

Abstract
Though many software packages have been developed to perform label-free quantification of proteins in complex biological samples using peptide intensities generated by LC–MS/MS, two critical issues are generally ignored in this field: (i) peptides have multiple elution patterns across runs in an experiment, and (ii) many peptides cannot be used for protein quantification. To address these two key issues, we have developed a novel alignment method to enable accurate peptide peak retention time determination and multiple filters to eliminate unqualified peptides for protein quantification. Repeatability and linearity have been tested using six very different samples, i.e., standard peptides, kidney tissue lysates, HT29-MTX cell lysates, depleted human serum, human serum albumin-bound proteins, and standard proteins spiked in kidney tissue lysates. At least 90.8% of the proteins (up to 1,390) had CVs ≤ 30% across 10 technical replicates, and at least 93.6% (up to 2,013) had R2 ≥ 0.9500 across 7 concentrations. Identical amounts of standard protein spiked in complex biological samples achieved a CV of 8.6% across eight injections of two groups. Further assessment was made by comparing mass spectrometric results to immunodetection, and consistent results were obtained. The new approach has novel and specific features enabling accurate label-free quantification.
Keywords: LC–S/MS, label-free quantification, alignment, retention time, unqualified peptides
INTRODUCTION
Currently, two main approaches are used to perform quantification of proteomic data from liquid chromatography–tandem mass spectrometry (LC–MS/MS)1. One method uses labeling techniques, while the other is label-free. Many different labeling methodologies have been published and reviewed in the literature2–5. These various approaches share the same principle, each with different strengths and weaknesses. 6,7 However, all of them suffer from several limitations: (i) additional sample processing steps in the experimental workflow, (ii) costs of the labeling reagents, (iii) variable labeling efficiency, and (iv) difficulty in analyzing low-abundance peptides in multiple samples, especially when numerous experimental groups are studied. Alternatively label-free quantification overcomes these drawbacks and is considered as a reliable, versatile, and cost-effective approach compared to label-based quantification8.
Label-free protein quantification by LC–MS/MS can be conducted by two fundamentally different strategies: spectral counting and peptide ion intensity. Spectral counting measures protein abundance by counting the number of spectra matched to peptides from a protein9. Although spectral counting has been applied in different biological complexes10,11, when low-end MS and limited LC separation (such as 1D) are applied, protein quantification using spectral counting is challenging, because (i) the enabling of dynamic exclusion of ions in data acquisition to obtain more MS/MS fragments of low-abundance peptides dramatically affects spectral acquisition, and (ii) coeluted peptides in liquid chromatography competing for MS/MS analysis influence the spectral acquisition. In contrast, peptide intensity measures protein abundance by extracting the mass spectrometric signal intensity of peptide precursor ions belonging to a particular protein. Therefore, peptide intensity is an excellent alternative for label-free protein quantification using data obtained from LC–MS/MS experiments.
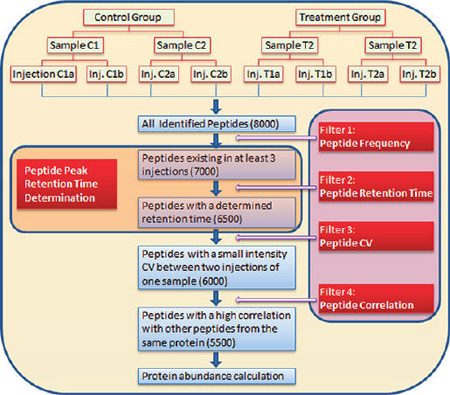
Numerous software packages for label-free quantification of LC–MS/MS-derived data based on peptide intensity have been developed and applied in proteomics research12. However, two critical issues are generally ignored in this field: (i) peptides have multiple elution patterns across runs in an experiment, and (ii) many peptides cannot be used for protein quantification. This is because a single chromatographic condition, e.g., one specific column with specific mobile phases and gradient, cannot be optimal for each of the thousands of peptides in a single injection of a complex sample. A peptide peak in the LC–MS/MS analysis has at least four features: mass-to-charge ratio (m/z), retention time, MS/MS spectrum, and intensity. Across multiple runs, m/z and MS/MS spectrum are consistent, while retention time and intensity (in technical replicates) are variable for many peptides. Therefore, accurate alignment of retention time and correct determination of peptide intensity are very important steps in label-free quantification. Here we describe a platform named IdentiQuantXL, developed to individually align the retention time of each peptide accurately and to apply multiple filters for exclusion of unqualified peptides to enhance label-free protein quantification (Figure 1).
Figure 1.
Typical workflow for label-free quantification using a novel alignment method and multiple filters for exclusion of unqualified peptides, illustrating a hypothetical experiment consisting of 2 groups, 2 samples in each group, and 2 injections in each sample. In the 8 injections, 8,000 peptides are identified. Some peptides exist in only one or two injections and thus are excluded (Filter 1: Peptide Frequency). Filtered by peptide frequency, 7,000 peptides are analyzed individually across all injections to obtain their peak retention time using a novel retention time determination method. Some peptides are excluded, because they lack a stable peak (Filter 2: Peptide Retention Time). Using the computed retention time, the peak area of 6,500 peptides is extracted. The coefficient of variation (CV) of each peptide between 2 injections in each sample is calculated, and peptides with a high CV are removed (Filter 3: Peptide CV). After Filter 3, 6,000 peptides remain. When multiple peptides are used to calculate a protein’s abundance, the correlation among them is calculated. A few peptides with poor correlation coefficients are excluded (Filter 4: Peptide Correlation). Using Filter 4, 5,500 peptidesare finally used to calculate protein abundance.
EXPERIMENTAL PROCEDURES
Study Design
Four types of experiments were performed to validate this approach. Study 1: Repeatability. A standard peptide solution (0.1 pmol/µL of Fibrinopeptide B, Angiotensin I and III), five different types of tryptic peptide samples (0.25 µg/µL) from kidney tissue lysates, HT29-MTX cell lysates, depleted human serum, human serum albumin-bound proteins, and 6 standard proteins (0.625–125 fmol, 0.0000481 –0.00203 µg, Supplementary Table 1A,B) spiked in 10 µg of kidney tissue lysates were injected in 10 replicates (5 replicates for the 6 standard proteins spiked samples) at 40 µL each into a Thermo-Finnigan linear ion-trap (LTQ) mass spectrometer coupled with a Surveyor autosampler and MS HPLC system (Thermo-Finnigan). Study 2: Linearity. Seven concentrations of 3 standard peptides (1.0, 0.50, 0.25, 0.10, 0.050, 0.025, and 0.010 pmol/µL of Fibrinopeptide B, Angiotensin I and III), serially diluted tissue/cell/serum protein/ albumin-bound samples (0.25, 0.125, 0.0625, 0.03125, 0.015625, 0.00078125, and 0.00390625 µg/µL), and 6 standard proteins (0.0390625–1,000 fmol, 0.000003–0.016239µg, Supplementary Table 1A,B) spiked in kidney tissue lysates were used. The tryptic peptides were injected in 5 replicates (2 replicates for the 6 standard proteins spiked samples) at 40 µL each into the LTQ mass spectrometer. Study 3: The quantification of an identical concentration (0.00125 µg/µL) of lysozyme spiked in human lymphocyte culture media containing secreted proteins in eight injections of four samples belonging to two groups. Study 4: A prototypical proteomics experiment that compared pituitary protein expression between wild-type and dwarfed mice, including 5 samples per group and 2 injections per sample. To summarize the samples used in the four studies, Supplementary Table 2 is presented.
Materials
dl-Dithiothreitol (DTT), urea, triethylphosphine, iodoethanol, and ammonium bicarbonate (NH4HCO3) were purchased from Sigma-Aldrich (St. Louis, MO, USA). LC–MS grade 0.1% formic acid in acetonitrile (ACN) and 0.1% formic acid in water (H2O) were purchased from Burdick & Jackson (Muskegon, MI, USA). Modified sequencing grade porcine trypsin was obtained from Princeton Separations (Freehold, NJ, USA). Lysozyme, myoglobin, β-lactoglobulin, fetuin, ovalbumin, transferrin, fibrinopeptide B, and angiotensin I and III were purchased from Sigma-Aldrich (St. Louis, MO, USA).
Sample Preparation
For the kidney samples, 259 mg of tissue was immersed in 2,072 µL of lysis buffer (8 Murea, 10 mM DTT solution, freshly prepared), thoroughly minced with needle-nose surgical scissors, and lysed using a ground-glass homogenizer. The homogenates were centrifuged at 100,000 × g for 20 min at 4 °C to remove insoluble materials. For the HT29-MTX cell samples, 1,600 µL of lysis buffer was added to the collected cells. For depleted serum samples, the depletion of high-abundance plasma proteins was performed as described previously13. The albumin-bound protein samples were tryptic peptides pooled from 16 human subjects studied in a previous experiment. The 6 standard proteins (0.0390625–1,000 fmol, 0.000003–0.016239 µg) were spiked in 10 µg of kidney tissue lysates in amounts shown in Supplementary Table 1A,B. For the lysozyme spiked samples, the media from lymphocyte cell cultures containing 100 µg of protein was used directly for the next step of sample preparation. Pituitary samples were processed in a manner identical to the kidney tissues. Protein reduction, alkylation, and digestion were carried out using a conventional method previously published by the authors13.
LC-MS/MS
The samples were injected onto a C18 microbore RP column (Zorbax SB-C18, 1.0 mm × 150 mm) at a flow rate of 50 µL/ min. The mobile phases A, B, and C were 0.1% formic acid in water, 50% ACN with 0.1% formic acid in water, and 80% ACN with 0.1% formic acid in water, respectively. The gradient elution profile was as follows: 10% B (90% A) for 5 min, 10–95% B (90–5% A) for 120 min, 100% C for 5 min, and 10% B (90% A) for 12 min. The data were collected in the “Data dependent MS/ MS” mode with the ESI interface using normalized collision energy of 35%. Dynamic exclusion settings were set to repeat count 1, repeat duration 30 s, exclusion duration 120 s, and exclusion mass width 0.75 m/z (low) and 2.0 m/z (high).
Peptide and Protein Identification
The acquired data were searched against the International Protein Index (IPI) database (ipi.RAT.v3.63, ipi.HUMAN.v3.69, and ipi.MOUSE.v3.77) using SEQUEST (v. Twenty-eight rev. 12) algorithms in Bioworks (v. 3.3). General parameters were set to peptide tolerance 2.0 amu, fragment ion tolerance 1.0 amu, enzyme limits set as “fully enzymatic - cleaves at both ends”, and missed cleavage sites set at 2.
Validation of Peptide and Protein Identification
The searched peptides and proteins were validated by PeptideProphet15 and ProteinProphet16 in the Trans-Proteomic Pipeline (TPP, v. 3.3.0) (http://tools.proteomecenter.org/software.php). Only proteins and peptides with protein probability ≥ 0.9000 and peptide probability ≥ 0.8000 were further analyzed.
Peptide Frequency Calculation
For each peptide, its protein ID, sequence, charge, scan time, m/z, injection, sample, and group information were extracted and collected. Peptides with identical sequence and charge were considered as a single entry (peptide) for further analysis. Peptide frequency was calculated, and only peptides that were identified in at least three injections were included. This represents the first filter in the approach.
Alignment To Determine Peptide Retention Time Using Clustering
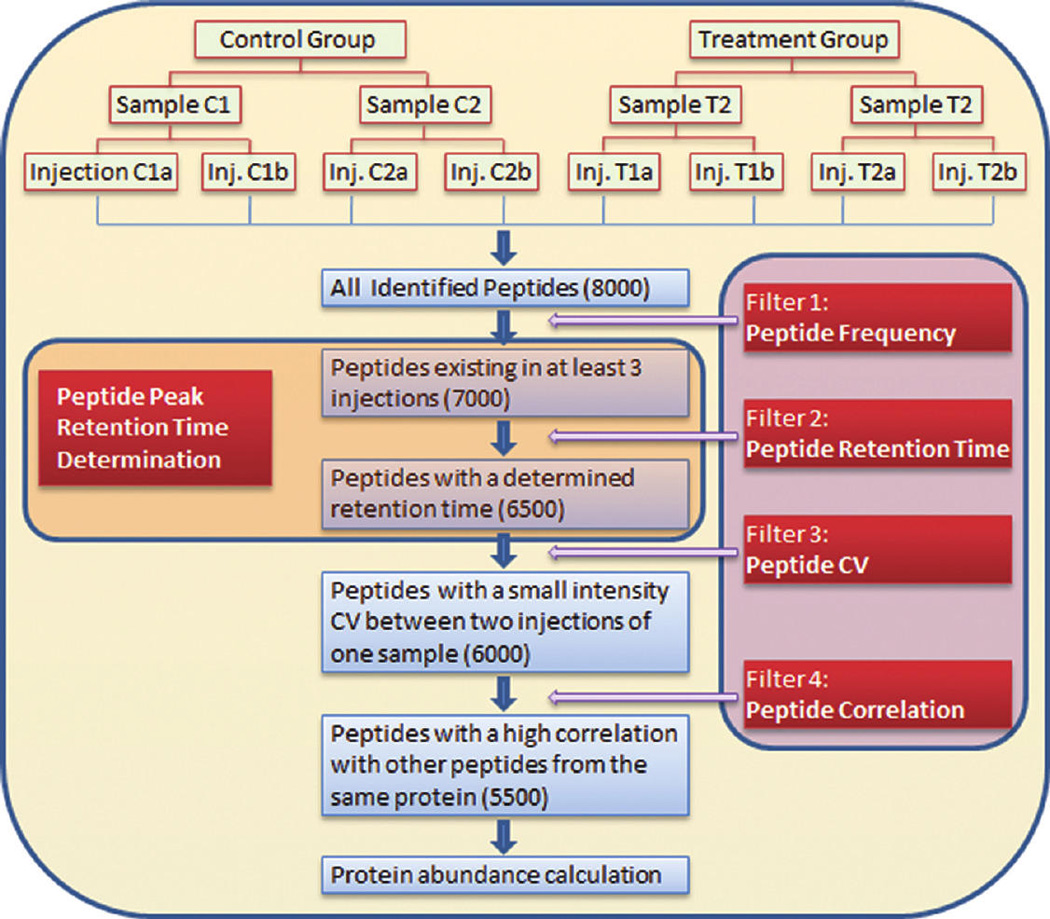
To align retention time, the retention time (scan time) range of each filtered peptide from all injections was calculated. For a peptide with range ≤3 min, a weighted mean was calculated using Tukey’s Biweight17 and used as the retention time for all injections. For a peptide with range >3 min, all obtained retention times were classified into three clusters based on elution patterns (Figure 2) using hierarchical clustering. Euclidean distance and single-linkage clustering were used for metrics and linkage criteria, respectively. The weighted mean retention time of each cluster was calculated and sorted from high to low (T1, T2, and T3). If T1–T3 ≤ 3 min, a weighted mean was calculated and used as retention time for all injections. If T1– T3 > 3 min, the frequency (F1, F2, and F3) and interquartile range (IQR1, 2, and 3) of each cluster were calculated and sorted. If a cluster with frequency (F) ≤ 0.25, or 0.25 < F ≤ 0.50 with IQR > 4, or 0.50 < F ≤ 0.75 with IQR > 6, or 0.75 < F ≤ 1.00 with IQR > 8, it was excluded. If F1 –F2 was more then 0.25, the cluster with F1 was chosen. If F1–F2 < 0.25 and F1–F3 ≥ 0.25, the cluster with a smaller retention time was chosen from the clusters with F1 and F2. If F1–F2 < 0.25 and F1– F3 < 0.25, the cluster with a smaller retention time was chosen from the three clusters. Peptides that did not meet the criteria were not assigned a retention time and were excluded. This represents the second filter in the approach. After the retention time was determined, a weighted mean m/z of each peptide was calculated. A tab delimited file was created to extract peptide intensity using MASIC18.
Figure 2.
Typical MS/MS spectrum distributions of six selected peptides across multiple injections. The x-axis is indicated according to run order. There is no difference or trend when the x-axis is indicated according to different samples (Supplementary Figure 1). (A) Multiple spectra are limited to a 3 min retention time range. (B) Most spectra are limited to a 3 min range; a few are scattered in a broad range. (C) Some spectra are concentrated in a narrow window of longer retention time, while other spectra are scattered in a wide range with a shorter retention time. (D) Some spectra are concentrated in a narrow window of shorter retention time, while other spectra are scattered in a wide range with a longer retention time. (E) Some spectra are concentrated in a narrow window of retention time, while other spectra are scattered in a wide range with a shorter and longer retention times. (F) All spectra are scattered in a wide range of retention time.
Extraction of Peptide Intensity
Peptide intensity extraction was performed by specifying “Acquisition time (minutes)” and “Limit the search to only custom m/z values (ignoring auto-fragmented m/z’s)” under Custom SIC Options in MASIC. The tab delimited file generated above was uploaded to read custom SIC search values.
Peptide CV
The coefficient of variation (CV) of each peptide in each sample was calculated. The CV values of a peptide from all samples were placed into four categories: unacceptable CV (>116%), high CV (71% < CV ≤ 116%), middle CV(47% < CV ≤ 71%), and low CV (≤47%). The frequency of each category was calculated. If its CV was >116% in any sample, the peptide was excluded. If a peptide had a “high CV” frequency >12.5%, it was filtered out. Likewise, if a peptide had a “low CV” frequency <12.5%, it too was filtered out. This represents the third filter in our approach.
Peptide Intensity Correlation
To calculate the correlation coefficient of peptides that correspond to a unique protein ID, Pearson’s correlation (for ≥3 variables) and uncentered Pearson’s correlation (for 2 variables) methods19 were applied. The intensity of each peptide was compared with all other peptides, and the average was used as the final correlation coefficient value. In the current studies, a peptide with a correlation coefficient <0.900 was excluded from protein quantification. This represents the fourth filter in the approach.
Protein Abundance Calculation
Protein abundance was calculated using the following equation:
Where Ap = protein abundance, I p = peptide intensity, and F p = frequency of peptide sharing. For a peptide shared by different proteins, the intensity of this peptide (I p) was divided by sharing frequency (Fp). The aim of this strategy is to decrease the impact of shared peptides.
RESULTS AND DISCUSSION
Peptide Elution Patterns
For a complex sample, a single chromatographic condition, e.g., one specific column with specific mobile phases and gradient, cannot be optimal for each of the thousands of peptides in a single sample injection. Peptides behave differently in such a system, and therefore each peptide may have one of six elution patterns: (i) it is completely eluted at one time point and very consistent in different sample injections analyzed; (ii) it is completely eluted at one time point but not consistently in all injections; (iii) its abundance is very high in some injections and cannot be bound completely to the column, so some amount of peptide is eluted before the main peak; (iv) it is bound to the column too tightly, mainly eluting at first, with a remainder eluting at higher organic concentration, after the main peak; (v) it has a pattern combining patterns (iii) and (iv); and (vi) it has poor chromatographic performance under the conditions and is eluted at indistinct time points across all injections. When Dynamic Exclusion is applied, six typical MS/MS spectrum distributions of various peptides are generated in one experiment, and these are shown in Figure 2A–F. The data were generated from injections of a human serum albumin-bound protein sample. Each figure illustrates the distribution of a specific peptide in all injections, and peptides have multiple MS/MS spectra in each injection. The x-axis is indicated according to run order. There is no difference or trend when the x-axis is indicated according to different samples (Supplementary Figure 1). In distribution A, all spectra are limited to a 3 min interval. In distribution B, most spectra are limited to 3 min range; a few are scattered in a broad range. In distribution C, some spectra are concentrated in a narrow window of longer retention time, while other spectra are scattered in a wide range with a shorter retention time. In distribution D, some spectra are concentrated in a narrow window of shorter retention time, while other spectra are scattered in a wide range with a longer retention time. In distribution E, some spectra are concentrated in a narrow window of retention time, while other spectra are scattered in a wide range with both shorter and longer retention times. Finally, in distribution F, all spectra are scattered in a wide range of retention time.
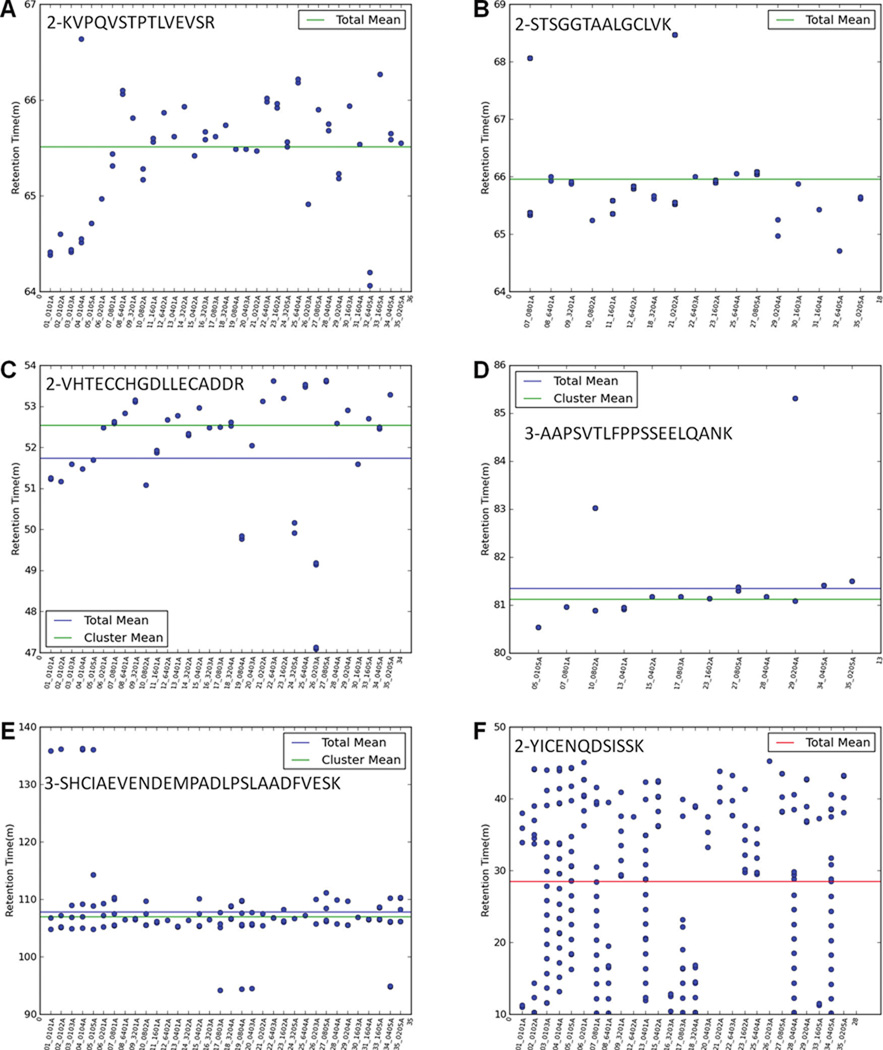
The distribution of peptides from kidney tissue lysates, HT29-MTX cell lysates, depleted human serum, human serum albumin-bound proteins, and 10 µg of kidney tissue lysates spiked with 6 standard proteins (0.0390625 – 1,000 fmol, 0.000003– 0.016239 µg) are summarized in Figure 3. The percentage of each distribution is sample-dependent. Our data show 77.4–92.1%, 4.2–12.8%, and 2.2–9.8% of peptides have distributions A and B; distributions C, D, and E; and distribution F, respectively.
Figure 3.
Percentage of each distribution of peptides from (A) kidney tissue lysates, (B) HT29-MTX cell lysates, (C) depleted human serum, (D) human serum albumin-bound proteins, and (E) 10 µg of kidney tissue lysates spiked with 6 standard proteins (0.3125–1,000 fmol, 0.000017–0.016239 µg). The percentage of each distribution is sample-dependent: 77.4–92.1%, 4.2–12.8%, and 2.2–9.8% of peptides have distributions A and B; distributions C, D, and E; and distribution F, respectively.
Alignment To Determine Peptide Retention Time
Currently, four approaches typically are used to align the retention times of peptides across multiple injections. In the most popular, researchers initially follow a workflow similar to that used in 2-DE image analysis20,21, aligning each peptide ion from different injections by plotting them as two-dimensional images22. Many software packages, such as OpenMS/TOPP23, MapQuant24, Msight20, msInspect25, and MZmine26, apply this approach. T his is a global and two-dimensional alignment (Retention Time and m/z). Considering the multiple peptide elution patterns, this alignment method only performs satisfactorily on peptides having elution patterns (i) and (ii) and incorrectly quantifies peptides having elution patterns (iii) –(vi). Furthermore, this is a pattern-based method and is only effective with high resolution data. For low resolution data, these software packages fail because no peaks can be extracted. To substantiate this, LC–MS data from the tryptic peptide samples used in this study were imported into MSight, the mass step was set at 0.05, 0.1, 0.2, and 0.5, and the result indicates that no spots were produced by MSight (Supplementary Figure 2). Clearly, m/z values obtained from low resolution instruments cannot be used for alignment in this way.
Because retention time alignment based solely on LC–MS features is not reliable for complex proteomic samples27, up- graded alignment methods such as SuperHirn28 and PEPPeR29 were generated, in which MS/MS scan boundaries become the basis for retention time comparisons in subsequent landmark matching. Even though this three-dimensional alignment (Retention Time, m/z, and MS/MS) significantly improves the alignment accuracy, it is still a global alignment. Like the first approach, false matching is generated when it is applied in elution patterns (iii) –(vi), and it is still a pattern-based method limited to the analysis of high resolution data.
The third approach is to align the “Base Peak” chromatograms, such as SIEVE from Thermo Scientific. The base peak chromatogram consists of only the most intense peak in each spectrum at every time point of the analysis. In an experiment where complex protein samples are analyzed, many peptides are eluted at the same time point but their intensities vary across multiple injections. Consequently, the “Base Peak” in each chromato-gram may be very different in some elution time windows (Supplementary Figure 3), rendering unreliable the accuracy of retention time determination by aligning only the base peak. This is another global and two-dimensional alignment (Retention Time and Base Peak Intensity). It is a shape-based method that can be applied to high and low resolution data, but it is inaccurate.
The last approach is an alignment using MS/MS scan times (identified peptides). DEAL-Q30,31 first aligns all identified peptides and constructs a regression model. The regression algorithm then is used to estimate the elution time of an unidentified peptide and perform peptide cross-assignment by mapping predicted elution time profiles across multiple LC–MS experiments. This is a global and three-dimensional alignment (Retention Time, m/z, and MS/MS), but it is an identity-based method. Therefore, it can be applied to high and low resolution data. However, it does not resolve those peptides having elution patterns (iii) –(vi). In their publication, the authors found that manual validation of all 11,940 peptide ions in six replicate LC–MS/MS runs revealed that 97.8% of the peptide ions were correctly aligned, and 93.3% were correctly validated by SCI. The 93.3% value is consistent with our observation that 77.4–92.1% of peptides have distributions A and B (elution patterns (i) and (ii)).
Some approaches, such as ProteinQuant32, do not align peptide retention time across multiple injections. In that approach, the first occurrence of a peptide MS/MS scan and its associated retention time (precursor MS/MS scan time) is collected in the master file. Within the user-designated retention time window associated with the precursor MS/MS scan time, it generates a base peak chromatogram for the precursor m/z value and then selects the maximum intensity peak According to the six elution patterns described earlier, the first occurrence of a peptide does not necessarily mean the peptide elution peak appears at that MS/MS scan time.
It is clear that accurate chromatographic alignment is essential in label-free quantification when peptide intensity extraction is performed, but it is also necessary to account for peptide elution patterns. Existing alignment methods apply a global alignment, an approach that totally ignores the impact of peptide elution patterns.
Individual Three Dimensional Alignment To Determine Peptide Retention Time Using a Clustering Method
To accurately determine peptide retention time, we have developed a novel alignment method to determine peptide peak retention time by clustering each identified peptide MS/MS scan time according to its elution pattern. For each peptide, the retention times of all MS/MS spectra from all injections were collected, and then the retention time distribution patterns were generated. Each peptide was processed independently, without considering other peptides’ retention times. Therefore, this is an individual alignment, in contrast to the global alignment methods described above. For distribution A (Figure 2), the variation is acceptable for all LC runs, no cluster is applied, and a weighted mean is calculated using Tukey’s Biweight as the retention time of the peptide across all injections. For distributions B–F (Figure 2), three clusters are generated, and the weighted mean (retention time), interquartile range (IQR) value, and frequency (of each cluster present in all injections) are calculated. The IQR value of a cluster is used to determine whether or not a peak exists at that retention time. If an IQR is small, it means a peak exists; if an IQR is large, it means the peak is absent or unacceptable (e.g., too broad). An IQR cutoff based on the cluster frequency is described in the Experimental Procedures. Only qualified clusters are applied to the comparison. For distribution B (Figure 2), the range of three clusters’ weighted means is limited to 3 min, signifying most spectra are within the 3 min interval and only a few are out of range. Therefore, the total weighted mean is calculated as the retention time for the peptide in every run. For distribution C (Figure 2), the highest cluster frequency indicates that this cluster is generated around the peak. Its weighted mean is thus used as the retention time for the peptide in every run. For distribution D (Figure 2), MS/MS spectra are acquired around the peak and then many times after a long time interval. In this case, the qualified cluster with shorter retention time is chosen. Distribution E (Figure 2) is the most complicated, and thus a multistep approximation is applied to determine the retention time of a peptide peak. First, the cluster frequency is used to exclude the MS/MS spectra eluting before the main peak. Then the main peak is selected because it has a shorter retention time. Finally, distribution F (Figure 2) is not suitable for retention time determination and is excluded because the peptide has poor chromatographic performance under the current experimental conditions. The application of the clustering method, with the aid of its frequency and IQR, can accurately determine the identified retention time. Because the determined retention time is identity-based, it can be applied to high and low resolution data. It is worth noting that this retention time is the weighted mean of the MS/MS acquisition time points that appear around the eluting peak. The mean retention time is the closest time point to the apex of the chromatographic peak of an identified peptide. Furthermore, MASIC is used to define the apex of the eluted peak by importing the mean retention time into the MASIC software. This approach is an individual and three-dimensional alignment (Retention Time, m/z, and MS/MS) and can be applied to both high and low resolution data.
Comparison of the key features of our novel method to other existing approaches is summarized in Table 1. Global alignment does not consider heterogeneous peptide elution patterns and only works well for elution patterns (i) and (ii). Individual alignment takes into account the various elution patterns and works well for all of them. A pattern-based method is limited to analyzing high resolution data. A shape-based method can analyze both high and low resolution data, but it is inaccurate. An identity-based method can be applied to high and low resolution data. Therefore, an individual, identity-based alignment method provides the most accurate retention time determination and widest application. Our alignment method incorporates these features. In addition, our platform applies multiple filters for exclusion of unqualified peptides to enhance label-free quantification.
Table 1.
Comparison of Key Features of IdentiQuantXL to Those of Other Software Packagesa
| features | SIEVE | Msight | PEPPeR | ProteinQuant | IDEAL-Q | IdentiQuantXL |
|---|---|---|---|---|---|---|
| considers elution patterns | no | no | no | none | no | yes |
| global alignment | yes | yes | yes | none | yes | no |
| individual alignment | no | no | no | none | no | yes |
| pattern-based | no | yes | yes | none | no | no |
| shape-based | yes | no | no | none | no | no |
| identity-based | no | no | no | none | yes | yes |
| dimensional | RT, BPIb | RT, m/z | RT, m/z, MS/MS | none | RT, m/z, MS/MS | RT, m/z, MS/MS |
| High resolution data | yes | yes | yes | yes | yes | yes |
| low resolution data | yes | no | no | yes | yes | yes |
| multiple filters | no | no | no | no | no | yes |
| accuracy (high resolution data) | − | ++ | +++ | ++ | +++ | +++++ |
| accuracy (low resolution data) | − | + | ++ | ++ | +++ | +++++ |
An individual, identity-based alignment method provides the most accurate retention time determination and wide application. Multiple filters enable the exclusion of unqualified peptides to enhance label-free quantification.
BPI = base peak intensity.
Peptide Frequency
Another of the two important issues in peptide intensity measurements relates to the use of every identified peptide for protein quantification. Many software packages have ignored this issue. To obtain accurate quantification results, several considerations prompt one to filter out some of the peptides. First, in a typical experiment, many peptides are identified only in some of the injections, even in only one or two. In our experiment in which a tryptic digest of whole rat kidney lysates was analyzed and included 35 injections, 7,361 peptides were identified. However, 1,883 (25.6%) of the peptides were identified only in 1 injection, 831 (11.3%) in each of 2 injections, 562 (7.6%) in each of 3 injections, and only 61 (0.8%) were identified in all 35 injections. The detailed distribution of identification is illustrated in Figure 4A,B. If a peptide is not analyzed by MS/MS in an injection, that does not mean it is absent from this injection32. A peptide identified only in one injection is likely to exist in all other injections in that experiment. However, confidence in this assumption is weakened because the mass spectrometer is very sensitive and numerous ions are fragmented; consequently non-peptide ions may be misidentified as peptides and other identified peptides may be false positive. Extraction of the intensities for nonpeptide ions or misidentified peptides with low identification frequency leads to erroneous quantification.
Figure 4.
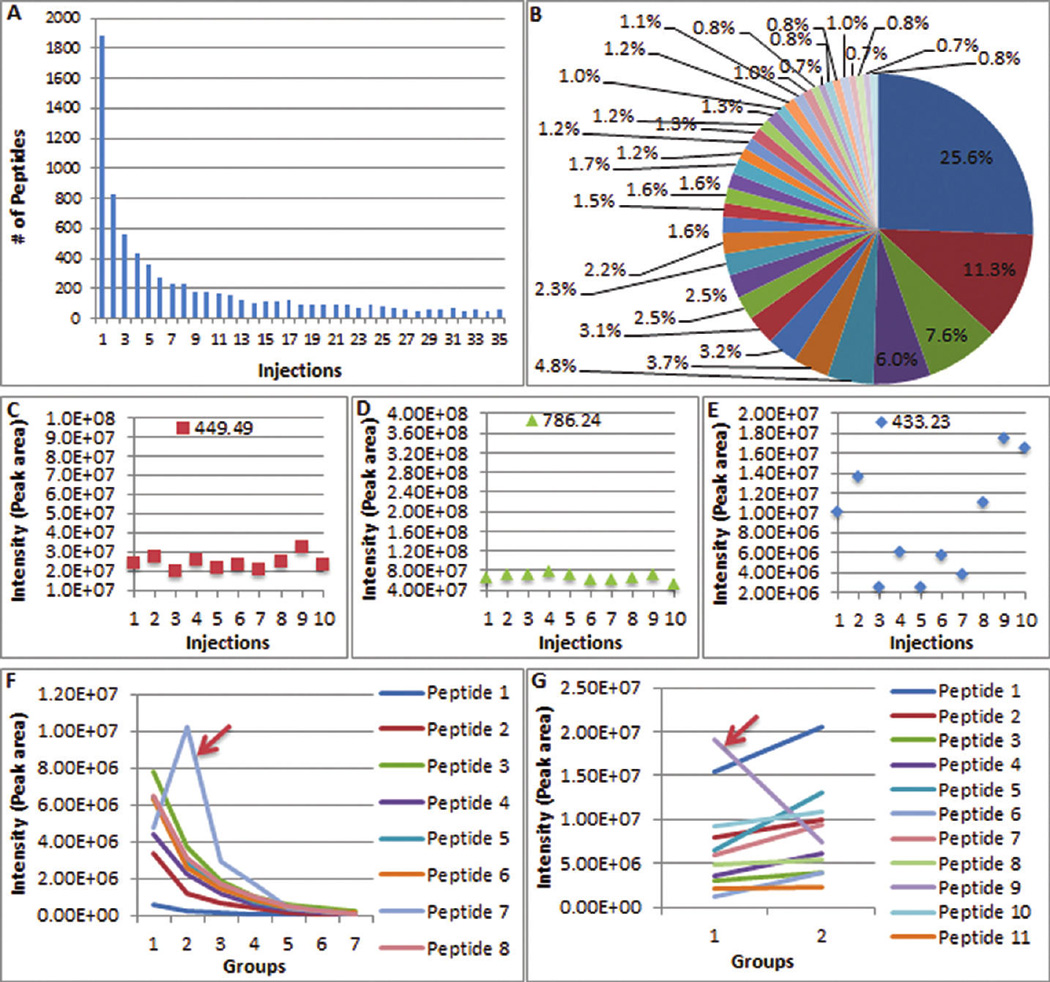
Multiple filters used in the approach for protein quantification. (A, B) Distribution of peptide identification in 35 injections of rat kidney sample lysates. In total, 7,361 peptides were identified. However, 1,883 (25.6%) of the peptides were identified in only 1 injection, and only 61 (0.8%) were identified in all 35 injections. Peptide Frequency is used as the first filter in this approach. Only peptides with identification frequencies higher than the cutoff are considered for the next step in the filtration process. (C–E) Peak areas of three standard peptides from 10 replicates (Angiotensin III, 449.49; Fibrinopeptide B, 786.24; and Angiotensin I,433.23). Data clearly show that some peptides cannot be used to calculate protein quantity. (F, G) Correlation of peptides from two proteins. The arrow indicates peptides significantly different from other peptides, their correlation coefficients are much lower than others, and they are excluded from subsequent protein quantification. Correlation represents the fourth and final filter in this approach.
To improve confidence in peptide quantification, we have developed a filter for peptide identification frequency whereby one can determine the peptide identification frequency criterion according to the characteristics of each experiment. Only peptides with identification frequencies higher than a specified cutoff are considered for the next step in the process.
Peptide Retention Time
A second consideration relates to the fact that chromatographic conditions cannot be optimal for all of the thousands of peptides detected in a single injection. Consequently, some peptides have an elution pattern like that shown in distribution F (Figure 2), namely, they do not form a quantifiable peak. Therefore, a retention time filter must be used to eliminate these peptides from further analysis.
Peptide CV
For the reasons mentioned above, a third filter has been developed to disregard peptides whose intensities measured by peak area are highly variable across technical replicates. In the current study, a mixture of three standard peptides, Angiotensin III, Fibrinopeptide B, and Angiotensin I, was injected into the LTQ. The intensities of each peptide from 10 replicates are shown in Figure 4C–E. The intensities of 4 pmol of Angiotensin III, Fibrinopeptide B, and Angiotensin I were 2.39 × 107 ± 0.36 × 107, 6.63 × 107 ± 0.75 × 107, and 8.90 × 106 ± 5.64 × 106 with CV 15.0%, 11.4%, and 63.4%, respectively. From these data, it is quite clear that not every peptide can be used to calculate protein abundance. The results indicate that Angiotensin I has a huge variation (63.4%) and is not qualified for protein quantification in the current study.
Peptide Intensity Correlation
Finally, when multiple peptides are used to calculate a protein’s abundance, the correlation among them may be poor. Examples from the current kidney experiment (see below) and an additional pituitary tissue lysate study are shown in Figure 4F,G. Peptide 7 in Figure 4F and peptide 9 in Figure 4G are significantly different from other peptides. The peptide sequences and protein names are provided in Supplementary Table 3A. Possible reasons for poor correlation are as follows: (i) some peptides have greater variation than others under the same chromatographic condition, generating a weak relationship among peptides; (ii) posttranslational modification (PTM) variably affects the relative abundance of unmodified peptides; and (iii) peptide sharing among diverse proteins causes inconsistent effects on some peptides. The correlation coefficients of peptide 7 in Figure 4F and peptide 9 in Figure 4G were calculated using Pearson’s correlation (for ≥3 variables) and uncentered Pearson’s correlation (for 2 variables) methods and are listed in Supplementary Table 3B. Peptides in Figure 4F had correlation coefficients of at least 0.926, except for peptide 7, which had a value of 0.595. Peptides in Figure 4G had correlation coefficients of at least 0.901, except for peptide 9 with a value of 0.814. Therefore, these two peptides were excluded for subsequent protein quantification.
Multiple Filters To Exclude Unqualified Peptides for Protein Quantification
From the results of the experiments described above, a fundamental concept emerged that not every peptide can be used to quantify a corresponding protein. Therefore, multiple filters (Peptide Frequency, Retention Time, Intensity CV, and Correlation) were developed to enhance protein quantification. To test the performance of this strategy, repeatability and linearity analyses were performed using (i) a simple sample consisting of 3 standard peptides, (ii) complex samples consisting of thousands of peptides, and (iii) 6 standard proteins spiked in complex samples (kidney tissue lysates). In some cases, correlation calculation (R2) is performed on the log–log scale. However, the variance of log ratios also depends on signal intensity. When raw data are considered, variation increases as intensity increases. When log-transformed data are considered, the variance is usually stable above a certain intensity, but the low intensity data can be highly variable.33 Therefore, R2 calculations using both log-transformed and raw data are presented.
In the repeatability test of the simple 3 peptide sample, all were identified in each of 10 injections, and their retention time range was less than 3 min. Two of the peptides (Angiotensin III and Fibrinopeptide B) had an intensity CV ≤ 15% and were qualified for the next step in the process (see Experimental Procedures for parameters). For these two peptides, extraordinary quantitative linearity was generated with R2 of 0.9964 and 0.9980 across 7 concentrations with 2 orders of magnitude (Figure 5A). The R2 calculation using raw data is shown in Supplementary Figure 4A. This result indicates that peptide intensity measurement is a reliable approach for protein quantification, when some peptides not qualified for quantification are excluded.
Figure 5.
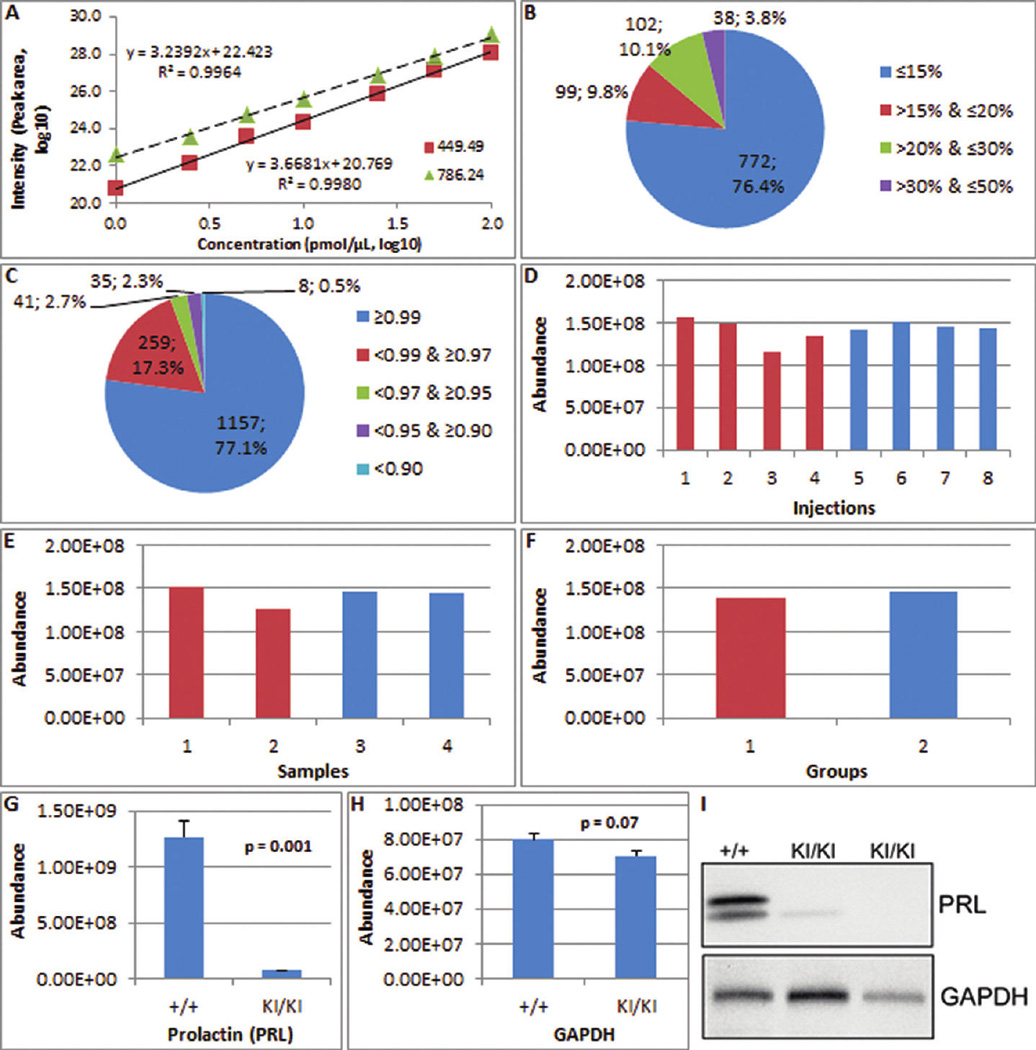
Performance of the approach in protein quantification. (A) The calibration curve of two standard peptides from 7 concentrations in 5 replicates each (Angiotensin III, 449.49; Fibrinopeptide B, 786.24). Extraordinary quantitative linearity was observed with R2 of 0.9964 and 0.9980 across 7 concentrations with 2 orders of magnitude. (B) The repeatability analysis of 1,011 proteins in the current kidney tissue lysates indicates 96.2% of proteins had CVs ≤30%. (C) The linearity analysis of 1,500 proteins in the kidney tissue lysates indicates 97.2% of proteins had an R2 of ≥ 0.9500. (D–F) Label-free quantification of identical concentrations of lysozyme with CV = 8.6%, 8.2%, and 3.2% for eight injections (D), four samples (E), and two groups (F), respectively. (G–I) The quantification of prolactin using this approach versus Western blot. Prolactin was 17.1-fold lower in dwarfed mice (KI/KI) compared to wild-type (+/+) using the label-free quantification strategy (G). The quantification of GAPDH reported as a control is 1.14-fold lower (H). Western blot of prolactin with GAPDH as a loading control (I) is reprinted with permission of the authors and shows very low or undetectable prolactin in the pituitaries of adult dwarfed mice compared with wild type controls34. Western blot analysis of prolactin is consistent with the fold change detected by the label-free quantification strategy.
In the repeatability test of a more complex sample (kidney tissue), 2,662 of 4,636 peptides were identified in at least 3 out of 10 injections, 2,647 of 2,662 peptides with an retention time were qualified for peak area extraction, and 2,599 of 2,647 peptides with an acceptable CV were used for protein quantification (see Experimental Procedures section for parameters). Using these three filters, 772 (76.4%), 99 (9.8%), 102 (10.1%), and 38 (3.8%) out of 1,011 proteins had a CV ≤ 15%, ≤ 20% and >15%, ≤ 30% and > 20%, and ≤ 50% and >30%, respectively, indicating that 96.2% of proteins had CVs ≤ 30% (Figure 5B).
In the repeatability test of 6 standard proteins (0.625 – 125 fmol, 0.000048–0.002030 µg, Supplementary Table 1A,B) spiked in 10 µg of kidney tissue lysate proteins, 1,837 of 5,526 peptides were identified in at least 3 out of 5 injections, 1,835 of 1,837 peptides with an retention time were qualified for peak area extraction, and 1,783 of 1,835 peptides with an acceptable CV were used for protein quantification. Using these three filters, 649 (67.1%), 127 (13.1%), 135 (14.0%), and 56 (5.8%) out of 967 proteins had a CV ≤ 15%, ≤ 20% and >15%, ≤ 30% and > 20%, and ≤ 50% and >30%, respectively, indicating that 94.2% of proteins had CVs ≤ 30% (Supplementary Figure 5A). The CVs of standard proteins were 8.6%, 9.3%, 12.2%, 10.3%, 8.0%, and 23.3% for lysozyme, myoglobin, β-lactoglobulin, fetuin, ovalbumin, and transferrin, respectively. The repeatability analysis results of the HT29-MTX cell lysate, depleted serum, and albumin-bound proteins experiments are provided in Supplementary Figure 5B –D. In each of the five unique experiments, at least 90.8% of the proteins had CVs ≤ 30%.
In the linearity test of the kidney tissue lysate, 4,647 of 7,361 peptides were identified in at least 3 of 35 injections (see Methods),4,474 of 4,647 peptides with an acceptable retention time were qualified for peak area extraction, 4,123 of 4,474 peptides with an acceptable CV were qualified for protein quantification, and 4,006 of 4,123 peptides with an acceptable correlation coefficient were used for protein quantification. Using these four filters, 1,157 (77.1%), 259 (17.3%), 41 (2.7%), 35 (2.3%), and 8 (0.5%) of 1,500 proteins had an R2 of ≥0.9900, ≥ 0.9700 and <0.9900, ≥ 0.9500 and <0.9700, ≥ 0.9000 and <0.9500, and <0.9000 across 7 concentrations (Figure 5C), respectively. The linearity analysis indicated 97.2% of proteins had an R2 ≥ 0.9500. The percentage of R2 distribution using raw data is shown in Supplementary Figure 4B.
In this analysis, the dilution ratios (fold differences) of the 7 kidney sample concentrations are known. The comparison between the known fold differences and the calculated fold differences by linear regression analysis across the different concentrations was used as an additional method to judge the performance of the approach. The fold differences calculated from the linear regressions between different concentrations and their R2 are listed in Table 2. All fold differences fall into the ±28% range, and the minimal R2 is 0.9376. Even the largest fold difference (64-fold) was successfully determined as 6.30 (log2 transformed), with an error of only 5%. The calculation of fold and R2 distribution using raw data is shown in Supplementary Table 4. The representative linear regressions of 1,500 proteins using log-transformed and raw data are shown in Figure 6 and Supplementary Figure 6, respectively. Other regressions using log-transformed and raw data are indicated in Supplementary Figure 7A,B.
Table 2.
Fold Differences from the Linear Regressions of 1,500 of Proteins at Different Concentrations and Their R2
| fold (log2) |
R2 |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| DF | 10a | 5 | 2.5 | 1.25 | 0.625 | 0.3125 | 10 | 5 | 2.5 | 1.25 | 0.625 | 0.3125 |
| 1 | 1.28 | 1.10 | 0.96 | 1.18 | 1.12 | 0.85 | 0.9759 | 0.9951 | 0.9936 | 0.9932 | 0.9887 | 0.9801 |
| 2 | 2.37 | 2.05 | 2.14 | 2.28 | 2.01 | 0.9718 | 0.9897 | 0.9870 | 0.9826 | 0.9630 | ||
| 3 | 3.23 | 3.22 | 3.27 | 3.19 | 0.9738 | 0.9818 | 0.9733 | 0.9555 | ||||
| 4 | 4.39 | 4.33 | 4.20 | 0.9673 | 0.9699 | 0.9435 | ||||||
| 5 | 5.42 | 5.24 | 0.9625 | 0.9409 | ||||||||
| 6 | 6.30 | 0.9376 | ||||||||||
10 µg sample, 5 µg sample, etc. DF = dilution factor.
Figure 6.
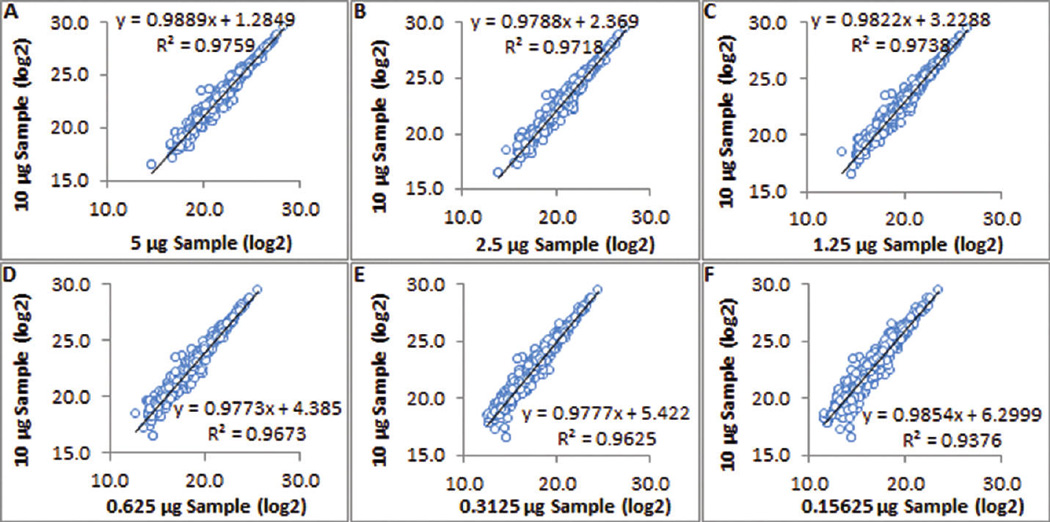
Linear regression analysis between 7 different concentrations of 1,500 proteins from kidney tissue lysates. All fold differences fall in the ±28% range, and the minimal R2 is 0.9376. The largest fold difference (64-fold) was accurately determined as 6.30 (log2 transformed), with an error of only 5%.
In the linearity test of 6 standard proteins (0.0390625 – 1,000 fmol, 0.000003–0.016239 µg, Supplementary Table 1A,B) spiked in 10 µg of kidney tissue lysates, 4,323 of 8,524 peptides were identified in at least 3 of 16 injections, 4,171 of 4,323 peptides with an acceptable retention time were qualified for peak area extraction, 4,060 of 4,171 peptides with an acceptable CVwere qualified for protein quantification, and 1,720 of 4,171 peptides with an acceptable correlation coefficient were used for protein quantification. With these four filters, the intensity and R2 of 6 standard proteins were calculated. In the range of 0.3125–1,000 fmol (0.000017–0.016239 µg), the R2 of 6 standard proteins were 0.9990, 0.9986, 0.9939, 0.9777, 0.9893, and 0.9763 for lysozyme, myoglobin, β-lactoglobulin, fetuin, ovalbumin, and transferrin (Figure 7), respectively. The calculation of R2 using raw data is shown in Supplementary Figure 8.
Figure 7.
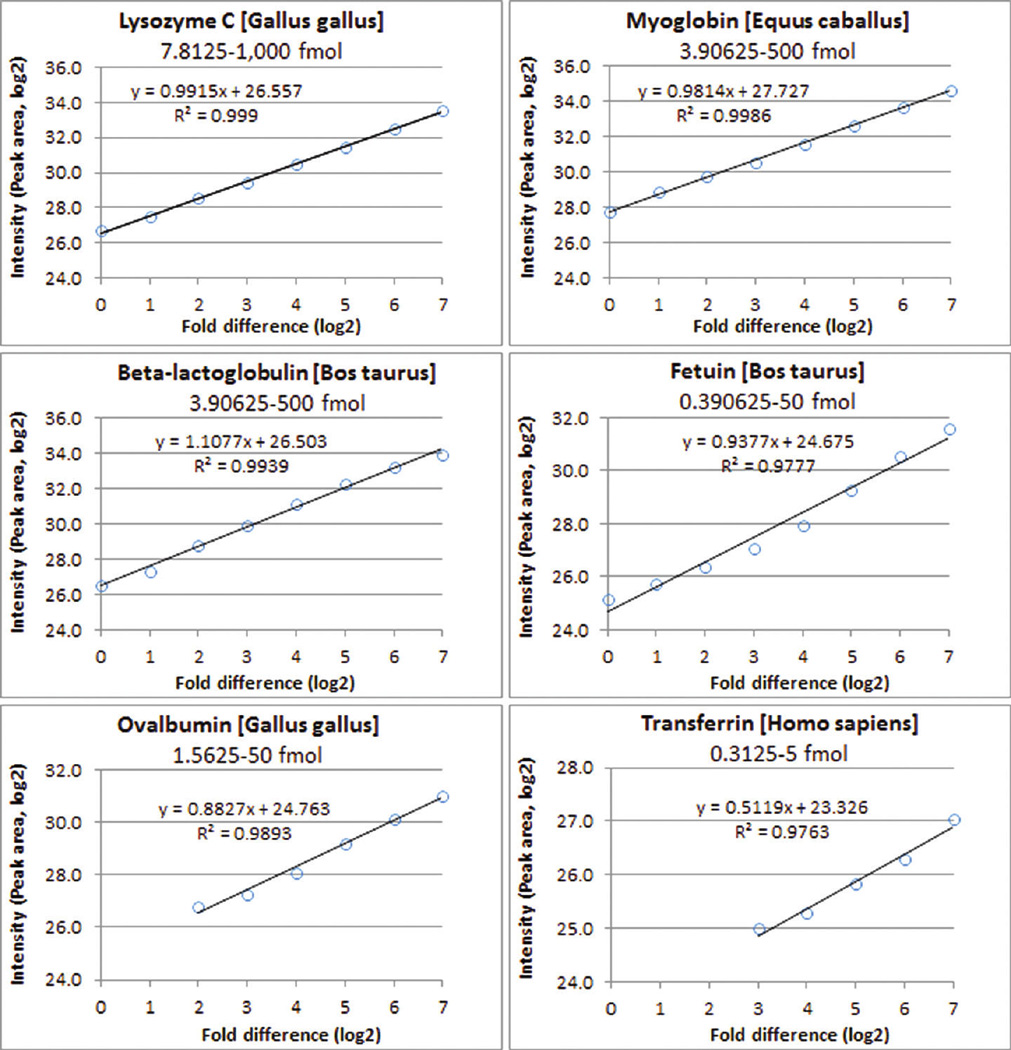
Linear regression analysis between 8 different concentrations of 6 standard proteins (0.3125–1,000 fmol, 0.000017–0.016239 µg) spiked in 10 µg of kidney tissue lysates. All 6 proteins have an excellent correlation in the range, and the minimal R2 is 0.9763.
The linearity analysis results from the HT29-MTX cell lysate, depleted serum, and albumin-bound protein experiments are provided in Supplementary Figure 9 and Supplementary Tables 5–7. Among these very different samples, the linearity analysis indicated 96.5%, 98.4%, and 93.6% of proteins had an R2 ≥ 0.9500, respectively. The linear regressions between different concentrations are indicated in Supplementary Figures 10–15.
Quantification of identical amounts of lysozyme spiked in human lymphocyte culture media containing secreted proteins was carried out to further evaluate the performance of the approach. In this experiment, eight injections of four complex biological media samples belonging to two groups were carried out. The label-free quantification of lysozyme is shown in Figure 5D–F. The resulting CV for lysozyme was 8.6%, 8.2%, and 3.2% for eight injections, four samples, and two groups, respectively, and indicates that this strategy was accurate in the quantification of a protein in a complex mixture.
After the validation of the approach, it was applied to a proteomics experiment that compared global pituitary protein expression between wild-type and a dwarfed mouse model of combined pituitary hormone deficiency (CPHD) containing a targeted knock-in mutation (Lhx3 W227ter), a model for human LHX3 W224ter disease.34 More than 4,000 proteins were quantified using the method. Expression of many pituitary proteins was decreased in the dwarfed mouse, consistent with results that were obtained by other methods. One of these proteins, prolactin, was decreased 17.1-fold in dwarfed mice compared to wild-type using the label-free quantification strategy (Figure 5G, H). Western blot experiments analyzing whole pituitaries have independently shown very low or undetectable prolactin in the pituitaries of adult dwarfed mice compared with wild type controls (Figure 5I).34 Again, this result demonstrates the utility of this label-free strategy in analyzing complex protein mixtures.
Optimal Filtering Thresholds for the Four Filters
After achieving successful quantification of proteins in different samples, an additional concern is the optimal threshold for each of the four filters. Because concrete values are not generally transferable and it is very difficult to set such parameters using a statistical analysis, unique experiments cannot use identical thresholds. In this platform, filter thresholds can be set at different values for different projects achieving optimal thresholds specific to the unique protocols/platforms used to generate the data.
A balanced approach to setting thresholds should be applied. Overly conservative parameters will reduce the number of proteins quantified, by filtering out those that vary only through minor experimental errors. Conversely, excessively liberal parameters will produce a large number of inaccurately quantified proteins. Because our platform applies the four filters in a sequential manner, inaccurate results generated using relatively liberal parameter settings in each previous step have an opportunity to be excluded in the next step, and so on. Therefore, reasonably liberal parameters can safely be applied, in general. A summary of the optimal filtering thresholds for the four filters is presented in Supplementary Table 8.
In general, a protein identified by two or more peptides is considered a more confident identification than one identified by only one peptide. Similarly, a peptide identified in multiple injections is considered a more confident identification than one from only one injection. Different laboratories use different software and cutoff values for peptide identification, and this affects the Peptide Frequency calculation. We suggest that peptide identifications from 2 or 3 injections minimum should be used as the threshold of Peptide Frequency.
In retention time determination, the threshold of 3 min is used as basic cutoff based on observations from many HPLC runs and the elution patterns shown in Figure 2. Although the development of a simpler approach more generally applicable to other platforms or protocols was attempted, such a method remains currently unavailable. Therefore, the rules to combine IQR with frequencies were developed.
The Peptide CV filter is a combination of the CV value and its frequency. Because at least two injections are needed when this filter is applied, the threshold of the CV value is calculated based on two injections. The concept is as follows: if a peptide has an unacceptable CV in one injection, it should be excluded from all injections; if a peptide has an excessively high CV, it should be excluded; and if a peptide has too few low CV values, it too is excluded.
The correlation coefficient (CC) filter is used to exclude extreme outliers. Though there is no requirement that qualified peptides have excellent correlation coefficients (such as CC ≥ 0.9), most poorly correlated peptides are excluded. Normally, when CC < 0.9, the threshold setting filters out 10–15% of the peptides. When outlier peptides are checked manually, this cutoff always excludes the pertinent, outlier peptides. Different experiments can apply different correlation coefficient cutoff because this cutoff is sample dependent. In our experience, CC ≥ 0.9 works well for data from two-group data.
In reality, optimal thresholds are difficult to achieve for all peptides, because over 10,000 peptides are processed simultaneously. Nonetheless, they may be obtained over the course of many trials. It also is important to concede that even the best software in the world cannot fix poor quality measurements; consequently, the quality of the LC–MS/MS data always is of the highest priority.
Comparison between IdentiQuantXL and SIEVE
In Figure 7 and Supplementary Figure 8, the linearity test of the 6 standard proteins (0.0390625–1,000 fmol, 0.000003–0.016239 µg, Supplementary Table 1A,B) spiked in 10 µg of kidney tissue lysates have been presented. Therefore, the fold differences between any two concentrations can be calculated easily. Two sets of comparisons were processed, 2:1 (128 vs 64) and 16:1 (128 vs 8). The results using IdentiQuantXL are shown in Table 3.
Table 3.
Comparison between IdentiQuantXL and SIEVE in Protein Quantification
| 2:1 (128 vs 64) |
16:1 (128 vs 8) |
|||
|---|---|---|---|---|
| protein | IdentiQuantXL | SIEVE | IdentiQuantXL | SIEVE |
| lysozyme C [Gallus gallus] | 2.0 (0.0%) | 1.8 (−10.0%) | 17.6 (10.0%) | 17.9 (11.9%) |
| myoglobin [Equus caballus] | 1.9 (−5.0%) | 1.7 (−15.0%) | 17.1 (6.9%) | 17.2 (7.5%) |
| β-lactoglobulin [Bos taurus] | 1.6 (−20.0%) | 1.6 (−20.0%) | 15.7 (−1.9%) | 8.2 (−48.8%) |
| fetuin [Bos taurus] | 2.1 (5.0%) | 2.2 (10.0%) | 23.3 (45.6%) | 34.6 (116.3%) |
| ovalbumin [Gallus gallus] | 1.8 (−10.0%) | 1.9 (−5.0%) | 13.6 (−15.0%) | 13.9 (−13.1%) |
| transferrin [Homo sapiens] | 1.7 (−15.0%) | NDa | 4.2 (−73.8%) | NDa |
No data reported.
The same data sets then were analyzed using the latest version of SIEVE software (1.3.744 SP1) developed by ThermoFisher Scientific, an automated LC–MS analysis platform for label-free quantitative differential expression analysis of proteins, peptides, and small molecules for quantitative measurements in conjunction with qualitative identification. In each data set, four raw data from two concentrations including two injections in each concentration were submitted to SIEVE. After the alignment, frames were created and submitted for identification. For the protein report, Maximum Percolator Peptide FDR was set as 1.00; the Minimum Xcorr for Charge 1, 2, 3, and >3 was 1.90, 2.20, 3.75, and 3.75, respectively, and Maximum Rank was set as 1 in SEQUEST Criteria. The results were exported to Excel, and the quantification of the 6 standard proteins is presented in Table 3.
Comparing the results from IdentiQuantXL and SIEVE in Table 3, it is clear that the performance of IdentiQuantXL is much better than SIEVE. For highly abundant proteins, both IdentiQuantXL and SIEVE generate excellent results, but IdentiQuantXL performs better than SIEVE. For low abundance proteins, IdentiQuantXL performs significantly better than SIEVE. Transferrin is the lowest abundance protein of the 6 standards. IdentiQuantXL accurately quantified the 2-fold difference as well as the 16-fold differences of transferrin, while SIEVE failed to report the quantification data. For β-lactoglobu-lin and fetuin in low abundance, the fold differences detected by SIEVE are highly inaccurate, while IdentiQuantXL quantified the fold changes accurately.
CONCLUSIONS
In LC–MS/MS analysis, a single chromatographic condition, e.g., one specific column with specific mobile phases and gradient, cannot be optimal for each of the thousands of peptides in a single sample injection of a complex sample. Accordingly, two important issues hinder the use of peptide intensity measurements: (i) peptides have multiple elution patterns across runs in an experiment, and (ii) many peptides cannot be used for protein quantification. However, these issues are generally ignored by many software packages that have been developed to perform label-free quantification of proteins in complex biological samples using peptide intensities.
In LC–MS/MS applications, the alignment of peptide retention time is challenging and critical to label-free quantification, and using peptides to quantify corresponding proteins is not always straightforward. Because many identified peptides are unqualified for use in protein quantification for reasons presented in this paper, the practice of unconstrained use of all peptides to calculate protein abundance generates inaccurate results. Therefore, we have developed a novel individual three-dimensional alignment method to determine peptide retention time and four novel filters, Peptide Frequency, Retention Time, Intensity CV, and Correlation, to enhance protein quantification.
By analyzing the repeatability and linearity of protein quantification in complex samples, this approach systematically demonstrated its excellent utility in a wide variety of concentrations. Repeatability and linearity have been tested using ion trapderived low resolution data from six very different samples, i.e., standard peptides, kidney tissue lysates, HT29-MTX cell lysates, depleted human serum, human serum albumin-bound proteins, and standard proteins spiked in kidney tissue lysates. In these unique experiments, at least 90.8% of the proteins (up to 1,390) had CVs ≤ 30% across 10 technical replicates, and at least 93.6% (up to 2,013) had R2 ≥ 0.9500 across 7 concentrations. By comparing known fold differences with calculated fold differences from linear regression analysis between different concentrations, this approach has successfully quantified up to a 64-fold difference in protein abundance in complex samples. The performance of our approach was verified using identical amounts of standard protein (lysozyme) spiked in complex biological samples (cell culture media containing secreted proteins) with a CV of 8.6% across eight injections. Further assessment was made by comparing mass spectrometric results to immunodetection. Prolactin, which was 17.1-fold lower in dwarfed mice versus wild-type using the label-free quantification strategy, was very low or undetectable using Western blot34. These results indicate that our new approach accurately quantifies numerous peptides and proteins in complex samples.
The results of repeatability testing, linearity assessment, fold difference comparison, standard protein inclusion, and Western blot analysis all clearly indicate this new approach performed accurately in the analysis of LC–MS data acquired by low resolution mass spectrometry and using the Dynamic Exclusion function. In addition, it has been applied successfully in the analysis of the aqueous humor proteome data acquired by high resolution mass spectrometry in patients with Fuchs endothelial corneal dystrophy35. While many software packages focus only on high resolution data, our approach is designed for both high and low resolution data. Consequently, it is very useful for data generated by low resolution mass spectrometers such as the LTQ, especially when the dynamic exclusion of ions in data acquisition is enabled to obtain more MS/MS fragments of low-abundance peptides to maximally identify proteins in a complex biological sample.
No specific “identification” and “quantification” runs are needed in this approach, as both LC–MS and MS/MS data are acquired in the same run, the benefits of which have been summarized by Andreev et al27. This approach responds to the realities and requirements of current systems biology and biomarker discovery research and is practical and relevant to a broad range of proteomic applications and mass spectrometric capabilities.
Supplementary Material
ACKNOWLEDGMENT
We are grateful to Drs. S. Rhodes, K. Prince (Indiana University School of Medicine, Indianapolis, IN), and K. Rithidech (State University of New York at Stony Brook, Stony Brook, NY) for allowing us to use the data generated from their samples, and we thank Dr. Antonius Koller (Stony Brook University Proteomics Center, Stony Brook, NY) for his critical reading of the manuscript. This work was supported by the following grants: NIEHS RC2ES018810, NIGMS R01GM085218 (FAW), and NCRR R01RR024236-01A1 (HT).
Footnotes
Supporting Information
This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Wong JW, Sullivan MJ, Cagney G. Computational methods for the comparative quantification of proteins in label-free LCn-MS experiments. Briefings Bioinf. 2008;9:156–165. doi: 10.1093/bib/bbm046. [DOI] [PubMed] [Google Scholar]
- 2.Gevaert K, Impens F, Ghesquiere B, Van Damme P, Lambrechts A, Vandekerckhove J. Stable isotopic labeling in proteomics. Proteomics. 2008;8:4873–4885. doi: 10.1002/pmic.200800421. [DOI] [PubMed] [Google Scholar]
- 3.Mueller LN, Brusniak MY, Mani DR, Aebersold R. An assessment of software solutions for the analysis of mass spectrometry based quantitative proteomics data. L Proteome Res. 2008;7:51–61. doi: 10.1021/pr700758r. [DOI] [PubMed] [Google Scholar]
- 4.Panchaud A, Affolter M, Moreillon P, Kussmann M. Experimental and computational approaches to quantitative proteomics: status quo and outlook. L.Proteomics. 2008;71:19–33. doi: 10.1016/j.jprot.2007.12.001. [DOI] [PubMed] [Google Scholar]
- 5.Elliott MH, Smith DS, Parker CE, Borchers C. Current trends in quantitative proteomics. J. Mass Spectrom. 2009;44:1637–1660. doi: 10.1002/jms.1692. [DOI] [PubMed] [Google Scholar]
- 6.Moritz B, Meyer HE. Approaches for the quantification of protein concentration ratios. L Mass Spectrom. 2003;3:2208–2220. doi: 10.1002/pmic.200300581. [DOI] [PubMed] [Google Scholar]
- 7.Bantscheff M, Schirle M, Sweetman G, Rick J, Kuster B. Quantitative mass spectrometry in proteomics: a critical review. Anal. Bioanal. Chem. 2007;389:1017–1031. doi: 10.1007/s00216-007-1486-6. [DOI] [PubMed] [Google Scholar]
- 8.Neilson KA, Ali NA, Muralidharan S, Mirzaei M, Mariani M, Assadourian G, Lee A, van Sluyter SC, Haynes PA. Less label, more free: approaches in label-free quantitative mass spectrometry. Proteomics. 2011;11:535–553. doi: 10.1002/pmic.201000553. [DOI] [PubMed] [Google Scholar]
- 9.Choi H, Fermin D, Nesvizhskii AI. Significance analysis of spectral count data in label-free shotgun proteomics. Mol. Cell. Proteomics. 2008;7:2373–2385. doi: 10.1074/mcp.M800203-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhu W, Smith JW, Huang CM. Mass spectrometry-based label-free quantitative proteomics. J. Biomed. Biotechnol. 2010;2010:840518. doi: 10.1155/2010/840518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Griffin NM, Yu J, Long F, Oh P, Shore S, Li Y, Koziol JA, Schnitzer JE. Label-free, normalized quantification of complex mass spectrometry data for proteomic analysis. Nat. Biotechnol. 2010;28:83–89. doi: 10.1038/nbt.1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang R, Barton A, Brittenden J, Huang JT, Crowther D. Evaluation of computational platforms for LS-MS based label-free quantitative proteomics: A global view. L.Proteomics Bioinf. 2010;3:260–265. [Google Scholar]
- 13.Lai X, Bacallao RL, Blazer-Yost BL, Hong D, Mason SB, Witzmann FA. Characterization of the renal cyst fluid proteome in autosomal dominant polycystic kidney disease (ADPKD) patients. Proteomics Clin.Appl. 2008;2:1140–1152. doi: 10.1002/prca.200780140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lai X, Liangpunsakul S, Crabb DW, Ringham HN, Witzmann FA. A proteomic workflow for discovery of serum carrier protein-bound biomarker candidates of alcohol abuse using LC–MS/ MS. Electrophoresis. 2009;30:2207–2214. doi: 10.1002/elps.200800775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Keller A, Nesvizhskii AI, Kolker E, Aebersold R. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem. 2002;74:5383–5392. doi: 10.1021/ac025747h. [DOI] [PubMed] [Google Scholar]
- 16.Nesvizhskii AI, Keller A, Kolker E, Aebersold RA. statistical model for identifying proteins by tandem mass spectrometry. Anal.Chem. 2003;75:4646–4658. doi: 10.1021/ac0341261. [DOI] [PubMed] [Google Scholar]
- 17.Beaton A, Tukey J. The fitting of power series, meaning polynomials, illustrated on band-spectroscopic data. Technometrics. 1974;16:147–185. [Google Scholar]
- 18.Monroe ME, Shaw JL, Daly DS, Adkins JN, Smith RD. MASIC: A software program for fast quantitation and flexible visualization of chromatographic profiles from detected LC-MS(/MS) features. Comput. Biol. Chem. 2008;32:215–217. doi: 10.1016/j.compbiolchem.2008.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fu Y, Yan SC, Huang TS. Correlation metric for generalized feature extraction. IEEE Trans.Pattern Anal.Mach.Intell. 2008;30:2229–2235. doi: 10.1109/TPAMI.2008.154. [DOI] [PubMed] [Google Scholar]
- 20.Palagi PM, Walther D, Quadroni M, Catherinet S, Burgess J, Zimmermann-Ivol CG, Sanchez JC, Binz PA, Hochstrasser DF, Appel RD. MSight: an image analysis software for liquid chromatography-mass spectrometry. Pjroteomics. 2005;5:2381–2384. doi: 10.1002/pmic.200401244. [DOI] [PubMed] [Google Scholar]
- 21.MacCoss MJ, Wu CC. Proteomic solutions for analytical challenges associated with alcohol research. Alcohol Res. Health. 2008;31:251–255. [PMC free article] [PubMed] [Google Scholar]
- 22.Berth M, Moser FM, Kolbe M, Bernhardt J. The state of the art in the analysis of two-dimensional gel electrophoresis images. Appl. Microbiol.Biotechnol. 2007;76:1223–1243. doi: 10.1007/s00253-007-1128-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kohlbacher O, Reinert K, Gropl C, Lange E, Pfeifer N, Schulz-Trieglaff O, Sturm M. TOPP–the OpenMS proteomics pipeline. Bioinformatics. 2007;23:e191–e197. doi: 10.1093/bioinformatics/btl299. [DOI] [PubMed] [Google Scholar]
- 24.Leptos KC, Sarracino DA, Jaffe JD, Krastins B, Church GM. MapQuant: open-source software for large-scale protein quantification. Proteomics. 2006;6:1770–1782. doi: 10.1002/pmic.200500201. [DOI] [PubMed] [Google Scholar]
- 25.May D, Law W, Fitzgibbon M, Fang Q, McIntosh M. Software platform for rapidly creating computational tools for mass spectrometry-based proteomics. L.Proteome Res. 2009;8:3212–3217. doi: 10.1021/pr900169w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Katajamaa M, Miettinen J, Oresic M. MZmine: toolbox for processing and visualization of mass spectrometry based molecular profile data. Bioinformatics. 2006;22:634–6. doi: 10.1093/bioinformatics/btk039. [DOI] [PubMed] [Google Scholar]
- 27.Andreev VP, Li LY, Cao L, Gu Y, Rejtar T, Wu SL, Karger BL. A new algorithm using cross-assignment for label-free quantitation with LC-LTQ-FT MS. J. Proteome Res. 2007;6:2186–2194. doi: 10.1021/pr0606880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mueller LN, Rinner O, Schmidt A, Letarte S, Bodenmiller B, Brusniak MY, Vitek O, Aebersold R, Muller M. SuperHirn–a novel tool for high resolution LC-MS-based peptide/protein profiling. Proteomics. 2007;7:3470–3480. doi: 10.1002/pmic.200700057. [DOI] [PubMed] [Google Scholar]
- 29.Jaffe JD, Mani DR, Leptos KC, Church GM, Gillette MA, Carr SA. PEPPeR, a platform for experimental proteomic pattern recognition. Mol,cell.Proteomics. 2006;5:1927–1941. doi: 10.1074/mcp.M600222-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tsou CC, Tsai CF, Tsui YH, Sudhir PR, Wang YT, Chen YJ, Chen JY, Sung TY, Hsu WL. IDEAL-Q, an automated tool for label-free quantitation analysis using an efficient peptide alignment approach and spectral data validation. Mol. Cell. Proteomics. 2010;9:131–144. doi: 10.1074/mcp.M900177-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang YT, Tsai CF, Hong TC, Tsou CC, Lin PY, Pan SH, Hong TM, Yang PC, Sung TY, Hsu WL, Chen YJ. An informatics-assisted label-free quantitation strategy that depicts phosphoproteomic profiles in lung cancer cell invasion. J. Proteome Res. 2010;9:5582–5597. doi: 10.1021/pr100394u. [DOI] [PubMed] [Google Scholar]
- 32.Mann B, Madera M, Sheng Q, Tang H, Mechref Y, Novotny MV. ProteinQuant Suite: a bundle of automated software tools for label-free quantitative proteomics. Rapid Commun. Mass Spectrom. 2008;22:3823–3834. doi: 10.1002/rcm.3781. [DOI] [PubMed] [Google Scholar]
- 33.Cui X, Kerr MK, Churchill GA. Transformations for cDNA microarray data. Stat. Appl. Genet. Mol. Biol. 2003;2 doi: 10.2202/1544-6115.1009. Article4. [DOI] [PubMed] [Google Scholar]
- 34.Colvin SC, Malik RE, Showalter AD, Sloop KW, Rhodes SJ. Model of pediatric pituitary hormone deficiency separates the endocrine and neural functions of the LHX3 transcription factor in vivo. Proc. Natl. Acad. Sci. U.S.A. 2011;108:173–178. doi: 10.1073/pnas.1009501108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Richardson MR, Segu ZM, Price MO, Lai X, Witzmann FA, Mechref Y, Yoder MC, Price FW. Alterations in the aqueous humor proteome in patients with Fuchs endothelial corneal dystrophy. Mol, vision. 2010;16:2376–2383. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.