

Abstract
Alloreactive memory T cells are present in virtually all transplant recipients due to prior sensitization or heterologous immunity and mediate injury undermining graft outcome. In mouse models, endogenous memory CD8 T cells infiltrate MHC-mismatched cardiac allografts and produce IFN-γ in response to donor class I MHC within 24 hours post-transplant. The current studies analyzed the efficacy of anti-LFA-1 mAb to inhibit early CD8 T cell cardiac allograft infiltration and activation. Anti-LFA-1 mAb given to C57BL/6 6 (H-2b) recipients of A/J (H-2a) heart grafts on days −1 and 0 completely inhibited CD8 T cell allograft infiltration, markedly decreased neutrophil infiltration, and significantly reduced intra-graft expression levels of IFN-γ-induced genes. Donor-specific T cells producing IFN-γ were at low/undetectable numbers in spleens of anti-LFA-1 mAb treated recipients until day 21. These effects combined to promote substantial prolongation (from day 8 to 27) in allograft survival. Delaying anti-LFA-1 mAb treatment until days 3 and 4 post-transplant did not inhibit early memory CD8 T cell infiltration and proliferation within the allograft. These data indicate that peri-transplant anti-LFA-1 mAb inhibits early donor-reactive memory CD8 T cell allograft infiltration and inflammation suggesting an effective strategy to attenuate the negative effects of heterologous immunity in transplant recipients.
Introduction
Transplantation of MHC-mismatched organs induces a vigorous alloimmune response that quickly mediates rejection of the graft unless checked by immunosuppression (1). In response to antigen-presenting cells emigrating from the allograft, donor-reactive CD4 and CD8 T cells are primed to develop to effector cells in secondary lymphoid organs. During this priming the reactive T cells upregulate the integrins and chemokine receptors that direct their trafficking to the allograft where they first interact with the graft vascular endothelium and migrate through this barrier into the tissue parenchyma to express the effector functions that mediate tissue injury and rejection of the graft (2, 3). In clinical transplantation priming of donor-reactive T cells is inhibited through the use of immunosuppressive drugs. Although this has decreased acute rejection of solid organ grafts, the use of these drugs is accompanied by nephrotoxicity that leads to renal tissue fibrosis, as well as increased incidences of infection and tumors (4). These adverse effects indicate the need to identify other strategies to inhibit the priming and/or function of donor-antigen reactive T effector cells.
The requirement for T cell trafficking to the allograft for cell-mediated rejection has raised the possibility of disrupting this trafficking as a strategy to prevent acute and chronic graft tissue injury and prolong graft survival. Antagonism of specific chemokines or their receptors that are expressed during rejection has, for the most part, been inefficient in disrupting leukocyte trafficking and the progression of acute cell-mediated rejection (5–8). In contrast, antagonism of integrin function has worked quite well. Lymphocyte function associated antigen-1 (LFA-1) is a β2 integrin required for T cell arrest on the vascular endothelium. Anti-LFA-1 antibodies are potent inhibitors of this arrest and T cell infiltration into inflammatory sites (9). In addition, LFA-1 is a key component of the immunological synapse and provides critical co-stimulatory signals during the activation of CD4 and CD8 T cells during interaction with antigen-presenting cells (10–16). Graft recipient treatment with anti-LFA-1 antibodies has been very effective in inhibiting acute rejection and prolonging the survival of allografts in rodent models (17–22).
Recent interest in transplantation has focused on the presence and impact of memory T cells with reactivity for donor antigens in candidate recipients prior to the transplant (23, 24). These memory T cells are generated in response viral and bacterial infections and through homeostatic proliferation (25–27). The presence of donor-reactive memory T cells in the peripheral blood of patients prior to transplant has a negative impact on the incidence of delayed graft function and long-term outcome of the allografts (28, 29). Studies in rodent models and in non-human primates have demonstrated the ability of donor-reactive memory T cells to subvert many immunosuppressive and tolerogenic strategies and promote rejection of allografts (30–34). Studies from this laboratory have documented the infiltration of endogenous effector memory CD8 T cells into class I MHC-mismatched cardiac allografts within 24 hrs post-transplantation in mouse models (35, 36). Within the allograft these memory CD8 T cells are activated to proliferate and to produce IFN-γ. Downstream consequences of this IFN-γ production are increased infiltration and activation of neutrophils in the allograft, which in turn, facilitate the recruitment of donor-antigen primed effector T cells into the graft. Thus, the presence of donor-reactive memory T cells in allograft recipients prior to transplant directly leads to graft injury and promotion of acute rejection. Given the importance of such memory T cells in transplantation, several strategies have been devised to attenuate their activity in graft recipients, including depletion with anti-CD52 mAb (alemtuzumab) or with anti-thymocyte globulin (ATG) (37, 38).
One strategy that might be effective in neutralizing the adverse effects of early memory T cell activation in response to allografts is to inhibit their infiltration into the graft. LFA-1 is expressed on both T and B cells as well as on neutrophils and macrophages and its expression is upregulated on effector and memory T cells (39–43). In the current study we tested the ability of a short-course of peri-transplant anti-LFA-1 mAb to inhibit the early infiltration and resulting inflammation mediated by memory CD8 T cells. The effects of this treatment on donor-specific effector T cell priming and on the induction of donor-specific antibody responses were also investigated. The results indicate that the short course treatment with anti-LFA-1 mAb is extremely effective in inhibiting the early allograft infiltration by memory CD8 T cells. In addition the treatment inhibits the priming of donor-reactive effector T cells for several weeks resulting in prolonged allograft survival. The treatment, however, was ineffective at inhibiting the induction of donor-specific antibody or its effects in a model of antibody-mediated graft rejection. The results indicate the efficacy of a short course of anti-LFA-1 mAb in attenuating early memory CD8 T cell mediated inflammatory events in allografts and in promoting allograft survival.
Materials and Methods
Mice
Colonies of C57BL/6 (H-2b) and A/J (H-2a) mice were purchased from Charles River Laboratories (Wilmington, MA). B6.2C/RAG-1−/−, B6.CCR5−/− and B6.CCR5−/−/B6, μMT−/− mice are maintained at our facility. Male mice, 8–10 weeks of age, were used in all experiments and all procedures involving animals were approved by the Institutional Animal Care and Use Committee at the Cleveland Clinic.
Cardiac transplantation and harvest
Heterotopic, intra-abdominal cardiac transplantation was performed following the method of Corry and coworkers (44). Total operative times averaged 45 min and hearts resumed spontaneous contraction immediately upon reperfusion. Graft survival was monitored by abdominal palpation of the graft and rejection was confirmed by laparotomy. At the time of graft harvest, the circulatory system was drained prior to removal of the heterotopic graft, which was immediately snap-frozen in liquid nitrogen or placed in media for digestion and purification of graft-infiltrating cells.
In vivo antibody treatments
Animals were treated on days −1 and 0 or on days 3 and 4 with 0.2 mg i.p. anti-LFA-1 mAb (rat IgG2b, clone FD441.8 from Bio X Cell, West Lebanon, NH) or control rat IgG (Sigma-Aldrich, St. Louis, MO). The dose of FD441.8 used was based on results from a limited preliminary allograft survival study.
Flow cytometry
Flow cytometric detection of graft-infiltrating cells was performed using a modification of the method published by Afanasyev and colleagues (45). Briefly, harvested tissues were weighed and then incubated for 1 h at 37°C in RPMI with Type II collagenase (Sigma-Aldrich). After incubation, tissue was pressed through a 40 μm filter and the collected cells were washed twice in RPMI, counted using a hemocytometer, and stained for common phenotypic surface markers (CD45, CD4, CD8, Gr1) using commercially available antibodies (BD Bioscience, San Jose, CA; eBioscience, San Diego, CA). Flow cytometry was performed using a FACSCalibur (BD Biosciences) cytometer and FlowJo analysis software (Tree Star Inc., Ashland OR). The forward scatter and FL1 (CD45+) channels were used to gate the leukocytes in the graft tissue followed by analysis of the specific leukocyte populations. For each sample, 200,000 events were accumulated. Total numbers of each leukocyte population were calculated by: (the total number of leukocytes in the sample counted using the hemocytometer) × (% of the leukocyte population in the CD45+ cells by flow cytometry)/100. The data are reported as number of each leukocyte population/mg graft tissue.
For analysis of donor-reactive CD8 T cell proliferation in recipient spleens, H-2Ld reactive CD8 T cells from 2C.RAG-1−/− mice were labeled with CFSE and aliquots of 106 cells were transferred to C57BL/6 mice 3 days before cardiac transplantation with or without treatment with anti-LFA-1 mAb. On day 7 post-transplant, aliquots of recipient spleen cells were stained with clonotypic antibody 1B2 and anti-CD8 mAb and CFSE staining in the gated double positive population was analyzed by histogram.
RNA purification and qRT-PCR
Snap-frozen grafts were crushed, homogenized, and RNA was isolated using fibrous tissue kits (Qiagen, Valencia, CA). Reverse transcription and real-time PCR were performed using commercially available reagents, probes, and a 7500 fast real-time thermocycler, all from Applied Biosystems (Foster City, CA).
ELISPOT
Donor-reactive T cells producing IL-2 and IFN-γ in the spleens of cardiac graft recipients were enumerated by ELISPOT assay as previously detailed (35, 36). Briefly, splenic responder cells and mitomycin C-treated self, donor and third-party stimulator cell populations were cocultured for 24 h at 37°C in serum-free HL-1 media in 96 well plates coated with anti-IFN-γ or anti-IL-2 capture mAb. To compare alloreactive CD4+ and CD8+ T cell priming, each cell population was purified from recipient spleen cell suspensions using positive selection columns (R&D Systems, Minneapolis, MN) and then these purified responder cells were stimulated with T cell depleted splenocytes from the graft donor or recipient strain. After culture, all cells were washed from the plate and biotinylated anti-IFN-γ or anti-IL-2 detecting mAb was added, followed by anti-biotin alkaline phosphatase. After development with the chromagen, the total number of spots per well was quantified using an ImmunoSpot Series 2 Analyzer (Cellular Technology Ltd., Shaker Heights, OH).
Immunohistochemistry
A midventricular portion of the cardiac graft was embedded in OCT compound (Sakura Finetek USA) and immediately frozen in liquid nitrogen after harvest, and 6 μm thick sections were prepared as previously described. Slides were stained with 10 μg/mL anti-CD8 mAb (53–6.7) or anti-Ly-6G mAb (RB6–8C5) in PBS with 1% BSA for 1 h at room temperature and then with biotinylated rat anti-rat IgG (Dako, Carpinteria, CA) diluted 1:100 in PBS with 1% BSA for 20 minutes at room temperature. The slides were developed with DAB for color change and counterstained with hematoxylin. Sections were washed with PBS followed by the addition of Vectashield to preserve fluorescence (Vector Laboratories). Images were captured and analyzed with Image-Pro Plus (Media Cybernetics, Silver Springs, MD). C4d staining was performed on methanol fixed tissue using polyclonal rabbit antibody to C4d and a secondary peroxidase-conjugated goat anti-rabbit antibody (Jackson ImmunoResearch Laboratories, West Grove, PA) (46).
Statistics
Graft survival between experimental groups was compared using Kaplan-Meier survival curves and Log-rank statistics. For other experiments statistical analyses were performed using the Mann–Whitney nonparametric test to analyze differences between experimental groups. A p value < 0.05 was considered significant. Error bars reflect Standard Error from the Mean for each group.
Results
Antagonism of LFA-1 decreases early inflammatory events in cardiac allografts
Previous studies from this laboratory have indicated intense infiltration of neutrophils and donor-reactive memory CD8 T cells into cardiac allografts within 24–48 hours following graft reperfusion and the activation of these leukocytes to express inflammatory functions in the graft (36, 47, 48). The requirement for LFA-1 engagement for the infiltration of these leukocytes into the allografts was tested by treating groups of C57BL/6 mice receiving complete MHC-mismatched A/J cardiac allografts or isografts with or without anti-LFA-1 mAb on the day before and the day of transplant. The grafts were retrieved 48 hours after reperfusion and the number of graft infiltrating CD8 T cells, neutrophils and macrophages was determined (Figure 1). As previously observed, infiltration of CD8 T cells into isografts at 48 hrs post-transplant was near background/naïve levels whereas marked infiltration of the CD8 T cells into allografts was observed. Treatment with anti-LFA-1 mAb decreased CD8 T cell infiltration into allografts to the low-absent levels observed in native hearts from naïve mice. Levels of neutrophil infiltration into allografts was 2–3 fold higher than into isografts and this infiltration into both iso- and allo-grafts was decreased by peri-transplant treatment with anti-LFA-1 mAb but was not reduced to the levels in native hearts from naïve animals. Anti-LFA-1 mAb had no effect on macrophage infiltration into isografts and only decreased macrophage infiltration into the allografts by about half.
Figure 1. Anti-LFA-1 mAb inhibits memory CD8+ T cell infiltration into cardiac allografts.
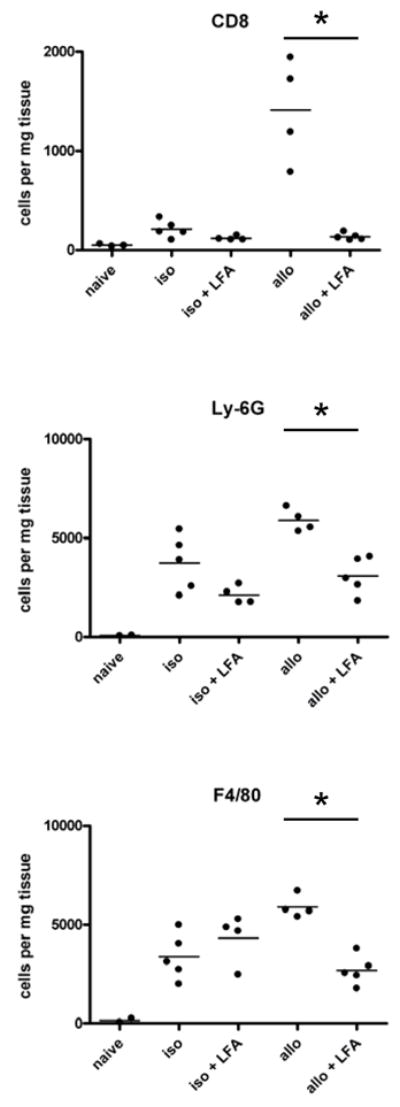
Groups of C57BL/6 mice (3–5/per group) were treated with or without 200 μg anti-LFA-1 mAb on days −1 and 0 and then received syngeneic or complete MHC mismatched A/J cardiac allografts on day 0. Grafts were harvested 48 h post-transplant, digested, and graft-infiltrating cells were analyzed by antibody staining and flow cytometry to determine the numbers of graft-infiltrating CD8 T cells, neutrophils and macrophages. *p ≤ 0.05.
Cardiac iso- and allo-graft levels of acute phase cytokines (TNFα, IL-1β, and IL-6) and neutrophil (CXCL1/KC) and macrophage (CCL2/MCP-1) chemoattractants were not significantly decreased in the grafts from anti-LFA-1 mAb vs. untreated recipients (Figure 2). In contrast, anti-LFA-1 mAb treatment decreased expression of genes associated with memory CD8 T cell infiltration and activation in the allografts (IFN-γ, CXCL9/Mig, CXCL10/IP-10, and ICOS) to the levels observed in native hearts from naïve mice.
Figure 2. Intragraft cytokine and chemokine mRNA expression in cardiac allografts from untreated vs. anti-LFA-1 mAb treated recipients.

Groups of C57BL/6 mice (3–5/per group) were treated with or without 200 μg anti-LFA-1 mAb on days −1 and 0 and then received syngeneic or complete MHC mismatched A/J cardiac allografts on day 0. Grafts were harvested 48 h post-transplant, whole cell RNA was isolated from graft homogenates, and quantitative real-time PCR was used to measure expression levels of mRNA encoding the indicated cytokines and chemokines. *p ≤ 0.05; n.s. not significantly different.
Antagonism of LFA-1 prolongs cardiac allograft survival
Recipient treatment with anti-LFA-1 mAb on the day before and the day of transplantation resulted in a marked prolongation of allograft survival when compared to control rat IgG treated recipients, from a median survival of 7.5 days to 27 days (Figure 3). This prolongation was accompanied by an almost complete absence in CD8 T cell and decreased neutrophil infiltration into the allografts when examined at day 7 post-transplant, the time when most of the allografts were rejected in the control IgG-treated recipients (Figure 4). However, CD8 T cell and neutrophil infiltration into allografts of anti-LFA-1 mAb treated recipients was apparent by day 21 post-transplant at similar levels to those observed during allograft rejection in the control IgG-treated recipients.
Figure 3. Short term, peri-transplant treatment with anti-LFA-1 mAb prolongs cardiac allograft survival.

Groups of C57BL/6 mice (4–5/per group) were treated with 200 μg control IgG or with anti-LFA-1 mAb either on days −1 and 0 or on days 3 and 4. The treated mice received syngeneic or complete MHC mismatched A/J cardiac allografts on day 0 and graft survival was followed daily by abdominal palpation and rejection confirmed by laparotomy. *p < 0.05 vs. allograft survival in all other groups.
Figure 4. Histological evaluation of cardiac allografts from control Ig and anti-LFA-1 mAb treated recipients.

Cardiac allografts were harvested at the indicated times post-transplant from recipients treated with control rat IgG or with anti-LFA-1 mAb on days −1 and 0. Prepared sections were stained with hematoxylin-eosin or with CD8- and Ly–6G-specific antibodies for immunohistochemical analyses. Sections shown are representative of 3–4 grafts analyzed in each group. Magnification, 200x.
Anti-LFA-1 mAb treatment decreases donor-reactive T cell priming
Since LFA-1/ICAM-1 interactions are important during T cell activation, the priming of donor-reactive T cells in anti-LFA-1 mAb treated allograft recipients was also tested as a potential mechanism of the prolonged allograft survival. First, unseparated spleen cells were tested for numbers of donor-reactive cells producing IFN-γ by ELISPOT assay at several different time points post-transplant in untreated and anti-LFA-1 mAb treated cardiac allograft recipients. Whereas high numbers of donor-reactive cells producing IFN-γ were observed in the spleens of control recipients on days 7 and 14 post-transplant, these cells were undetectable at these time points in the anti-LFA-1 mAb treated recipients and were only detectable at low numbers on day 21 post-transplant (Figure 5). Similar results were observed when CD4 and CD8 T cells were purified from recipient spleen cell suspensions and tested for numbers of donor-reactive cells producing IFN-γ (Figure 5).
Figure 5. Peritransplant anti-LFA-1 mAb inhibits donor-reactive CD8+ and CD4+ T cell development to IFN-γ producing cells.

C57BL/6 mice were treated with or without 200 μg anti-LFA-1 mAb on days −1 and 0 and then received complete MHC mismatched A/J cardiac allografts on day 0. On the indicated day post-transplant, aliquots of unseparated spleen cells, enriched CD4+ or enriched CD8+ T cells from the recipients were cultured overnight with syngeneic or donor stimulator cells on anti-IFN-γ mAb coated filters. Numbers of donor-reactive T cells producing IFN-γ were determined by ELISPOT. Numbers of spots in co-cultures with syngeneic stimulators were always < 3 spots/5 × 105 stimulator cells. The data shown are representative of two independent experiments.
Delayed treatment with anti-LFA-1 mAb does not prevent endogenous memory CD8 T cell infiltration into allografts
The results to this point indicated inhibitory effects of anti-LFA-1 mAb on the infiltration of memory CD8 T cells into MHC-mismatched cardiac allografts as well as on the priming of donor-reactive CD4 and CD8 T cells, both, which are likely to contribute to the prolonged allograft survival observed. To attempt to distinguish the impact of LFA-1 antagonism on the infiltration of memory CD8 T cells and development of donor-specific T cells to IFN-γ producing cells in the recipient spleen, cardiac allograft recipients were treated with anti-LFA-1 mAb on days 3 and 4 post-transplant, times when the priming of the donor-reactive T cells in the recipient spleen has presumably been initiated. Treatment with anti-LFA-1 mAb delayed rejection to 15 days but was not as effective as treatment at day −1 and 0 (Figure 3). Consistent with this prolonged allograft survival, the delay of anti-LFA-1 mAb treatment until day 3 and 4 post-transplant still inhibited the development of donor-reactive CD4 and CD8 T cells to IFN-γ producing cells when tested on day 7 post-transplant (Figure 6). In contrast to the inhibition of donor-reactive T cell development in recipients given anti-LFA-1 mAb on days −1 and 0, however, these T cells were apparent at near the levels observed in untreated recipients by day 14 post-transplant (Figure 6). Examination of allografts harvested from recipients treated with anti-LFA-1 mAb on days 3 and 4 post-transplant indicated intense CD8 T cells and neutrophil infiltration into the allografts at day 7 post-transplant (data not shown).
Figure 6. Treatment with anti-LFA-1 mAb on days 3 and 4 post-transplant inhibits donor-reactive CD8+ and CD4+ T cell development to IFN-γ producing cells.

C57BL/6 mice were treated with or without 200 μg anti-LFA-1 mAb on days 3 and 4 and then received complete MHC mismatched A/J cardiac allografts on day 0. (A) On days 7 and 14 post-transplant, aliquots of unseparated spleen cells, enriched CD4+ or enriched CD8+ T cells from the recipients were cultured overnight with syngeneic or donor stimulator cells on anti-IFN-γ mAb coated filters. Numbers of donor-reactive T cells producing IFN-γ were determined by ELISPOT. Numbers of spots in co-cultures with syngeneic stimulators were always < 3 spots/5 × 105 stimulator cells. The data shown are representative of two independent experiments. (B) On day 7 post-transplant cardiac allografts were harvested from recipients treated with anti-LFA-1 mAb on days 3 and 4 and prepared sections were stained with hematoxylin-eosin or with CD8-specific antibodies for immunohistochemical analyses. Sections shown are representative of 3–4 grafts analyzed in each group. Magnification, 200x.
To further investigate effects of anti-LFA-1 mAb given on days 0 and −1 vs. on days 3 and 4 on the priming and functional development of donor-reactive T cell priming, two approaches were taken. First, the induction of donor-specific T cells producing IL-2 on day 7 post-transplant was tested using ELISPOT analyses (Figure 7). Cardiac allograft recipients treated with anti-LFA-1 mAb on days −1 and 0 had low levels of T cells producing IL-2 in cultures stimulated with allograft donor and third-party allogeneic stimulators that were similar to those observed in spleens from naïve animals. In contrast, recipients treated with anti-LFA-1 mAb on days 3 and 4 had similar numbers of donor-reactive T cells producing IL-2 as control IgG treated recipients. As a second approach, transgenic A/J-reactive 2C CD8 T cells were labeled with CFSE and transferred to C57BL/6 mice that then received A/J cardiac allografts two days later. The recipients were treated with control IgG or anti-LFA-1 mAb on days −1 and 0 or with anti-LFA-1 mAb on days 3 and 4 post-transplant. On day 7 post-transplant the proliferation of the 2C cells was assessed (Figure 8). Robust proliferation of the 2C cells was observed in allograft recipients that were treated with control IgG and this proliferation was absent in recipients treated with anti-LFA-1 mAb on days −1 and 0 as well as in recipients of isografts. In contrast, proliferation of the 2C cells in allograft recipients treated with anti-LFA-1 mAb on days 3 and 4 post-transplant was similar to that observed in the control IgG -treated allograft recipient. Overall, the results indicated that anti-LFA-1 mAb given at the time of transplant inhibited the development of donor-reactive T cells to IL-2 and to IFN-γ producing cells whereas delay of anti-LFA-1 mAb treatment to days 3 and 4 post-transplant did not affect donor-reactive T cell priming to IL-2 producing cells but did delay further development to IFN-γ producing cells.
Figure 7. Peri-transplant anti-LFA-1 mAb but not treatment on days 3 and 4 inhibits donor-reactive T cell priming to IL-2 producing cells.

C57BL/6 mice were treated with 200 μg control IgG or with anti-LFA-1 mAb either on days −1 and 0 or on days 3 and 4. The treated mice received syngeneic or complete MHC mismatched A/J cardiac allografts on day 0. On day 7, aliquots of splenic T cells from the recipients were cultured overnight with syngeneic, A/J donor, or third-party DBA/1 stimulator cells on anti-IL-2 mAb coated filters. Numbers of T cells producing IL-2 were determined by ELISPOT. The data shown are representative of two independent experiments.
Figure 8. Peri-transplant but not delayed anti-LFA-1 mAb inhibits the proliferation of donor-reactive CD8 T cells in the recipient spleen.

Ld-reactive 2C TCR transgenic CD8 T cells were labeled with CFSE and 2 × 106 aliquots were transferred into groups of wild-type C57BL/6. Two day after the 2C cell transfer, the C57BL/6 mice received syngeneic or Ld-expressing A/J cardiac allografts. The allograft recipients were treated with 200 μg control rat IgG or anti-LFA-1 mAb either on days −1 and 0 or on days 3 and 4. On day 7 post-transplant recipient spleen cell suspensions were prepared, stained with anti-CD8 and 1B2 mAb, and analyzed by flow cytometry. The gated CD8+1B2+ 2C cells were analyzed for expression of CFSE in the FL1 channel to assess proliferation of the donor-reactive T cells. The data shown are representative of two independent experiments.
LFA-1 antagonism does not inhibit donor-reactive antibody production
The ability of a short course of anti-LFA-1 mAb given at the time of transplant to inhibit early inflammatory events and donor-reactive T cell priming led us to ask if such treatment would also be effective in inhibiting the induction of donor-reactive antibody responses. Previous studies from this lab have demonstrated the dysregulated antibody responses produced in B6.CCR5−/− recipients of MHC-mismatched cardiac and renal allografts and the resulting antibody-mediated rejection of the allografts (5, 49). Therefore, groups of B6.CCR5−/− mice were treated with control IgG or anti-LFA-1 mAb on days −1 and 0 and received MHC-mismatched cardiac allografts. Unlike the marked delay of T cell mediated-rejection observed in wild-type recipients, treatment of B6.CCR5−/− allograft recipients with anti-LFA-1 mAb only extended graft survival 3–4 days (Figure 9A). Treatment with anti-LFA-1 mAb did not diminish the titers of donor-specific antibody induced in the CCR5-deficient allograft recipients (data not shown) using a previously reported flow based assay to determine serum donor-reactive antibody titers (5). Histological examination of rejecting allografts indicated no decrease in cell infiltration into the allografts in CCR5−/− recipients treated with anti-LFA-1 mAb on days −1 and 0 post-transplant and no decrease in C4d deposition in the rejected allografts (Figure 9B).
Figure 9. Anti-LFA-1 monotherapy does not induce long-term allograft survival in CCR5−/− recipients.

Groups of 4–5 B6.CCR5−/− or B6.μMT−/−/CCR5−/− mice were treated with 200 μg control rat IgG or with anti-LFA-1 mAb on days −1 and 0. (A) The treated mice received complete MHC mismatched A/J cardiac allografts on day 0 and graft survival was followed daily by abdominal palpation and rejection confirmed by laparotomy. p < 0.05 vs. survival in control IgG CCR5−/− and vs. untreated μMT−/−/CCR5−/− recipients. (B) Prepared sections were stained with hematoxylin-eosin or with C4d-specific antibodies for immunohistochemical analyses. Sections shown are representative of 3–4 grafts analyzed in each group. Magnification, 200x.
To directly test the effects of LFA-1 antagonism on antibody- vs. T cell-mediated rejection in CCR5−/− recipients, groups of B6.μMT−/−/CCR5−/− recipients of the MHC-mismatched cardiac allografts were treated with or without anti-LFA-1 mAb on days −1 and 0 post-transplant. In contrast to the absence of an effect on allograft survival in B6.CCR5−/− recipients treated with anti-LFA-1 mAb, treatment of recipients that cannot make antibody (B6.μMT−/−/CCR5−/− mice) resulted in a marked extension of cardiac allograft survival (Figure 9A).
Discussion
Early models of immune responses to allografts proposed that acute rejection was mediated by donor-reactive T cells primed from naïve precursor cells by donor- and recipient-derived antigen-presenting cells (50). Following development to effector cells, the T cells were recruited to the allograft and activated by the donor alloantigens to express the effector functions mediating graft tissue injury and rejection. It is now appreciated that memory T cells generated in response to viruses, bacteria, and lymphopenic environments can be reactive with allogeneic class I and class II MHC molecules and that such memory T cells have a detrimental effect on the outcome of an allograft (25–27, 30, 51). In clinical studies, the pre-transplant presence of donor-reactive T cells producing IFN-γ predicts poor renal allograft outcome (28, 29). In support of the detrimental effect of this heterologous immunity on allograft survival, the activities of effector memory CD8 T cells generated in response to virus infection accelerate rejection of skin and cardiac allografts in rodent models (30, 33). Recent studies from this laboratory have demonstrated the infiltration of endogenous CD8 T cells with an effector memory phenotype into MHC-mismatched cardiac allografts within 24 hours of the transplant in mice (35, 36). The infiltrating memory CD8 T cells are activated by graft class I MHC molecules to proliferate and produce IFN-γ. This endogenous memory CD8 T cell induced inflammation plays a role in acute rejection by promoting the recruitment of effector T cells primed from naïve precursors. Thus, mice housed in cages with environmentally restricted exposure may have fewer endogenous effector CD8 memory T cells than adult humans but these T cells are clearly present in housed mice and are a factor in the rejection of solid organ allografts. An important goal of this and many laboratories has been to develop strategies to neutralize the infiltration and/or activities of these effector CD8 memory T cells.
Since memory T cells express high levels of LFA-1 (41, 43), we tested the efficacy of a short-course of peri-transplant monotherapy with anti-LFA-1 mAb in disrupting early post-transplant activity of endogenous memory CD8 T cells in response to MHC-mismatched cardiac grafts. This strategy was found to be extremely effective in inhibiting the infiltration of these memory CD8 T cells into the cardiac allografts and was accompanied by the absence of T cell-derived cytokine production in the allografts. One of the inflammatory mechanisms mediated by IFN-γ is the increased infiltration and activation of neutrophils into the allografts vs. that observed in isografts (48). In contrast to the almost complete inhibition of CD8 T cell infiltration into the cardiac allografts, peri-transplant treatment with anti-LFA-1 mAb decreased neutrophil infiltration to levels observed in the isografts. This is consistent with our previous results that neutrophils are initially recruited into cardiac iso- and allo-grafts through innate immune mechanisms that are independent of the memory CD8 T cells and that the activation of the CD8 T cells amplifies the infiltration and activation of neutrophils to mediate tissue injury within the allograft (48, 52).
In addition to abrogating the infiltration of the memory CD8 T cells into the allografts, the short course of anti-LFA-1 mAb treatment was also very effective in inhibiting the priming of donor-reactive CD4 and CD8 T cells in peripheral lymphoid organs. Studies from many laboratories have reported the inhibition of antigen-specific CD4 or CD8 T cell priming by anti-LFA-1 antibodies (10, 12, 15, 16). When given the day before and the day of cardiac allograft transplantation, the anti-LFA-1 mAb inhibited priming of donor-reactive CD4 and CD8 T cells to IL-2- and to IFN-γ-producing cells until about day 21 post-transplant and was associated with the absence of cell infiltration into the allografts until that time when the grafts began to experience rejection. These results suggest that effector T cell priming from naïve donor-reactive T cell precursors is inhibited until the time near the observation of rejection and that the inhibitory effect of the anti-LFA-1 mAb is not just restricted to the inhibited development of the T cells to IFN-γ producing cells. Interestingly, waiting until day 3 and 4 post-transplant to initiate administration of the anti-LFA-1 mAb also inhibited the appearance of donor-reactive CD4 and CD8 T cells producing IFN-γ but did not inhibit donor-reactive T cell priming to IL-2 producing cells when assessed at day 7 post-transplant. Furthermore, proliferation of donor-reactive CD8 T cells in the allograft recipient spleen was almost completely absent in the spleens of allograft recipients treated with anti-LFA-1 mAb at the time of transplant whereas the delayed treatment with anti-LFA-1 mAb did not inhibit the proliferation of these T cells. These results are consistent with recent studies indicating that the initial activation of antigen-reactive T cells with antigen-presenting dendritic cells results in an initial activation of the T cells to produce IL-2 and undergo clonal expansion and that subsequent interactions with the antigen-presenting dendritic cells is required for development of the activated T cells to particular functional phenotypes such as IFN-γ or IL-17 production (53–56). The implication of these results is that the use of anti-LFA-1 mAb may limit or arrest the functional development of T cells during an ongoing alloimmune response as well as inhibiting infiltration of primed effector T cells into the allograft.
Recent results from this lab have shown the early infiltration of memory T cells into the allografts and their rapid activation to proliferate and to produce IFN-γ (35, 36). ICOS interactions are required for activation of the memory CD8 T cells to express effector function within the allograft but not to infiltrate the graft and undergo proliferation. It is worth noting the presence at day 7 post-transplant of many CD8 T cells in the allografts of recipients treated with anti-LFA-1 mAb on days 3 and 4. In addition, the levels of IFN-γ mRNA in the allografts from recipients treated with the delayed anti-LFA-1 mAb are similar to those observed in allografts from control treated recipients on day 7 post-transplant indicating the activation of the infiltrating CD8 T cells (data not shown). Since this proliferation occurs within the first 1–3 days post-transplant, it is likely that the majority of CD8 T cells in the allograft at day 7 in these anti-LFA-1 mAb treated recipients are derived from these endogenous memory CD8 T cells. These results suggest that the anti-LFA-1 mAb may have no obvious effect on the presence or activity of the memory CD8 T cells once they infiltrate the allograft and begin to proliferate within the graft.
Peri-transplant treatment with anti-LFA-1 mAb was not effective in inhibiting anti-donor antibody responses and allograft rejection in CCR5−/− recipients. LFA-1 expression on B-lymphocytes has been reported and in vitro studies have indicated the ability of anti-LFA-1 antibodies to inhibit B cell activation to produce antibody through direct and indirect means (39, 40, 42, 57). Furthermore, studies have indicated the ability of anti-LFA-1 mAb given with additional immunosuppressive agents to inhibit the generation of antibody responses in vivo including in response to xeno- and allo-grafts (19, 57, 58). The current studies tested the effects of the anti-LFA-1 mAb in a model of antibody-mediated rejection in CCR5-deficient recipients where rapid rejection of the allograft is dependent on the anti-donor antibody produced (5, 49). Although the antibody response to allografts is dysregulated in CCR5−/− recipients, the generation of the antibody response in these recipients is dependent on CD4 T helper cells. Similar to the efficacy in treated wild-type allograft recipients, peri-transplant anti-LFA-1 mAb did prolong allograft survival in μMT−/−/CCR5−/− recipients that are unable to produce antibodies. These results indicate the ability of peri-transplant treatment with anti-LFA-1 mAb to inhibit the induction of cell- but not antibody-mediated allograft rejection. The results further suggest that LFA-1 function may not be as critical for the activation and/or function of T follicular helper cells and further studies are warranted to directly test this. In addition, the results indicate that the use of anti-LFA-1 antibody therapy may not be an effective immunosuppressive strategy to prevent anti-donor antibody responses in sensitized patients with high panel reactive antibody (PRA).
The results of this study have demonstrated that the consequences of a short course of anti-LFA-1 mAb given at the time of the cardiac transplant are a marked inhibition of endogenous memory CD8 T cell infiltration into the allograft, inhibition of donor-reactive T cell priming from naïve precursors for approximately three weeks, and a marked prolongation in allograft survival. Clinical studies have demonstrated the efficacy of a humanized IgG1 anti-LFA-1 mAb, efalizumab, for treatment of psoriasis (59, 60). A low dose of efalizumab in conjunction with a steroid-free immunosuppressive strategy was recently found to be effective in promoting long-term survival of clinical islet grafts and insulin independence of the treated patients (61). This antibody was also recently tested in a phase I/II clinical trial in 38 renal transplant patients where the antibody was given weekly at either a high (2 mg/kg) or low (0.5 mg/kg) dose for 12 weeks post-transplant with standard triple immunosuppression and was found to be effective in prolonging survival of the grafts (62). However, in the high dose anti-LFA-1 mAb treatment arm of the renal transplant patients who were also treated with a full dose of cyclosporine A, 3 of 9 patients developed post-transplant lymphoproliferative disease whereas there was no occurrence of this disease in the low dose efalizumab or in lower dose cyclosporine A treatment arms. These results indicate an increased chance of PTLD occurrence when anti-LFA-1 mAb is used in conjunction with high levels of standard immunosuppression. In spite of its clinical effectiveness, the occurrence of progressive multifocal myeloencephalopathy (PML) in 4 of more than 40,000 psoriatic patients treated with efalizumab prompted removal of the antibody for clinical use in 2009. The results of the current study indicate the effectiveness of short, peri-transplant administration of anti-LFA-1 mAb in promoting graft survival and suggest that anti-LFA-1 mAb may not have to be given on a chronic basis to yield beneficial effects in preventing the adverse effects of endogenous memory T cells and donor-reactive T cell priming in transplant recipients.
Acknowledgments
We thank the staff of the Cleveland Clinic Biological Resources Unit for excellent care of the animals used in this study. The authors thank Drs. Anna Valujskikh, and Flavio Vincenti for their helpful comments during the course of this work and preparation of the manuscript.
This work was supported by grants from the NIH (RO1 AI40459 and RO1 AI74740) and from the Roche Organ Transplant Research Foundation (60495086) to RLF.
Footnotes
Disclosures
The authors have no financial conflict of interest to declare.
References
- 1.Lechler RI, Sykes M, Thomson AW, Turka LA. Organ transplantation-how much of the promise has been realized? Nat Med. 2005;11:605–613. doi: 10.1038/nm1251. [DOI] [PubMed] [Google Scholar]
- 2.El-Sawy T, Fahmy NM, Fairchild RL. Chemokines: directing leukocyte infitration into allografts. Curr Opin Immunol. 2002;14:562–568. doi: 10.1016/s0952-7915(02)00382-5. [DOI] [PubMed] [Google Scholar]
- 3.Hancock WW. Chemokine receptor-dependent alloresponses. Immunol Rev. 2003;196:37–50. doi: 10.1046/j.1600-065x.2003.00084.x. [DOI] [PubMed] [Google Scholar]
- 4.Sayegh MH, Remuzzi G. Clinical update: immunosuppression minimization. Lancet. 2007;369:1676–1678. doi: 10.1016/S0140-6736(07)60762-4. [DOI] [PubMed] [Google Scholar]
- 5.Amano H, Bickerstaff A, Orosz CG, Novick AC, Toma H, Fairchild RL. Absence of recipient CCR5 promotes early and increased allospecific antibody responses to cardiac allografts. J Immunol. 2005;174:6499–6508. doi: 10.4049/jimmunol.174.10.6499. [DOI] [PubMed] [Google Scholar]
- 6.Kwun J, Hazinedaroglu SM, Schadde E, Kayaoglu HA, Fechner J, Hu HZ, et al. Unaltered graft survival and intragraft lymphocytes infiltration in the cardiac allograft of CxcR3−/− mouse recipients. Am J Transplant. 2008;8:1593–1603. doi: 10.1111/j.1600-6143.2008.02250.x. [DOI] [PubMed] [Google Scholar]
- 7.Rosenblum JM, Zhang QW, Siu G, Collins TL, Sullivan T, Dairaghi DJ, et al. CXCR3 antagonism impairs the development of donor-reactive, IFN-gamma-producing effectors and prolongs allograft survival. Transplantation. 2009;87:360–369. doi: 10.1097/TP.0b013e31819574e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zerwes H-G, Li, Kovarik J, Streiff M, Hofmann M, Roth L, et al. The chemokine receptor Cxcr3 is not essential for acute cardiac allograft rejection in mice and rats. Am J Transplant. 2008;8:1604–1613. doi: 10.1111/j.1600-6143.2008.02309.x. [DOI] [PubMed] [Google Scholar]
- 9.Nicolls MR, Gill RG. LFA-1 (CD11a) as a therapeutic target. Am J Transplant. 2006;6:27–36. doi: 10.1111/j.1600-6143.2005.01158.x. [DOI] [PubMed] [Google Scholar]
- 10.Chou YK, Edwards DM, Weinberg AD, Vandenbark AA, Kotzin BL, Fontenot AP, et al. Activation pathways implicate anti-HLA-DP and anti-LFA-1 antibodies as lead candidates for intervention in chronic berylliosis. J Immunol. 2005;174:4316–4324. doi: 10.4049/jimmunol.174.7.4316. [DOI] [PubMed] [Google Scholar]
- 11.Freiberg BA, Kupfer H, Maslanik W, Delli H, Kappler J, Zaller DM, et al. Staging and resetting T cell activation in SMACs. Nat Immunol. 2002;3:911–917. doi: 10.1038/ni836. [DOI] [PubMed] [Google Scholar]
- 12.Gorochov G, Gross G, Waks T, Eshhar Z. Anti-leukocyte function-associated antigen-1 antibodies inhibit T-cell activation following low-avidity and adhesion-independent interactions. Immunology. 1993;79:548–555. [PMC free article] [PubMed] [Google Scholar]
- 13.Grakoui A, Bromley SK, Sumen C, Davis MM, Shaw AS, Allen PM, et al. The immunological synapse: a molecular machine controlling T cell activation. Science. 2002;285:221–227. doi: 10.1126/science.285.5425.221. [DOI] [PubMed] [Google Scholar]
- 14.Lee K-H, Holdorf AD, Dustin ML, Chan AC, Allen PM, Shaw AS. T cell receptor signaling precedes immunological synapse formation. Science. 2002;295:1539–1542. doi: 10.1126/science.1067710. [DOI] [PubMed] [Google Scholar]
- 15.Rutigliano JA, Johnson TR, Hollinger TN, Fischer JE, Aung S, Graham BS. Treatment with anti-LFA-1 delays the CD8+ cytotoxic -T-lymphocyte response and viral clearance in mice with primary respiratory syncytial virus infection. J Virol. 2004;78:3014–3023. doi: 10.1128/JVI.78.6.3014-3023.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Springer TA, Dustin ML, Kishimoto TK, Marlin SD. The lympyhocyte function-associated LFA-1, CD2, and LFA-3 molecules: cell adhesion receptors of the immune system. Annu Rev Immunol. 1987;107:223–252. doi: 10.1146/annurev.iy.05.040187.001255. [DOI] [PubMed] [Google Scholar]
- 17.Isobe M, Yagita H, Okumura K, Ihara A. Specific acceptance of cardiac allograft after treatment with antibodies to ICAM-1 and LFA-1. Science. 1992;255:1125–1129. doi: 10.1126/science.1347662. [DOI] [PubMed] [Google Scholar]
- 18.Lunsford KE, Koester MA, Eiring AM, Horne PH, Gao D, Bumgartner GL. Targeting LFA-1 and CD154 suppresses the in vivo activation and development of cytolytic (CD4-independent) CD8+ T cells. J Immunol. 2005;175:7855–7866. doi: 10.4049/jimmunol.175.12.7855. [DOI] [PubMed] [Google Scholar]
- 19.Metzler B, Gfeller P, Bigaud M, Li J, Wiezorek G, Heusser C, et al. Combinations of anti-LFA-1, everolimus, anti-CD40 ligand, and allogeneic bone marrow induce central transplantation tolerance through hemopoietic chimerism, including protection from chronic heart allograft rejection. J Immunol. 2004;173:7025–7036. doi: 10.4049/jimmunol.173.11.7025. [DOI] [PubMed] [Google Scholar]
- 20.Murakawa T, Kerklo MM, Zamora MR, Wei Y, Gill RG, Henson PM, et al. Simultaneous LFA-1 and CD40 ligand antagonism prevents airway remodeling in orthotopic airway transplantation: implications for the role of respiratory epithelium as a modulator of fibrosis. J Immunol. 2005;174:3869–3879. doi: 10.4049/jimmunol.174.7.3869. [DOI] [PubMed] [Google Scholar]
- 21.Nicolls MR, Coulombe M, Beilke J, Gelhaus HC, Gill RG. CD4-dependent generation of dominant transplantation tolerance induced by simultaneous perturbation of CD154 and LFA-1 pathways. J Immunol. 2002;169:4831–4839. doi: 10.4049/jimmunol.169.9.4831. [DOI] [PubMed] [Google Scholar]
- 22.NIcolls MR, Coulombe M, Yang H, Bolwerk A, Gill RG. Anti-LFA-1 therapy induces long-term islet allograft acceptance in the absence of IFN-g or IL-4. J Immunol. 2000;164:3627–3634. doi: 10.4049/jimmunol.164.7.3627. [DOI] [PubMed] [Google Scholar]
- 23.Page AJ, Ford ML, Kirk AD. Memory T-cell-specific therapeutics in organ transplantation. Curr Opin Organ Transplant. 2009;14:643–649. doi: 10.1097/MOT.0b013e328332bd4a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Valujskikh A. Targeting T-cell memory: where do we stand? Curr Opin Organ Transplant. 2008;13:344–349. doi: 10.1097/MOT.0b013e3283061126. [DOI] [PubMed] [Google Scholar]
- 25.Adams AB, Pearson TC, Larsen CP. Heterologous immunity: an overlooked barrier to tolerance. Immunol Rev. 2003;196:147–160. doi: 10.1046/j.1600-065x.2003.00082.x. [DOI] [PubMed] [Google Scholar]
- 26.Brehm MA, Markees TG, Daniels KA, Greiner DL, Rossini AA, Welsh RM. Direct visualization of cross-reactive effector and memory allo-specific CD8 T cells generated in response to viral infections. J Immunol. 2003;170:4077–4086. doi: 10.4049/jimmunol.170.8.4077. [DOI] [PubMed] [Google Scholar]
- 27.Selin LK, Brehm MA, Naumov YN, Cornberg M, Kim SK, Clute SC, et al. Memory of mice and men: CD8+ T-cell cross-reactivity and heterlogous immunity. Immunol Rev. 2006;211:164–181. doi: 10.1111/j.0105-2896.2006.00394.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Augustine JJ, Siu DS, Clemente MJ, Schulak JA, Heeger PS, Hricik DE. Pre-transplant IFN-gamma ELISPOTs are associated with post-transplant renal function in African American renal transplant recipients. Am J Transplant. 2005;5:1971–1975. doi: 10.1111/j.1600-6143.2005.00958.x. [DOI] [PubMed] [Google Scholar]
- 29.Heeger PS, Greenspan NS, Kuhlenschmidt S, Dejelo C, Hricik DE, Schulak JA, et al. Pretransplant frequency of donor-specific, IFN-gamma-producing lymphcytes is a manifestation of immunologic memory and correlates with the risk of post-transplant rejection episodes. J Immunol. 1999;163:2267–2275. [PubMed] [Google Scholar]
- 30.Adams AB, Williams MR, Jones TR, Shirasugi N, Durham MM, Kaech SM, et al. Heterologous immunity provides a potent barrier to transplantation tolerance. J Clin Invest. 2003;111:1887–1895. doi: 10.1172/JCI17477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen Y, Heeger PS, Valujskikh A. In vivo helper functions of alloreactive memory CD4+ T cells remain intact despite donor-specific transfusion and anti-CD40 ligand therapy. J Immunol. 2004;172:5456–5466. doi: 10.4049/jimmunol.172.9.5456. [DOI] [PubMed] [Google Scholar]
- 32.Koyama I, Nadazdin O, Boskovic S, Ochial T, Smith RN, sykes M, et al. Depletion of CD8 memory T cells for induction of tolerance of a previously transplanted kidney allograft. Am J Transplant. 2007;7:1055–1061. doi: 10.1111/j.1600-6143.2006.01703.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Welsh RM, markees TG, Woda BA, Daniels KA, Brehm MA, Mordes JP, et al. Virus-induced abrogation of transplantation tolerance induced by donor-specific transfusion and anti-CD154 antibody. J Virol. 2000;74:2210–2218. doi: 10.1128/jvi.74.5.2210-2218.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang Q, Chen Y, Fairchild RL, Heeger PS, Valujskikh A. Lymphoid sequestration of alloreactive memory CD4 T cells promotes cardiac allograft survival. J Immunol. 2006;176:770–777. doi: 10.4049/jimmunol.176.2.770. [DOI] [PubMed] [Google Scholar]
- 35.Schenk AD, Gorbacheva V, Rabant M, Fairchild RL, Valujskikh A. Effector functions of donor-reactive CD8 memory T cells are dependent on ICOS induced during division in cardiac grafts. Am J Transplant. 2009;9:64–73. doi: 10.1111/j.1600-6143.2008.02460.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schenk AD, Nozaki T, Rabant M, Valujskikh A, Fairchild RL. Donor-reactive CD8 memory T cells infiltrate cardiac allografts within 24-h posttransplant in naive recipients. Am J Transplant. 2008;8:1652–1661. doi: 10.1111/j.1600-6143.2008.02302.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Louis S, Audrain M, Cantarovich D, Schaffrath B, Hafmann K, Janssen U, et al. Long-term cell monitoring of kidney recipients after an antilymphocyte globulin induction with and without steroids. Transplantation. 2007;83:712–721. doi: 10.1097/01.tp.0000255683.66156.d3. [DOI] [PubMed] [Google Scholar]
- 38.Weaver TA, Charafeddine AH, Agarwal A, Turner AP, Russell M, Leopardi FV, et al. Alefacept promotes co-stimulation blockade based allograft survival in nonhuman primates. Nat Med. 2009;15:746–749. doi: 10.1038/nm.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fischer A, Durandy A, Sterkers G, Griscelli C. Role of the LFA-1 molecule in cellular interactions required for antibody production in humans. J Immunol. 1986;136:3198–3203. [PubMed] [Google Scholar]
- 40.Howard DR, Eaves AC, Takei F. Lymphocyte function-associated antigen (LFA-1) is involved in B cell activation. J Immunol. 1986;136:4013–4018. [PubMed] [Google Scholar]
- 41.Lee WT, Vitetta ES. The differential expression of homing and adhesion molecules on virgin and memory T cells in the mouse. Cell Immunol. 1991;132:215–222. doi: 10.1016/0008-8749(91)90020-c. [DOI] [PubMed] [Google Scholar]
- 42.Moy VT, Brian AA. Signaling by lymphocyte function-associated antigen 1 (LFA-1) in B cells: enhanced antigen presentation after stimulation through LFA-1. J Exp Med. 1992;175:1–7. doi: 10.1084/jem.175.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sanders ME, Makgoba MW, Sharrow SO, Stephany D, Springer TA, Young HA, et al. Human memory T lymphocytes express increases levels of three cell adhsion molecules (LFA-3, CD2, and LFA-1) and three other molecules (UCHL1, CDw29, and Pgp-1) and have enhanced IFN-gamma production. J Immunol. 1988;140:1401–1407. [PubMed] [Google Scholar]
- 44.Corry RJ, Winn HJ, Russell PS. Primarily vascularized allografts of hearts in mice. Transplantation. 1973;16:343–350. doi: 10.1097/00007890-197310000-00010. [DOI] [PubMed] [Google Scholar]
- 45.Afanasyeva M, Georgakopoulos D, Belardi DF, Ramsundar AC, Barin JG, Kass DA, et al. Quantitative analysis of myocardial inflammation by flow cytometry in murine autoimmune myocarditis: correlation with cardiac function. Am J Pathol. 2004;164:807–815. doi: 10.1016/S0002-9440(10)63169-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Murata K, Fox-Talbot K, Qian Z, Takahashi K, Stahl GL, Baldwin WM, 3rd, et al. Synergistic depostion of C4d by complement-activating and non-activating antibodies in cardiac transplants. Am J Transplant. 2007;7:2605–2614. doi: 10.1111/j.1600-6143.2007.01971.x. [DOI] [PubMed] [Google Scholar]
- 47.El-Sawy T, Belperio JA, Strieter RM, Remick DG, Fairchild RL. Inhibition of polymorphonuclear leukocyte-mediated graft damage synergizes with short-term costimulatory blockade to prevent cardiac allograft rejection. Circulation. 2005;112:320–331. doi: 10.1161/CIRCULATIONAHA.104.516708. [DOI] [PubMed] [Google Scholar]
- 48.El-Sawy T, Miura M, Fairchild R. Early T cell response to allografts occurring prior to alloantigen priming up-regulates innate mediated inflammation and graft necrosis. Am J Pathol. 2004;165:147–157. doi: 10.1016/s0002-9440(10)63283-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nozaki T, Amano H, Bickerstaff A, Orosz CG, Novick AC, Tanabe K, et al. Antibody-mediated rejection of cardiac allografts in CCR5-deficient recipients. J Immunol. 2007;179:5238–5245. doi: 10.4049/jimmunol.179.8.5238. [DOI] [PubMed] [Google Scholar]
- 50.LaRosa DF, Rahman AH, Turka LA. The innate immune system in allograft rejection and tolerance. J Immunol. 2007;178:7503–7509. doi: 10.4049/jimmunol.178.12.7503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Burrows SR, Khanna R, Burrows JM, Moss DJ. An alloresponse in humans is dominated by cytotoxic T lymphocytes (CTL) cross-reactive with a single Epstein-Barr virus CTL epitope: implications for graft-versus-host disease. J Exp Med. 1994;179:1155–1161. doi: 10.1084/jem.179.4.1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Morita K, Miura M, Paolone DR, Engeman TM, Kapoor A, Remick DG, et al. Early chemokine cascades in murine cardiac grafts regulate T cell recruitment and progression of acute allograft rejection. J Immunol. 2001;167:2979–2984. doi: 10.4049/jimmunol.167.5.2979. [DOI] [PubMed] [Google Scholar]
- 53.Chang JT, Palanivel Vr, Kinjyo I, Schambach F, Intlekofer AM, Banerjee A, et al. Asymmetric T lymphocyte division in the initiation of adaptive immune responses. Science. 2007;315:1687–1691. doi: 10.1126/science.1139393. [DOI] [PubMed] [Google Scholar]
- 54.Mempel TR, Henrickson SE, von Andrian UH. T-cell priming by dendritic cells in lymph nodes occurs in three distinct phases. Nature. 2004;427:154–159. doi: 10.1038/nature02238. [DOI] [PubMed] [Google Scholar]
- 55.Sabatos CA, Doh J, Chakravarti S, Freidman RS, Pandurangi PG, Tooley AJ, et al. A synaptic basis for paracrine interleukin-2 signaling during homotypic T cell interaction. Immunity. 2008;29:238–248. doi: 10.1016/j.immuni.2008.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stemberger C, Huster KM, Koffler M, Anderl F, Schiemann M, Wagner H, et al. A single naive CD8+ T cell precursor can develop into diverse effector and memory subsets. Immunity. 2007;27:985–997. doi: 10.1016/j.immuni.2007.10.012. [DOI] [PubMed] [Google Scholar]
- 57.Owens T. A role for adhesion molecules in contact-dependent T help for B cells. Eur J Immunol. 1991;21:979–983. doi: 10.1002/eji.1830210418. [DOI] [PubMed] [Google Scholar]
- 58.Rayat GR, Gill RG. Indefinite survival of neonatal porcine islet xenografts by simulataneous targeting of LFA-1 and CD154 or CD45RB. Diabetes. 2005;54:443–451. doi: 10.2337/diabetes.54.2.443. [DOI] [PubMed] [Google Scholar]
- 59.Gottlieb AB, Hamilton T, Caro I, Kwon P, Compton PG, Leonardi CL. Long-term continuous efalizumab therapy in patients with moderate to severe chronic plaque psoriasis: updated results from an ongoing trial. J Am Acad Dermatol. 2006;54:S154–163. doi: 10.1016/j.jaad.2005.12.018. [DOI] [PubMed] [Google Scholar]
- 60.Lebwohl M, Tyring SK, Hamilton TK, Toth D, Glazer S, Tawfik NH, et al. A novel targeted T-cell modulator, efalizumab, for plaque psoriasis. N Engl J Med. 2003;349:2004–2013. doi: 10.1056/NEJMoa030002. [DOI] [PubMed] [Google Scholar]
- 61.Posselt AM, Bellin MD, Tavakol M, Tzot GL, Frassetto LA, Masharani U, et al. Islet transplantation in type 1 diabetics using an immunosuppressive protocol based on the anti-LFA-1 antibody efalizumab. Am J Transplant. 2010;10:1870–1880. doi: 10.1111/j.1600-6143.2010.03073.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vincenti F, Mendez R, Pescovitz M, Rajagopalan PR, Wlilkinson AH, Butt K, et al. A phase I/II randomized open-label multicenter trial of efalizumab, a humanized anti-CD11a, anti-LFA-1 in renal transplantation. Am J Transplant. 2007;7:1770–1777. doi: 10.1111/j.1600-6143.2007.01845.x. [DOI] [PubMed] [Google Scholar]