decreased time to loss of righting reflex (−0.59), increased burst-suppression ratio (r=0.52), and increased emergence time (r=0.54); an interaction-effect of isoflurane dose was observed (burst-suppression ratio: p<0.001). We conclude that the sleep loss caused by ablation of VLPO neurons sensitizes animals to the general anesthetic effects of isoflurane, but that the sedation produced by VLPO neurons themselves is not required for deep isoflurane anesthesia.
Keywords: hypothalamus, isoflurane rat, sleep, sleep deprivation
1. Introduction
General anesthesia is a drug-induced, reversible condition with five components: hypnosis (loss of consciousness), amnesia, analgesia, immobility (no movement in response to pain stimuli), and hemodynamic stability with control of the stress response (Evers and Crowder, 2006). About 2–5% of the population is placed under general anesthesia per year, but a detailed understanding of the neural basis for this condition is still lacking. Anesthesia and natural sleep share some molecular mechanisms. First, most commonly used general anesthetics potentiate gamma-amino-butyric acid (GABA)-induced chloride currents (Franks, 2008) and GABA is the principle neurotransmitter used by sleep-promoting neurons in the ventrolateral preoptic nucleus (VLPO). Sleep requires the inhibition of multiple pathways in the arousal system and available evidence suggests that the VLPO is a major source of sleep-active neurons that provide GABAergic, inhibitory input to many elements of the arousal system (Sherin et al., 1996), including the tuberomammillary nucleus (TMN), locus coeruleus, and orexinergic neurons (Saper et al., 2010). In addition, many compounds that enhance GABAergic transmission promote sleep at low doses (Kales et al., 1968; Kay et al., 1972), but induce anesthesia at high doses (Eikermann et al., 2009).
It has recently been shown that GABA-potentiating general anesthetic drugs engage hypothalamic sleep-promoting neurons in the VLPO (Lu et al., 2008; Nelson et al., 2002). Drugs as diverse as isoflurane, pentobarbital, and chloral hydrate at general anesthetic doses activate VLPO neurons, as judged by expression of the immediate early gene cFos (Lu et al., 2008). Although activation of VLPO neurons would presumably potentiate the drug effects (by causing more GABA release onto arousal neurons), whether this activity plays an important role in the effects of these drugs at general anesthetic doses remained unclear. If it did, rats with VLPO lesions may exhibit lighter anesthesia at controlled end tidal levels of isoflurane. Furthermore, ablation of the VLPO may be expected to prolong the time to onset of anesthesia and shorten time to recovery. On the other hand, if VLPO neurons are not required for isoflurane general anesthesia, anesthetic effects may be potentiated by accumulated sleepiness that occurs in VLPO lesioned animals (Lu et al., 2000). Therefore, we tested whether ablation of VLPO neurons would cause increased or decreased sensitivity to isoflurane anesthesia by measuring the effects of isoflurane in terms of clinical and electroencephalographic metrics of onset latency, depth, and recovery from anaesthesia in animals with VLPO lesions vs. sham lesions.
2. Results
2.1. Lesion extent
As intended, the degree of cell loss in the VLPO varied in these experiments from none (n=3) to nearly complete (n=3), with the majority of cases (n= 19) having partial lesions with varying degrees of involvement of surrounding structures including the median preoptic nucleus and the nucleus of the diagonal band (Figure 1). Rats with saline injections had no damage (n=8).
Figure 1.

VLPO lesions. A and B) Photomicrographs of the VLPO in two representative cases where the VLPO was virtually intact (left) and lost over 90% of its neurons (right). Note that the VLPO is located at the base of the hypothalamus at the level of the optic chiasm (ox) just medial to the nucleus of the diagonal band (DB). 3v denotes the third ventricle. The box (300×300 microns) outlines the counting region.
2.2. The effects of lesions on natural sleep
Both the number of VLPO neurons lost as well as lesion size were associated with sleep loss (r=0.77 and r=0.62, respectively). However, multiple regression analysis revealed that loss of VLPO neurons and not lesion size predicted the cumulative sleep loss, indicating that loss of VLPO neurons rather than an unspecific brain damage represents the biologically relevant mechanism of sleep deprivation observed in our study. The VLPO lesions produced a significant increase in wakefulness (to 55.3% +/− 1.5, n=25, ranging from 47.9 to 70.4%) compared to sham lesioned animals (46.6% +/− 0.9, n=8, ranging from 43.4 to 51.1%).
2.2. The effects of VLPO lesions on general anesthesia
The transition from the awake to the anesthetized state was marked by typical EEG waveforms progressing from desynchronized to elevated slow wave activity to synchronized bursting alternating with silence (burst suppression pattern) (Fig. 2). At steady-state 2% isoflurane, the burst suppression ratio (BSR) in VLPO-lesioned rats was 0.75 +/− 0.02 n=25, indicating electrographic silence 75% of the time (Fig. 3B, D). The BSR at 1% isoflurane averaged 0.25 +/− 0.03; n=25 (Figure 3A, C). For comparison, the BSRs in sham-lesioned rats were 0.66 +/− 0.12 and 0.10 +/− 0.04 in 2 and 1% isoflurane, respectively (n=6). The depth of anesthesia as estimated by the burst suppression ratio was correlated with the degree of VLPO lesion as taken by cell counts (p=0.0001; linear mixed model, see Statistics. The greater the cell loss in VLPO, the greater percentage of time the EEG was in the isoelectric state, indicating a deeper level of anesthesia. This effect was dose-dependent; an interaction-effect between isoflurane dose and number of remaining VLPO neurons was detected for burst-suppression ratio (p=0.0001), indicating that the effects of VLPO lesion on EEG signs of depth of isoflurane-induced unconsciousness were manifested only at shallow levels of anesthesia (1% isoflurane; Figure 3). The correlations between VLPO neuron number and latency to loss of righting reflex (LORR; r=0.377; p=.063; Figure 4) and to the first silent second (r= 0.381; p=.060) were not significant but were trending in the direction of VLPO neuron loss contributing to anaesthesia induction. There was no correlation between VLPO lesions and the onset time of slow wave activity (r= 0.77, p=0.715).
Figure 2.
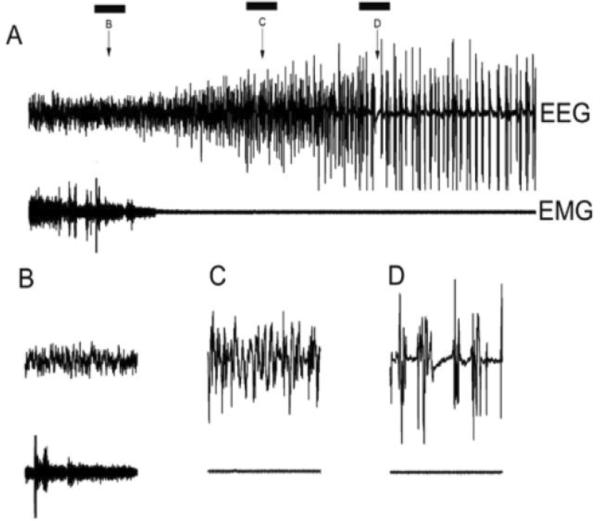
Changes in EEG and EMG during anesthesia induction. A) isoflurane administration began at the beginning of the record. The top trace shows the progression of EEG from the desynchronized pattern of wakefulness (B) to synchronized (C) to a burst suppression pattern (D). The bottom trace shows the EMG reflecting the loss of muscle tone during induction. The bars indicate 10 sec epochs that are shown with an expanded time base in the bottom panel. Note that the segment in “D” contains the first silent second which occurred with a latency of 114 sec in this case (R4029).
Figure 3.

The relationship between the number of VLPO neurons and the depth of anesthesia.
Top: Correlation between the number of remaining VLPO neurons following lesions and the burst suppression ratio at steady state isoflurane A) 1% and B) 2%. Bottom: Sample EEG traces (1 min duration) during 1% (C) and 2% (D) isoflurane.
Figure 4.

The relationship between the number of VLPO neurons and the speed of induction and recovery from anesthesia. Plotted is the VLPO neuron number vs. time to loss (A) and recovery (B) of righting reflex.
We measured emergence time from a steady-state level of 1% isoflurane. The number of remaining VLPO neurons was associated with time to emergence as measured by recovery of righting reflex (r= −0.488; p=0.013).
Overall, these correlations were not consistent with the concept that VLPO neurons play a role in induction of general anesthesia with isoflurane, but rather suggested the opposite. We thus explored the idea that the observed changes were related to sleep loss induced by VLPO lesions. We found that the amount of sleep loss was correlated with BSR (again, only at 1% isoflurane, r=0.571; p=0.003) (figure 5), time to LORR and RORR (r= −0.585, p=0.002; r=0.587, p=0.002 respectively) (Fig 6). Finally, since LORR and RORR include motor components and thus may not necessarily reflect consciousness per se, we explored the relationship between amount of sleep loss and electrographic metrics of induction and emergence time. We found that latency to silent second (see fig 2) was not significantly correlated with sleep loss (r= −0.265, p=0.201). However, emergence of an activated cortical EEG (figure 7) was correlated with sleep loss (r=0.547, p=0.019, n=18; Fig 8).
Figure 5.
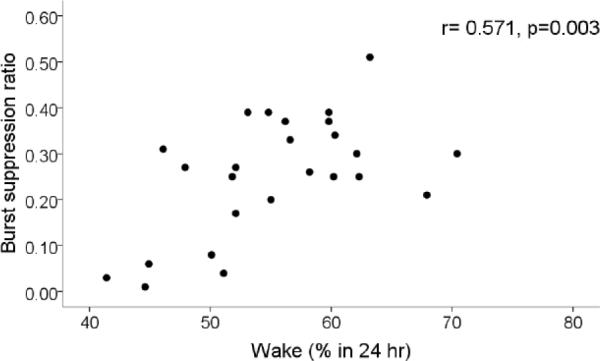
Relationship between sleep loss and depth of anesthesia. Plotted is the burst suppression ratio at steady-state 1% isoflurane anesthesia vs. the percentage of time spent in the waking state. Note that a greater degree of insomnia correlates with deeper anesthesia.
Figure 6.

Relationship between sleep loss and induction and emergence time. Plotted is the latency to loss (A) and recovery (B) of righting following isoflurane administration reflex vs the percentage of time spent in the waking state. Note that the less the rats slept the sooner they lost righting reflex and the longer it took for them to regaining righting reflex.
Figure 7.
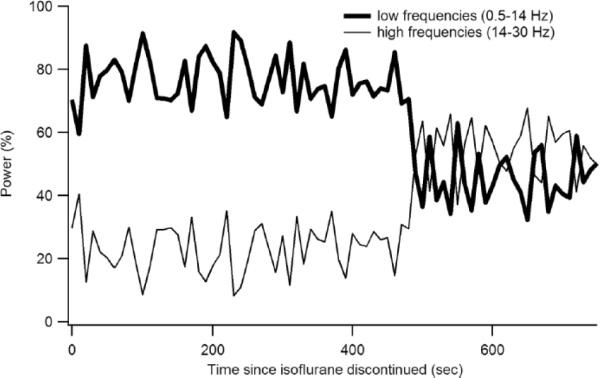
Time course of the return of high frequency EEG power at recovery from anesthesia. Shown is the relative low frequency (0.5–14 Hz) and high frequency (14–30 Hz) EEG power following discontinuation of isoflurane in one rat (R3924). Note the abrupt transition that we took as electrographic emergence (Fig 8).
Figure 8.
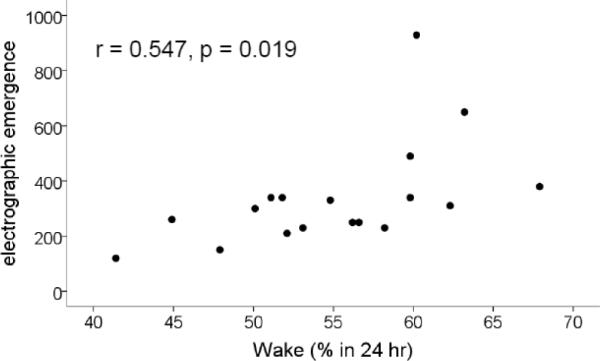
Relationship between sleep loss and the latency to electrographic emergence. Plotted is the latency to the abrupt loss of the ratio of low to high power in the EEG (electrographic emergence) vs the time spent awake. Note that the more time the rats spent awake, the longer it took for EEG recovery from isoflurane anesthesia.
3. Discussion
We found that rats with VLPO lesions were more sensitive to isoflurane. Moreover, the greater the cell loss in VLPO, the greater percentage of time the EEG was in the isoelectric state (at 1% steady-state isoflurane) and the longer the time to recovery. However, these same parameters correlated with greater sleep loss (due to the VLPO lesions). These results indicate that VLPO neurons are not required for isoflurane to induce general anesthesia, but that the sleep loss caused by damage to VLPO neurons may increase the sensitivity to anesthesia. Loss of consciousness is an essential component of general anesthesia; sleep is a behavioral state also marked by lack of awareness. Although sleep and general anesthesia clearly differ in important ways (e.g., sleep can be interrupted by behavioral stimulation, but anesthesia cannot), it has been speculated that overlapping neural mechanisms may underlie these states. Specifically, it has been proposed that the activation of sleep-promoting neurons in the VLPO by administration of GABA-enhancing anesthetic agents (Lu et al., 2008; Nelson et al., 2002), may contribute to inhibition of the arousal system. For example, VLPO neurons that project to and inhibit the histaminergic tuberomamillary nucleus and other arousal-promoting neurons (Sherin et al., 1998) are activated by general anesthetics, and injection of a GABA antagonist into the TMN decreases the duration of LORR induced by i.v. propofol (Nelson et al., 2002). However, whether increased firing of VLPO neurons during general anesthesia participates significantly in the sedative effects of the drugs is not known. Alternatively, GABA-A enhancing drugs, at doses that produce general anesthesia, may have such profound inhibitory effects on both the arousal systems and their targets that the activation of VLPO neurons may not pay an important role in their actions.
In our experiments, we tested the hypothesis that the firing of VLPO neurons would play an important role in inducing general anesthetic effects. Previous work from our laboratory has shown that although the lesions created by cell-specific toxins may be complex and involve several cell groups, the correlation of cell loss in a particular cell group with physiological effects provides a powerful tool for dissection the functional significance of specific cell groups (Chou et al., 2003; Lu et al., 2000; Lu et al., 2006). In the current study, we used a cohort of rats with lesions of variable size. While both the number of VLPO neurons lost as well as the total size of the lesion were associated with sleep loss, our backward stepwise regression analysis revealed that VLPO neuron number and not lesion size predicted the cumulative sleep loss, indicating that loss of VLPO neurons rather than an unspecific brain damage represents the biologically relevant mechanism of sleep deprivation observed in our study.
We found that rats with decreasing numbers of surviving VLPO neurons exhibited deeper, rather than lighter, anesthesia and delayed, rather than quicker, emergence. We attribute this to the sleep loss caused by the VLPO lesions, as previous studies have shown that sleep deprivation reduces the dosage threshold and increases the speed of onset of anesthesia and prolongs time to emergence (Figs 5–7 and (Tung et al., 2002)). We additionally show that the depth of anesthesia is dependent on prior sleep and that this effect is absent during very deep levels of anesthesia. On the other hand, even though VLPO neurons are engaged by isoflurane at 2%, they clearly are not necessary for the generation of either high levels of delta activity or burst-suppression EEG patterns caused by this agent. Thus, at anesthetic doses of isoflurane and perhaps other drugs capable of causing general anesthesia, the main mechanism of action is likely to be direct and profound pharmacological inhibition of arousal systems as well as cerebral cortical and motor system neurons, rather than potentiation of the more modest physiological VLPO effect on the arousal system.
It is likely that adenosine, a key mediator of homeostatic sleep drive(Porkka-Heiskanen et al., 1997) contributes to effects of sleep loss on the anesthesia. Tung et al found that effects of acute sleep loss on time to loss of and recovery of righting reflex in rats are partially reversed by adenosine antagonists (Tung et al., 2005). Interestingly, adenosine excites VLPO neurons via disinhibition (Chamberlin et al., 2003) and this may be one means by which it promotes sleep. However, our data make this an unlikely mechanism for affecting anesthesia-induced hypnosis. Instead, we suspect that elevated adenosine levels may act by presynaptic inhibition via the A1 adenosine receptor on excitatory glutamatergic systems in both the cerebral cortex and the arousal systems (Brambilla et al., 2005), which in turn result in deeper and longer lasting general anesthesia. Adenosine is known to affect multiple arousal centers, including the basal forebrain (Arrigoni et al., 2006) (Strecker et al., 2000), the lateral hypothalamus, where orexin neurons are inhibited by adenosine (Rai et al., 2010), and the TMN (Oishi et al., 2008). Persuasive data suggest that arousal neuron activity counteracts general anesthesia and facilitates emergence from anesthesia; orexin-KO mice exhibit delayed recovery from anesthesia (Kelz et al., 2008) and disinhibition of the TMN by gabazine decreases duration of general anesthesia (Nelson et al., 2002). Taken together with the findings that anesthetics directly inhibit arousal-promoting hypothalamic neurons (Zecharia et al., 2009), these data suggest that adenosine may potentiate the effects of certain GABAergic anesthetics at these sites.
In conclusion, at doses of isoflurane sufficient to cause burst-suppression patterns in the EEG, the participation of VLPO neurons is not necessary for the induction of anesthesia. On the other hand, in animals with substantial sleep loss due to VLPO lesions, there is increased susceptibility to general anesthesia and prolonged recovery time, and this correlates with the amount of sleep loss. These findings reinforce the importance of the sleep state of the individual patient pre-anesthesia in the time to onset and recovery from anesthesia.
4. Experimental Procedures
4.1. VLPO lesions and recording electrode implantation
Adult male Sprague-Dawley rats (n=33) weighing between 325 and 360 gms were included in this study. Rats were maintained in standard laboratory conditions including 12:12 hr light:dark cycle (lights on at 7:00) and 23±1°C room temperature. The rats had ad libitum access to food and water. Care of the rats met National Institutes of Health standards, as set forth in the Guide for the Care and Use of Laboratory Animals, and all protocols were approved by the BIDMC and Harvard Medical School Institutional Animal Care and Use Committees.
The rats were anesthetized with chloral hydrate (350 mg/kg) and 200 nanoliters of orexinsaporin (n=25; Advanced targeting systems, CA, USA) or saline (0.9%; n=8) was injected into the VLPO bilaterally using a fine glass micropipette as explained previously (Lu et al., 2000). We used orexin-saporin as a non-selective neurotoxin as it has been shown that when injected in high doses it nonspecifically kills all the neurons in the injected region (Fuller et al., 2011). The advantage of this toxin compared to excitotoxins such as ibotenic acid is that it acts slowly and with fewer unwanted side effects due to transient neuron activation. The coordinates (with reference to Bregma) for the VLPO were anteroposterior, −0.6 mm; dorsoventral, −8.5 mm; and mediolateral, ±1.0 mm, with the incisor bar set at −3.3 mm. Following the injections, the rats were implanted with 4 electrodes for recording electroencephalogram (EEG) and two electrodes for recording neck electromyogram (EMG) as mentioned earlier (Lu et al., 2000). Wounds were closed and rats given postoperative analgesia (flunixin; 2.5 mg/kg, sc). Following the surgical procedure, the rats were maintained for at least 10 days and 48-hr polysomnographic recordings were made subsequently. For sleep recordings, the animals were first connected via flexible recording cables to a commutator, which in turn was connected to a Grass polygraph and computer and they were habituated to the cables for one day. Continuous recording of the EEG/EMG and time-lock video began after the habituation period and continued uninterrupted for 48 hours on each occasion.
4.2. Isoflurane testing protocol
After recovering from surgery, rats were tested for sensitivity to isoflurane as follows. Rats were placed into a modified cage that was ventilated with compressed air (4 l/min). A cable was screwed to the connector on the rat's headset and connected to a commutator which allowed free movement of the rat without tangling the cable. EEG and EMG data were recorded throughout the experiment (AxoScope 9.0 at 2 kHz). EEG signals were low-pass filtered at 100Hz and EMG signals bandpass-filtered at 100–1000 Hz. Chamber CO2 and isoflurane levels were continuously monitored and the former remained virtually zero throughout the study. Before isoflurane administration, rats were acclimated to the recording environment for at least one hour. Isoflurane was delivered with a precision vaporizer (3%; 4 l/min in 100% O2) and the rats visually monitored both by an assessor and by a video camera. Note that the experimenter was blinded to the extent of the VLPO lesion as this was only determined by post mortem histology. After rats lost their righting response, i.e. lost postural muscle tone, sank to the ground, and exhibited a rhythmic breathing pattern, the isoflurane level was reduced to 2% for 45 min and subsequently to 1% for an additional 45 min. Isoflurane administration was then discontinued. Emergence from anesthesia was taken as the time the rat regained its footing. Because loss and recovery of righting reflex (LORR and RORR, respectively) represent composite endpoints that may reflect effects of anesthesia not only on consciousness but also on motor control, we additionally assessed electrographic metrics that should reflect mainly hypnotic effects of anesthesia: onset to slow wave pattern and the first silent second of burst suppression for induction and the recovery of high frequency activity for emergence (see below).
4.3. Histology
To quantify cell loss in VLPO, rats were deeply anesthetized with chloral hydrate (700 mg/kg, ip) and transcardially perfused with formalin. Brains were removed, postfixed overnight in 20% sucrose and 40 micron frozen sections were made through the hypothalamus, mounted on glass microscope slides, and stained for Nissl substance with 0.25% thionin (Figure 1).
The numbers of remaining neurons in the VLPO cluster in three representative sections through the nucleus were counted by an investigator (SY) who was blind to the behavioral testing results. The VLPO cluster, defined by a counting box (300 × 300 microns) placed at the base of the brain just medial to the Diagonal band of Broca. All Nissl-stained neuronal nuclei were counted on each side of 3 consecutive sections of a 1-in-4 series spaced 160 microns apart (Lu et al., 2002). Total lesion size was measured on Nissl-stained sections with ImageJ (http://rsbweb.nih.gov/ij/index.html).
4.4. Data Analysis
4.4.1 Spontaneous sleep
The polysomnography signals (EEG/EMG) of each rat were divided into 12-sec epochs and visually scored using Sleep Sign for Animals (Kissei Comtec Co., LTD., Nagano, Japan) into one of the three sleep-wake stages - wake, non rapid eye movement sleep (NREM) sleep or rapid eye movement (REM) sleep as per the established criteria (Lu et al., 2000; Chou et al., 2003). Scoring was done before histological examination and so the scorer was unaware of the extent of the lesions. The percentage of time spent in wake, NREM sleep and REM sleep per 24 hrs were calculated.
4.4.2 Isoflurane effects on EEG
Data were analyzed with SleepSign, Clampfit and Excel. To calculate anesthetic depth we used Clampfit to measure the portion of the time that the EEG was in the isoelectric state- the burst suppression ratio (BRS). During a 10 min segment of 1% and 2% steady-state end-tidal isoflurane anesthesia (30–40 min) periods of isoelectric EEG were identified visually and the cumulative time of isoelectric EEG as a percentage of the 10 min sample time was calculated. We used electroencephalographic signals to calculate objective metrics of induction and emergence. Spectral analysis of EEG data was performed in 4 sec epochs with Sleep Sign; results were exported to Excel for quantifying. We measured the time from the onset of isoflurane administration to the onset of burst suppression which was taken as the time of the first interval of isoelectric EEG that was at least one second in duration (Fig 2). The time to emergence was taken as the time at which beta power (14–30Hz) became higher than lower frequency power (0.5–14 Hz; FIG 7).
4.4.3 Statistics
The data were analyzed statistically with SPSS software (ver 18.0; Chicago, IL). Using a hierarchical sequence, repetitive measurements of different variables of isoflurane-induced unconsciousness could be tested with an alpha error of 5% without adjustment for multiple testing. The following sequence was defined: first, in the cohort of orexin-saporin injected rats we tested whether the burst suppression ratio during steady-state anesthesia was associated with the number of VLPO neurons. Possible differences in latency to the onset of loss of righting reflex (LORR), slow wave activity, burst suppression, the time to re-emergence of desynchronized EEG and recovery of righting reflex (RORR) after termination of anesthesia were tested subsequently only if the hypothesis of no relationship of VLPO cell loss with burst suppression ratio was rejected. We used a linear mixed model for repeated measures (compound symmetry repeated covariance type), including isoflurane concentration as a repeated, independent variable, VLPO neuron number as fixed, independent variable and the dependent variable was BSR. As for sample size estimation, we expected a correlation of VLPO lesion score and BSR of 0.55. Accordingly, we calculated that a sample size of 25 rats would provide us with an 80% power to detect an effect of VLPO lesion on the primary outcome variables (alpha-error: 5%). Multiple regression analysis was used to evaluate whether the independent variables VLPO neuron loss and lesion size predict sleep loss and sensitivity to isoflurane anesthesia.
Highlights
Anesthesia and sleep share some characteristics
Sleep-promoting VLPO neurons are active during anesthesia
Sleep loss increases anesthesia sensitivity
Lesion of VLPO does not interfere with anesthesia induction, maintenance or emergence
Acknowledgments
This work was supported by grants from the National Institute of Health (Bethesda, MD, USA, P50 HL060292, HL095491, AG09975 and NS072337), and a grant from the German Research Council (DFG-EI684/2-1).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arrigoni E, Chamberlin NL, Saper CB, McCarley RW. Adenosine inhibits basal forebrain cholinergic and noncholinergic neurons in vitro. Neuroscience. 2006;140:403–13. doi: 10.1016/j.neuroscience.2006.02.010. [DOI] [PubMed] [Google Scholar]
- Brambilla D, Chapman D, Greene R. Adenosine mediation of presynaptic feedback inhibition of glutamate release. Neuron. 2005;46:275–83. doi: 10.1016/j.neuron.2005.03.016. [DOI] [PubMed] [Google Scholar]
- Chamberlin NL, Arrigoni E, Chou TC, Scammell TE, Greene RW, Saper CB. Effects of adenosine on gabaergic synaptic inputs to identified ventrolateral preoptic neurons. Neuroscience. 2003;119:913–8. doi: 10.1016/s0306-4522(03)00246-x. [DOI] [PubMed] [Google Scholar]
- Chou TC, Scammell TE, Gooley JJ, Gaus SE, Saper CB, Lu J. Critical Role of Dorsomedial Hypothalamic Nucleus in a Wide Range of Behavioral Circadian Rhythms. J. Neurosci. 2003;23:10691–10702. doi: 10.1523/JNEUROSCI.23-33-10691.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eikermann M, Fassbender P, Zaremba S, Jordan AS, Rosow C, Malhotra A, Chamberlin NL. Pentobarbital dose-dependently increases respiratory genioglossus muscle activity while impairing diaphragmatic function in anesthetized rats. Anesthesiology. 2009;110:1327–34. doi: 10.1097/ALN.0b013e3181a16337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evers AS, Crowder CM. Cellular and molecular mechanisms of anesthesia. In: Barash PG, Cullen BF, Stoelting RK, editors. In Clinical Anesthesia Vol. Lippincott, Williams; Wilkins New York: 2006. pp. 111–132. [Google Scholar]
- Franks NP. General anaesthesia: from molecular targets to neuronal pathways of sleep and arousal. Nat Rev Neurosci. 2008;9:370–86. doi: 10.1038/nrn2372. [DOI] [PubMed] [Google Scholar]
- Fuller P, Sherman D, Pedersen NP, Saper CB, Lu J. Reassessment of the structural basis of the ascending arousal system. Journal of Comparative Neurology. 2011;519:933–56. doi: 10.1002/cne.22559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kales A, Kales JD, Jacobson A, Walter RD, Wilson TE. Effects of methyprylon and pentobarbital on sleep patterns. Electroencephalogr Clin Neurophysiol. 1968;24:397. [PubMed] [Google Scholar]
- Kay DC, Jasinski DR, Eisenstein RB, Kelly OA. Quantified human sleep after pentobarbital. Clin Pharmacol Ther. 1972;13:221–31. doi: 10.1002/cpt1972132221. [DOI] [PubMed] [Google Scholar]
- Kelz MB, Sun Y, Chen J, Cheng Meng Q, Moore JT, Veasey SC, Dixon S, Thornton M, Funato H, Yanagisawa M. An essential role for orexins in emergence from general anesthesia. Proc Natl Acad Sci U S A. 2008;105:1309–14. doi: 10.1073/pnas.0707146105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Greco MA, Shiromani P, Saper CB. Effect of lesions of the ventrolateral preoptic nucleus on NREM and REM sleep. J Neurosci. 2000;20:3830–42. doi: 10.1523/JNEUROSCI.20-10-03830.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Bjorkum AA, Xu M, Gaus SE, Shiromani PJ, Saper CB. Selective activation of the extended ventrolateral preoptic nucleus during rapid eye movement sleep. J Neurosci. 2002;22:4568–76. doi: 10.1523/JNEUROSCI.22-11-04568.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Jhou TC, Saper CB. Identification of Wake-Active Dopaminergic Neurons in the Ventral Periaqueductal Gray Matter. J. Neurosci. 2006;26:193–202. doi: 10.1523/JNEUROSCI.2244-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Nelson LE, Franks N, Maze M, Chamberlin NL, Saper CB. Role of endogenous sleep-wake and analgesic systems in anesthesia. J Comp Neurol. 2008;508:648–62. doi: 10.1002/cne.21685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson LE, Guo TZ, Lu J, Saper CB, Franks NP, Maze M. The sedative component of anesthesia is mediated by GABA(A) receptors in an endogenous sleep pathway. Nat Neurosci. 2002;5:979–84. doi: 10.1038/nn913. [DOI] [PubMed] [Google Scholar]
- Oishi Y, Huang ZL, Fredholm BB, Urade Y, Hayaishi O. Adenosine in the tuberomammillary nucleus inhibits the histaminergic system via A1 receptors and promotes non-rapid eye movement sleep. Proc Natl Acad Sci U S A. 2008;105:19992–7. doi: 10.1073/pnas.0810926105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porkka-Heiskanen T, Strecker RE, Thakkar M, Bjorkum AA, Greene RW, McCarley RW. Adenosine: a mediator of the sleep-inducing effects of prolonged wakefulness. Science. 1997;276:1265–8. doi: 10.1126/science.276.5316.1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rai S, Kumar S, Alam MA, Szymusiak R, McGinty D, Alam MN. A1 receptor mediated adenosinergic regulation of perifornical-lateral hypothalamic area neurons in freely behaving rats. Neuroscience. 2010;167:40–48. doi: 10.1016/j.neuroscience.2010.01.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saper CB, Fuller PM, Pedersen NP, Lu J, Scammell TE. Sleep state switching. Neuron. 2010;68:1023–42. doi: 10.1016/j.neuron.2010.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherin JE, Shiromani PJ, McCarley RW, Saper CB. Activation of ventrolateral preoptic neurons during sleep. Science. 1996;271:216–9. doi: 10.1126/science.271.5246.216. [DOI] [PubMed] [Google Scholar]
- Sherin JE, Elmquist JK, Torrealba F, Saper CB. Innervation of histaminergic tuberomammillary neurons by GABAergic and galaninergic neurons in the ventrolateral preoptic nucleus of the rat. J Neurosci. 1998;18:4705–21. doi: 10.1523/JNEUROSCI.18-12-04705.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strecker RE, Morairty S, Thakkar MM, Porkka-Heiskanen T, Basheer R, Dauphin LJ, Rainnie DG, Portas CM, Greene RW, McCarley RW. Adenosinergic modulation of basal forebrain and preoptic/anterior hypothalamic neuronal activity in the control of behavioral state. Behav Brain Res. 2000;115:183–204. doi: 10.1016/s0166-4328(00)00258-8. [DOI] [PubMed] [Google Scholar]
- Tung A, Szafran MJ, Bluhm B, Mendelson WB. Sleep deprivation potentiates the onset and duration of loss of righting reflex induced by propofol and isoflurane. Anesthesiology. 2002;97:906–11. doi: 10.1097/00000542-200210000-00024. [DOI] [PubMed] [Google Scholar]
- Tung A, Herrera S, Szafran MJ, Kasza K, Mendelson WB. Effect of sleep deprivation on righting reflex in the rat is partially reversed by administration of adenosine A1 and A2 receptor antagonists. Anesthesiology. 2005;102:1158–64. doi: 10.1097/00000542-200506000-00015. [DOI] [PubMed] [Google Scholar]
- Zecharia AY, Nelson LE, Gent TC, Schumacher M, Jurd R, Rudolph U, Brickley SG, Maze M, Franks NP. The Involvement of Hypothalamic Sleep Pathways in General Anesthesia: Testing the Hypothesis Using the GABAA Receptor {beta}3N265M Knock-In Mouse. J. Neurosci. 2009;29:2177–2187. doi: 10.1523/JNEUROSCI.4997-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]