

Abstract
One of the criteria for inferring a species tree from a collection of gene trees, when gene tree incongruence is assumed to be due to incomplete lineage sorting (ILS), is Minimize Deep Coalescence (MDC). Exact algorithms for inferring the species tree from rooted, binary trees under MDC were recently introduced. Nevertheless, in phylogenetic analyses of biological data sets, estimated gene trees may differ from true gene trees, be incompletely resolved, and not necessarily rooted. In this article, we propose new MDC formulations for the cases where the gene trees are unrooted/binary, rooted/non-binary, and unrooted/non-binary. Further, we prove structural theorems that allow us to extend the algorithms for the rooted/binary gene tree case to these cases in a straightforward manner. In addition, we devise MDC-based algorithms for cases when multiple alleles per species may be sampled. We study the performance of these methods in coalescent-based computer simulations.
Key words: algorithms, coalescence, dynamic programming, graph theory, phylogenetic trees
1. Introduction
Biologists have long acknowledged that the evolutionary history of a set of species—the species tree—and that of a genomic region from those species—the gene tree—need not be congruent (Maddison, 1997). While many processes can cause gene/species tree incongruence, such as horizontal gene transfer and gene duplication/loss, we focus in this article on incomplete lineage sorting (ILS), which is best understood under the coalescent model (Nei, 1986; Tajima, 1983; Takahata, 1989), as we illustrate in Figure 1.
FIG. 1.

Gene/species tree incongruence due to ILS. Given species tree ST, with constant population size throughout and time t in coalescent units (number of generations divided by the population size) between the two divergence events, each of the three gene tree topologies gt1, gt2, and gt3 may be observed, with probabilities 1 − (2/3)e−t, (1/3)e−t, and (1/3)e−t, respectively.
Use of DNA sequence data for inferring phylogenetic relationships among species is central in biology; however, the evolution of DNA is complex, and this complexity is almost never fully taken into account in phylogenetic inference from DNA sequence data. Of particular importance in phylogenomic analyses, the evolution of the DNA regions used for phylogenetic inference (often genes, but not always) need not be congruent with the evolution of the species. This is the classic gene tree/species tree problem (Maddison, 1997; Degnan and Rosenberg, 2009a). Errors in estimating gene trees (due, for example, to inadequate sequence length, improper phylogeny estimation methods, or insufficient computational resources) can produce estimated gene trees that differ from the true gene trees, and so from the species tree even when the true gene tree and species tree are identical. In addition, however, many biological factors can cause true gene trees to be different from the true species trees. For example, horizontal gene transfer, gene duplication/loss, and ILS can result in gene/species tree incongruence. In this article, we focus on ILS as the sole biological cause of such incongruence (Fig. 1).
ILS is best understood under the coalescent model (Degnan and Rosenberg, 2006; Degnan and Salter, 2005; Hudson, 1983; Nei, 1986, 1987; Rosenberg, 2002; Tajima, 1983; Takahata, 1989). The coalescent model views gene lineages moving backward in time, eventually coalescing down to one lineage. In each time interval between species divergences (e.g., t in Fig. 1), lineages entering the interval from a more recent time period may or may not coalesce—an event whose probability is determined largely by the population size and branch lengths.
Thus, a gene tree is viewed as a random variable conditional on a species tree. For the species tree ((AB)C), with time t between species divergences, the three possible outcomes for the gene tree topology random variable, along with their probabilities, are shown in Figure 1.
The population genetics community has long recognized the existence of gene tree discordance (Hudson, 1983; Nei, 1986; Tajima, 1983). It has also been known that this phenomenon can sometimes cause severe errors to be made by phylogenetic inference procedures (Pamilo and Nei, 1988; Takahata, 1989; Wu, 1991). However, for most species, it has until recently been difficult to gather data on more than a single part of the genome (Driskell et al., 2004). With the advent of technologies that make it possible to obtain large amounts of sequence data from multiple species, multi-locus data are becoming widely available, highlighting the issue of gene tree discordance (Degnan and Rosenberg, 2009b; Kuo et al., 2008; Pollard et al., 2006; Rokas et al., 2003; Syring et al., 2005; Than et al., 2008b).
Several methods have been introduced for inferring a species tree from a collection of gene trees under ILS-based incongruence. Summary statistics, such as the majority-rule consensus (Degnan et al., 2009; Kuo et al., 2008) and democratic vote (Dawkins, 2004; Degnan and Rosenberg, 2006; Wu, 1991, 1992), are fast to compute and provide a good estimate of the species tree in many cases. However, the accuracy of these methods suffers under certain conditions. Further, these methods do not provide explicit reconciliation scenarios; rather, they provide summaries of the gene trees. Recently, methods that explicitly model ILS were introduced, such as Bayesian inference (Edwards et al., 2007; Liu and Pearl, 2007), maximum likelihood (Kubatko et al., 2009), and the maximum parsimony criterion Minimize Deep Coalescence (MDC) (Maddison, 1997; Maddison and Knowles, 2006; Than et al., 2008b). We introduced the first exact algorithms for inferring species trees under the MDC criterion from a collection of rooted, binary gene trees (Than and Nakhleh, 2009, 2010). Nevertheless, in phylogenetic analyses of biological data sets, estimated gene trees may differ from the true gene trees, may be incompletely resolved, and may not be rooted. Requiring gene trees to be fully resolved may result in gene trees with wrong branching patterns (e.g., those branches with low bootstrap support) that masquerade as true gene/species tree incongruence, thus resulting in over- and possibly under-estimation of deep coalescences.
Here we propose an approach to estimating species trees from estimated gene trees that avoids these problems. Instead of assuming that all gene trees are correct (and hence fully resolved, rooted trees), we consider the case where all gene trees are modified so that they are reasonably likely to be unrooted, edge-contracted versions of the true gene trees. For example, the reliable edges in the gene trees can be identified using statistical techniques, such as bootstrapping, and all low-support edges can be contracted. In this way, the MDC problem becomes one in which the input is a set of gene trees that may not be rooted and may not be fully resolved, and the objective is a rooted, binary species tree and binary rooted refinements of the input gene trees that optimizes the MDC criterion. We provide exact algorithms and heuristics for inferring species trees for these cases. Further, Than and Nakhleh had extended the MDC criterion and devised an algorithm for cases where multiple alleles (or no alleles) per species may be sampled. Here, we establish that the algorithm is exact, in that it finds a species tree that minimizes the amount of deep coalescences when multiple alleles may be involved in the analysis.
We have implemented several of these algorithms and heuristics in our PhyloNet software package (Than et al., 2008a), which is publicly available at http://bioinfo.cs.rice.edu/phylonet, and we evaluate the performance of these algorithms and heuristics on synthetic data.
2. Preliminary Material
Clades and clusters
Throughout this section, unless specified otherwise, all trees are presumed to be rooted binary trees, bijectively leaf-labelled by the elements of 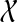 (that is, each
(that is, each 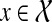 labels one leaf in each tree). We denote by
labels one leaf in each tree). We denote by 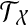 the set of all binary rooted trees on leaf-set
the set of all binary rooted trees on leaf-set 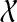 . We denote by V(T), E(T), and L(T) the node-set, edge-set, and leaf-set, respectively, of T. For v a node in T, we define parent(v) to be the parent of v in T, and Children(v) to be the children of v. A clade in a tree T is a rooted subtree of T, which can be identified by the node in T rooting the clade. For a given tree T, we denote the subtree of T rooted at v by CladeT(v), and when the tree T is understood, by Clade(v). The clade for node v is Clade(v), and since nodes can have children, the children of a clade Clade(v) are the clades rooted at the children of v. The set of all clades of a tree T is denoted by Clades(T). The set of leaves in CladeT(v) is called a cluster and denoted by ClusterT(v) (or more simply by Cluster(v) if the tree T is understood). The clusters that contain either all the taxa or just single leaves are called trivial, and the other clusters are called non-trivial. The cluster of node v is Cluster(v). As with clades, clusters can also have children. If Y is a cluster in a tree T, then the clade for Y within T, denoted by CladeT(Y), is the clade of T induced by Y. The set of all clusters of T is denoted by Clusters(T). We say that edge e in gt is outside cluster Y if it satisfies
. We denote by V(T), E(T), and L(T) the node-set, edge-set, and leaf-set, respectively, of T. For v a node in T, we define parent(v) to be the parent of v in T, and Children(v) to be the children of v. A clade in a tree T is a rooted subtree of T, which can be identified by the node in T rooting the clade. For a given tree T, we denote the subtree of T rooted at v by CladeT(v), and when the tree T is understood, by Clade(v). The clade for node v is Clade(v), and since nodes can have children, the children of a clade Clade(v) are the clades rooted at the children of v. The set of all clades of a tree T is denoted by Clades(T). The set of leaves in CladeT(v) is called a cluster and denoted by ClusterT(v) (or more simply by Cluster(v) if the tree T is understood). The clusters that contain either all the taxa or just single leaves are called trivial, and the other clusters are called non-trivial. The cluster of node v is Cluster(v). As with clades, clusters can also have children. If Y is a cluster in a tree T, then the clade for Y within T, denoted by CladeT(Y), is the clade of T induced by Y. The set of all clusters of T is denoted by Clusters(T). We say that edge e in gt is outside cluster Y if it satisfies 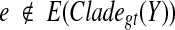 , and otherwise that it is inside Y. Given a set A ⊆ L(T), we define MRCAT (A) to be the most recent (or least) common ancestor of the taxa in A. Finally, given trees t and T, both on
, and otherwise that it is inside Y. Given a set A ⊆ L(T), we define MRCAT (A) to be the most recent (or least) common ancestor of the taxa in A. Finally, given trees t and T, both on  , we define H : V(t) → V(T) by HT(v) = MRCAT(Clustert(v)).
, we define H : V(t) → V(T) by HT(v) = MRCAT(Clustert(v)).
We extend the definitions of Clades(T) and Clusters(T) to the case where T is unrooted by defining Clades(T) to be the set of all clades of all possible rootings of T, and Clusters(T) to be the set of all clusters of all possible rootings of T. Thus, the sets Clades(T) and Clusters(T) depend upon whether T is rooted or not.
Given a cluster 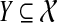 of T, the parent edge of Y within T is the edge incident with the root of the clade for Y, but which does not lie within the clade. When T is understood by context, we will refer to this as the parent edge of Y.
of T, the parent edge of Y within T is the edge incident with the root of the clade for Y, but which does not lie within the clade. When T is understood by context, we will refer to this as the parent edge of Y.
A set 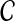 of clusters is said to be compatible if there is a rooted tree T on leaf-set S such that
of clusters is said to be compatible if there is a rooted tree T on leaf-set S such that 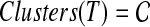 . By Semple and Steel (2003), the set
. By Semple and Steel (2003), the set 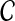 is compatible if and only if every pair A and B of clusters in
is compatible if and only if every pair A and B of clusters in 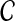 are either disjoint or one contains the other.
are either disjoint or one contains the other.
Valid coalescent histories and extra lineages
Given gene tree gt and species tree ST, a valid coalescent history is a function f: V(gt) → V(ST) such that the following conditions hold:
if w is a leaf in gt, then f (w) is the leaf in ST with the same label; and,
if w is a vertex in Cladegt(v), then f (w) is a vertex in CladeST( f (v)).
Note that, these two conditions together imply that f (v) is a node on the path between the root of ST and the MRCA in ST of Clustergt(v). Given a gene tree gt and a species tree ST and given a function f defining a valid coalescent history of gt within ST, the number of lineages on each edge in ST can be computed by inspection.
Notice that, in a rooted tree, each edge (u, v) is uniquely associated with, or identified by, its head node, v. Further, when multiple coalescence events occur within a branch in the species tree, the order in which these events occur does not matter under the MDC criterion. Based on these two observations, we always map coalescence events to nodes. Let e = (u, v) be an edge in the species tree, and x and y be the two children of v. If lx lineages enter edge e from the edge (v, x) and ly lineages enter edge e from the edge (v, y), and q coalescence events are mapped to node v under a given valid coalescent history, then the number of lineages in edge e is lx + ly − q (Fig. 2).
FIG. 2.

Under the coalescent, time flows backward from the leaves towards the root. In a valid coalescent history, the number of lineages in edge e = (u, v) equals the sum of the numbers of lineages entering e, when going backward in time, from edges (v, x) and (v, y), minus the number of coalescence events that occur at node v.
An optimal valid coalescent history is one that results in the minimum number of lineages over all valid coalescent histories. We denote the number of extra lineages on an edge 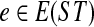 (one less than the number of lineages on e) in an optimal valid coalescent history of gt within ST by XL(e, gt), and we denote by XL(ST, gt) the total number of extra lineages within an optimal valid coalescent history of gt within ST, i.e.,
(one less than the number of lineages on e) in an optimal valid coalescent history of gt within ST by XL(e, gt), and we denote by XL(ST, gt) the total number of extra lineages within an optimal valid coalescent history of gt within ST, i.e., 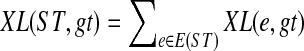 (Fig. 3). Finally, we denote by
(Fig. 3). Finally, we denote by 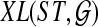 the total number of extra lineages, or MDC score, over all gene trees in
the total number of extra lineages, or MDC score, over all gene trees in  , so
, so 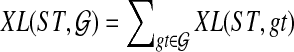 . Given gene tree gt and species tree ST, finding the valid coalescent history that yields the smallest number of extra lineages is achievable in polynomial time, as we now show. Given cluster A in gt and cluster B in ST, we say that A is B-maximal if (1) A ⊆ B and (2) for all
. Given gene tree gt and species tree ST, finding the valid coalescent history that yields the smallest number of extra lineages is achievable in polynomial time, as we now show. Given cluster A in gt and cluster B in ST, we say that A is B-maximal if (1) A ⊆ B and (2) for all 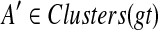 , if A ⊂ A′ then A′⊈B. We set kB(gt) to be the number of B-maximal clusters within gt. Finally, we say that cluster A is ST-maximal if there is a cluster
, if A ⊂ A′ then A′⊈B. We set kB(gt) to be the number of B-maximal clusters within gt. Finally, we say that cluster A is ST-maximal if there is a cluster 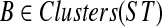 such that
such that 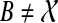 and A is B-maximal.
and A is B-maximal.
FIG. 3.

Illustration of optimal and non-optimal reconciliations of a rooted, binary gene tree gt with a rooted, binary species tree ST, which yield 1 and 4 extra lineages, respectively.
Theorem 1
(Than and Nakhleh, 2009). Let gt be a gene tree, ST be a species tree, both binary rooted trees on leaf-set X. Let B be a cluster in ST and let e be the parent edge of B in ST. Then kB(gt) is equal to the number of lineages on e in an optimal valid coalescent history. Therefore, XL(e, gt) = kB(gt) − 1, and 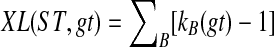 , where B ranges over the clusters of ST. Furthermore, a valid coalescent history f that achieves this total number of extra lineages can be produced by setting f(v) = HST(v) (i.e., f(v) = M RCAST(Clustergt(v))) for all v.
, where B ranges over the clusters of ST. Furthermore, a valid coalescent history f that achieves this total number of extra lineages can be produced by setting f(v) = HST(v) (i.e., f(v) = M RCAST(Clustergt(v))) for all v.
In other words, we can score a candidate species tree ST with respect to a set 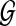 of rooted binary trees with
of rooted binary trees with  . Finally,
. Finally,
Corollary 1
Given collection  of k gene trees and species tree ST, each tree labelled by the species in
of k gene trees and species tree ST, each tree labelled by the species in  , we can compute the optimal coalescent histories relating each gene tree to ST so as to minimize the total number of extra lineages in O(nk) time, and the MDC score of these optimal coalescent histories in O(nk) time, where
, we can compute the optimal coalescent histories relating each gene tree to ST so as to minimize the total number of extra lineages in O(nk) time, and the MDC score of these optimal coalescent histories in O(nk) time, where 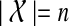 .
.
The analysis of the running time follows from the following two lemmas:
Lemma 1
Given a rooted gene tree gt and a rooted binary species tree ST, we can compute all HST(v) (letting v range over V (gt)) in O(n) time.
Proof
We begin by preprocessing both gt and ST in O(n) time so that each subsequent MRCA query takes constant time (Harel and Tarjan, 1984; Bender and Farach-Colton, 2000). Then, for each node 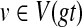 , we can compute HST(v) in O(1) time. Thus, we can compute the optimal coalescent history relating gt to ST in O(n) time.
, we can compute HST(v) in O(1) time. Thus, we can compute the optimal coalescent history relating gt to ST in O(n) time.
Lemma 2
Given a rooted gene tree gt and a rooted binary species tree ST, and assuming that HST(v), for each 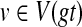 has been computed, we can compute XL(ST, gt) in O(n) time.
has been computed, we can compute XL(ST, gt) in O(n) time.
Proof
We define the function 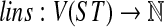 as follows. For a vertex
as follows. For a vertex 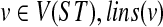 is the number of lineages in the parent edge of node v given an optimal valid coalescent history (i.e., under the HST mapping).
is the number of lineages in the parent edge of node v given an optimal valid coalescent history (i.e., under the HST mapping).
Denote by coalv the number of gene tree nodes mapped to node v under the HST mapping. That is, 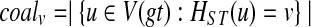 . The function lins can be computed recursively as
. The function lins can be computed recursively as
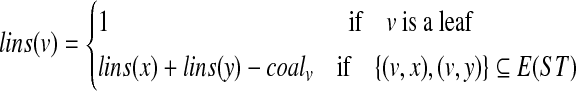 |
(1) |
We now prove that (1) for every node 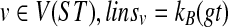 where B = Cluster(v) in ST, and (2) lins is computable in O(n) time.
where B = Cluster(v) in ST, and (2) lins is computable in O(n) time.
The proof of (1) uses strong induction on the height of node v, where the height of v is the length of the longest path from v to any leaf in Cluster(v). For the base case, let v be a node of height 1, and its two children (which are leaves) x and y be labeled lx and ly, respectively. For this base case, lins(x) = lins(y) = coalx = coaly = 1. If {lx, ly} is a cluster in gt, then kB(gt) = 1, in which case the algorithm sets lins(v) to lins(x) + lins(y) − coalv = 1 + 1 − 1 = 1, since coalv = 1. If {lx, ly} is not a cluster in gt, then kB(gt) = 2, and the algorithm sets lins(v) to lins(x) + lins(y) − coalv = 1 + 1 − 0 = 2, since no coalescence event occurs at node v. Therefore, kB(gt) = lins(v) for nodes v of height 1 in ST.
Now assume that kB(gt) = lins(v) for every node v of height at most p, and let w be a node in ST of height p + 1, with B = cluster(w). Let x and y be w's two children, with Bx = Cluster(x) and By = Cluster(y). Clearly, the height of x and y is smaller than p. By the induction hypothesis, kBx(gt) = lins(x) and kBy(gt) = lins(y). By Theorem 1, kB(gt) is equal to the number of lineages on the parent edge of B in ST, which is the sum of the numbers of lineages on the parent edges of Bx and By, minus the number of coalescence events that occur at node w, i.e., kB(gt) = kBx(gt) + kBy(gt) − coalw. By the strong induction hypothesis, this is identical to lins(x) + lins(y) − coalw. By Equation (1), we have lins(x) + lins(y) −coalw = lins(w). This completes the proof that kB(gt) = lins(w).
For the second part, notice that computing the values of lins for all nodes in ST can be achieved by a bottom-up algorithm that traverses each node 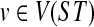 exactly once. For each node v, the values of lins(x) and lins(y) of its children x and y are already computed, and the value of coalv is already computed via the HST mapping. Thus, the algorithm takes O(n) time. ▪
exactly once. For each node v, the values of lins(x) and lins(y) of its children x and y are already computed, and the value of coalv is already computed via the HST mapping. Thus, the algorithm takes O(n) time. ▪
2.1. MDC on rooted binary gene trees: the single-allele case
The MDC problem, formulated by Maddison (1997), is equivalent to finding a species tree that minimizes the total number of extra lineages over all gene trees in 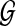 . Thus, the MDC problem can be stated as follows: given a set
. Thus, the MDC problem can be stated as follows: given a set 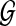 of rooted, binary gene trees, we seek a species tree ST such that
of rooted, binary gene trees, we seek a species tree ST such that 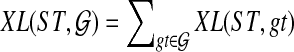 is minimized.
is minimized.
MDC is conjectured to be NP-hard, and no polynomial-time exact algorithm is known for this problem. However, it can be solved exactly using several techniques, as we now show.
Algorithms for MDC
The material in this section is from Than and Nakhleh (2009). The simplest technique to compute the optimal species tree with respect to a set 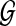 of gene trees is to compute a minimum-weight clique of size n − 2 (where
of gene trees is to compute a minimum-weight clique of size n − 2 (where 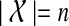 ) in a graph which we now describe. Let
) in a graph which we now describe. Let 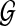 be the set of gene trees in the input to MDC, and let
be the set of gene trees in the input to MDC, and let 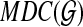 be the graph with one vertex for each non-trivial subset of
be the graph with one vertex for each non-trivial subset of 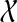 (so
(so 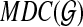 does not contain trivial clusters), edges between A and B if the two clusters are compatible (and so A ∩ B = 0̸, A ⊂ B, or B ⊂ A). A clique inside this graph therefore defines a set of pairwise compatible clusters, and hence a rooted tree on
does not contain trivial clusters), edges between A and B if the two clusters are compatible (and so A ∩ B = 0̸, A ⊂ B, or B ⊂ A). A clique inside this graph therefore defines a set of pairwise compatible clusters, and hence a rooted tree on 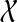 . We set the weight of each node A to be
. We set the weight of each node A to be 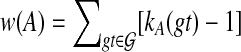 . We seek a clique of size n − 2, and among all such cliques we seek one of minimum weight. By construction, the clique will define a rooted, binary tree ST such that
. We seek a clique of size n − 2, and among all such cliques we seek one of minimum weight. By construction, the clique will define a rooted, binary tree ST such that 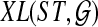 is minimized.
is minimized.
The graph 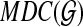 contains 2n − n − 1 vertices, where
contains 2n − n − 1 vertices, where 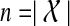 , and is therefore large even for relatively small n. We can constrain this graph size by restricting the allowable clusters to a smaller set,
, and is therefore large even for relatively small n. We can constrain this graph size by restricting the allowable clusters to a smaller set, 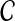 , of subsets of
, of subsets of 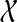 . For example, we can set
. For example, we can set 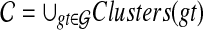 (minus the trivial clusters), and we can define
(minus the trivial clusters), and we can define 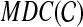 to be the subgraph of
to be the subgraph of 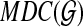 defined on the vertices corresponding to
defined on the vertices corresponding to 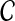 . However, the cliques of size n − 2 in the graph
. However, the cliques of size n − 2 in the graph 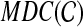 may not have minimum possible weights; therefore, instead of seeking a minimum weight clique of size n − 2 within
may not have minimum possible weights; therefore, instead of seeking a minimum weight clique of size n − 2 within 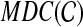 , we will set the weight of node A to be w′(A) = Q −w(A), for some very large Q, and seek a maximum weight clique within the graph.
, we will set the weight of node A to be w′(A) = Q −w(A), for some very large Q, and seek a maximum weight clique within the graph.
Finally, we can also solve the problem exactly using dynamic programming. For 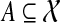 and binary rooted tree T on leaf-set A, we define
and binary rooted tree T on leaf-set A, we define
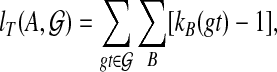 |
where B ranges over all clusters of T. We then set
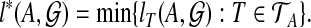 |
By Theorem 1, 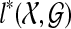 is the minimum number of extra lineages achievable in any species tree on
is the minimum number of extra lineages achievable in any species tree on 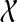 , and so any tree T such that
, and so any tree T such that 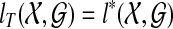 is a solution to the MDC problem on input
is a solution to the MDC problem on input 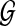 . We now show how to compute
. We now show how to compute 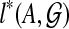 for all
for all 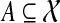 using dynamic programming. By backtracking, we can then compute the optimal species tree on
using dynamic programming. By backtracking, we can then compute the optimal species tree on 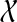 with respect to the set
with respect to the set 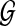 of gene trees.
of gene trees.
Consider a binary rooted tree T on leaf-set A that gives an optimal score for 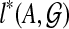 , and let the two subtrees off the root of T be T1 and T2 with leaf sets A1 and A2 = A − A1, respectively. Then, letting B range over the clusters of T, we obtain
, and let the two subtrees off the root of T be T1 and T2 with leaf sets A1 and A2 = A − A1, respectively. Then, letting B range over the clusters of T, we obtain
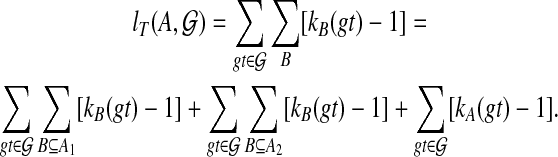 |
If for i = 1 or 2, 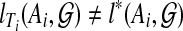 , then we can replace Ti by a different tree on Ai and obtain a tree T′ on A such that
, then we can replace Ti by a different tree on Ai and obtain a tree T′ on A such that 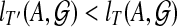 , contradicting the optimality of T. Thus,
, contradicting the optimality of T. Thus, 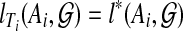 for i = 1, 2, and so
for i = 1, 2, and so 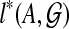 is obtained by taking the minimum over all sets
is obtained by taking the minimum over all sets 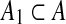 of
of 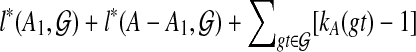 . In other words, we have proven the following:
. In other words, we have proven the following:
Lemma 3
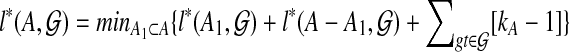 .
.
This lemma suggests the dynamic programming algorithm:
Order the subsets of
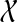 by cardinality, breaking ties arbitrarily.
by cardinality, breaking ties arbitrarily.Compute kA(gt) for all
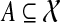 and
and 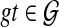 .
.For all singleton sets A, set
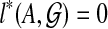 .
.For each subset with at least two elements, from smallest to largest, compute
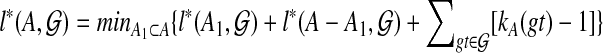 .
.Return
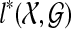 .
.
There are 2n − 1 subproblems to compute (one for each set A) and each takes O(2nn) time (there are at most 2n subsets A1 of A, and each pair A, A1 involves computing kA for each 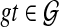 , which costs O(n) time). Hence, the running time is O(n22n) time. A tighter bound of O(2n + 3n) can also be achieved. This follows from the fact that for each value of i, for 1 ≤ i ≤ n, we have
, which costs O(n) time). Hence, the running time is O(n22n) time. A tighter bound of O(2n + 3n) can also be achieved. This follows from the fact that for each value of i, for 1 ≤ i ≤ n, we have  sets of size i, and for each of these sets, we need to consider all its subsets (there are 2i of them) and score the number of extra lineages.
sets of size i, and for each of these sets, we need to consider all its subsets (there are 2i of them) and score the number of extra lineages.
However, Than and Nakhleh (2009, 2010) showed that using only the clusters of the gene trees would produce almost equally good estimates of the species tree.
3. MDC On Estimated Gene Trees: The Single-Allele Case
Estimating gene trees with high accuracy is a challenging task, particularly in cases where branch lengths are very short (which are also cases under which ILS is very likely to occur). As a result, gene tree estimates are often unrooted, unresolved, or both. To deal with these practical cases, we formulate the problems as estimating species trees and completely resolved, rooted versions of the input trees to optimize the MDC criterion. We show that the clique-based and DP algorithms can still be applied.
3.1. Unrooted, binary gene trees
When reconciling an unrooted, binary gene tree with a rooted, binary species tree under parsimony, it is natural to seek the rooting of the gene tree that results in the minimum number of extra lineages over all possible rootings (Fig. 4). In this case, the MDC problem can be formulated as follows: given a set 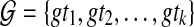 of gene trees, each of which is unrooted, binary, with leaf-set
of gene trees, each of which is unrooted, binary, with leaf-set 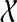 , we seek a species tree ST and set
, we seek a species tree ST and set 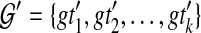 , where
, where 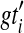 is a rooted version of gti, so that
is a rooted version of gti, so that 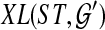 is minimum over all such sets
is minimum over all such sets 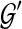 .
.
FIG. 4.

Illustration of optimal and non-optimal reconciliations of an unrooted, binary gene tree gt with a rooted, binary species tree ST, which yield 1 and 3 extra lineages, respectively.
Given a species tree and a set of unrooted gene trees, it is easy to compute the optimal rootings of each gene tree with respect to the given species tree, since there are only O(n) possible locations for the root in an n leaf tree, and for each possible rooting we can compute the score of that solution in O(n2) time. Thus, it is possible to compute the optimal rooting and its score in O(n3) time. Here we show how to solve this problem more efficiently—finding the optimal rooting in O(n) time, and the score for the optimal rooting in O(n2) time, thus saving a factor of n. We accomplish this using a small modification to the techniques used in the case of rooted gene trees.
We begin by extending the definition of B-maximal clusters to the case of unrooted gene trees, for B a cluster in a species tree ST, in the obvious way. Recall that the set Clusters(gt) depends on whether gt is rooted or not, and that kB(gt) is the number of B-maximal clusters in gt. We continue with the following:
Lemma 4
Let gt be an unrooted binary gene tree on 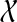 and let ST be a rooted binary species tree on
and let ST be a rooted binary species tree on 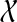 . Let
. Let 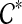 be the set of ST-maximal clusters in gt. Let e be any edge of gt such that
be the set of ST-maximal clusters in gt. Let e be any edge of gt such that 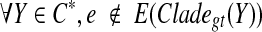 (i.e., e is not inside any subtree of gt induced by one of the clusters in
(i.e., e is not inside any subtree of gt induced by one of the clusters in 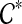 ). Then the tree gt′ produced by rooting gt on edge e satisfies (1)
). Then the tree gt′ produced by rooting gt on edge e satisfies (1) 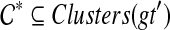 , and (2)
, and (2) 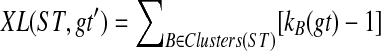 , which is the best possible. Furthermore, there is at least one such edge e in gt.
, which is the best possible. Furthermore, there is at least one such edge e in gt.
Proof
We begin by showing that there is at least one edge e that is outside Y for all 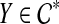 . Pick a cluster
. Pick a cluster 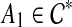 that is maximal (i.e., it is not a subset of any other cluster in
that is maximal (i.e., it is not a subset of any other cluster in 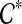 ); we will show that the parent edge of A1 is outside all clusters in
); we will show that the parent edge of A1 is outside all clusters in 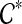 . Suppose e is inside cluster
. Suppose e is inside cluster 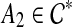 . Since A1 is maximal, it follows that A2 ⊈ A1. However, if the parent edge of A2 is not inside A1, then either A2 is disjoint from A1 or A2 contains A1, neither of which is consistent with the assumptions that A1 is maximal and the parent edge of A1 is inside A2. Therefore, the parent edge of A2 must be inside A1. In this case, A1 ∩ A2 ≠ 0̸ and
. Since A1 is maximal, it follows that A2 ⊈ A1. However, if the parent edge of A2 is not inside A1, then either A2 is disjoint from A1 or A2 contains A1, neither of which is consistent with the assumptions that A1 is maximal and the parent edge of A1 is inside A2. Therefore, the parent edge of A2 must be inside A1. In this case, A1 ∩ A2 ≠ 0̸ and 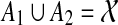 . Let Bi be the cluster in ST such that Ai is Bi-maximal, i = 1,2. Then B1 ∩ B2 ≠ 0̸, and so without loss of generality B1 ⊆ B2. But then A1 ∪ A2 ⊆ B1 ∪ B2 = B2 and so
. Let Bi be the cluster in ST such that Ai is Bi-maximal, i = 1,2. Then B1 ∩ B2 ≠ 0̸, and so without loss of generality B1 ⊆ B2. But then A1 ∪ A2 ⊆ B1 ∪ B2 = B2 and so 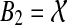 . But
. But 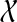 is the only
is the only 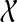 -maximal cluster, contradicting our hypotheses. Hence, the parent edge of any maximal cluster in
-maximal cluster, contradicting our hypotheses. Hence, the parent edge of any maximal cluster in 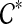 is not inside any cluster in
is not inside any cluster in 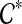 .
.
We now show that rooting gt on any edge e that is not inside any cluster in 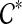 satisfies
satisfies 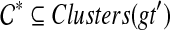 . Let e be any such edge, and let gt′ be the result of rooting gt on e. Under this rooting, the two children of the root of gt′ define subtrees T1, with cluster A1, and T2, with cluster A2. Now, suppose
. Let e be any such edge, and let gt′ be the result of rooting gt on e. Under this rooting, the two children of the root of gt′ define subtrees T1, with cluster A1, and T2, with cluster A2. Now, suppose 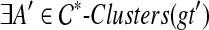 . Since
. Since 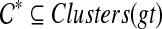 , it follows that A′ is the complement of a cluster
, it follows that A′ is the complement of a cluster 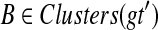 . If B is a proper subset of either A1 or A2, then the subtree of gt induced by A′ contains edge e (since
. If B is a proper subset of either A1 or A2, then the subtree of gt induced by A′ contains edge e (since 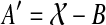 ), contradicting how we selected e. Hence, it must be that B = A1 or B = A2. However, in this case, A′ is also equal to either A1 or A2, and hence
), contradicting how we selected e. Hence, it must be that B = A1 or B = A2. However, in this case, A′ is also equal to either A1 or A2, and hence 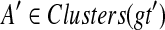 , contradicting our hypothesis about A′.
, contradicting our hypothesis about A′.
We finish the proof by showing that XL(ST, gt′) is optimal for all such rooted trees gt′, and that all other locations for rooting gt produce a larger number of extra lineages. By Theorem 1, 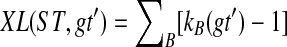 , as B ranges over the clusters of ST. By construction, this is exactly
, as B ranges over the clusters of ST. By construction, this is exactly 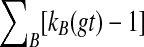 , as B ranges over the clusters of ST. Also note that for any rooted version gt* of gt, kB(gt*) ≥ kB(gt), so that this is optimal. Now consider a rooted version gt* in which the root is on an edge that is inside some subtree of gt induced by
, as B ranges over the clusters of ST. Also note that for any rooted version gt* of gt, kB(gt*) ≥ kB(gt), so that this is optimal. Now consider a rooted version gt* in which the root is on an edge that is inside some subtree of gt induced by 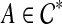 . Let gt* have subtrees T1 and T2 with clusters A1 and A2, respectively. Without loss of generality, assume that A1 ⊂ A, and that A2 ∩ A ≠ 0̸. Since
. Let gt* have subtrees T1 and T2 with clusters A1 and A2, respectively. Without loss of generality, assume that A1 ⊂ A, and that A2 ∩ A ≠ 0̸. Since 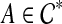 , there is a cluster
, there is a cluster 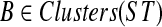 such that A is B-maximal. But then A1 is B-maximal. However, since A − A1 ≠ 0̸, there is also at least one B-maximal cluster Y ⊂ A within T2. Hence, kB(gt*) > kB(gt). On the other hand, for all other clusters B′ of ST,
such that A is B-maximal. But then A1 is B-maximal. However, since A − A1 ≠ 0̸, there is also at least one B-maximal cluster Y ⊂ A within T2. Hence, kB(gt*) > kB(gt). On the other hand, for all other clusters B′ of ST, 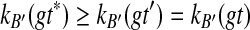 . Therefore, XL(ST, gt*) > XL(ST, gt′). In other words, any rooting of gt on an edge that is not within a subtree induced by a cluster in
. Therefore, XL(ST, gt*) > XL(ST, gt′). In other words, any rooting of gt on an edge that is not within a subtree induced by a cluster in 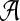 is optimal, while any rooting of gt on any other edge produces a strictly larger number of extra lineages. ▪
is optimal, while any rooting of gt on any other edge produces a strictly larger number of extra lineages. ▪
This theorem allows us to compute the optimal rooting of an unrooted binary gene tree with respect to a rooted binary species tree, and hence gives us a way of computing the score of any candidate species tree with respect to a set of unrooted gene trees:
Corollary 2
Let ST be a species tree and 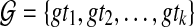 be a set of unrooted binary gene trees. Let
be a set of unrooted binary gene trees. Let 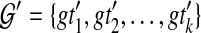 be a set of binary gene trees such that
be a set of binary gene trees such that 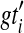 is a rooted version of gti for each
is a rooted version of gti for each 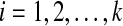 , and which minimizes
, and which minimizes 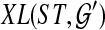 . Then
. Then 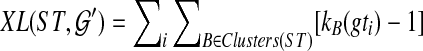 . Furthermore, the optimal
. Furthermore, the optimal 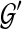 can be computed in O(nk) time, and the score of
can be computed in O(nk) time, and the score of 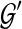 computed in O(n2k) time.
computed in O(n2k) time.
Solving MDC given unrooted, binary gene trees
Let 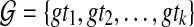 , as above. We define the MDC-score of a candidate (rooted, binary) species tree ST by
, as above. We define the MDC-score of a candidate (rooted, binary) species tree ST by  ; by Corollary 2, the tree ST* that has the minimum score will be an optimal species tree for the MDC problem on input
; by Corollary 2, the tree ST* that has the minimum score will be an optimal species tree for the MDC problem on input 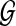 . As a result, we can use all the techniques used for solving MDC given binary rooted gene trees, since the score function is unchanged.
. As a result, we can use all the techniques used for solving MDC given binary rooted gene trees, since the score function is unchanged.
3.2. Rooted, non-binary gene trees
When reconciling a rooted, non-binary gene tree with a rooted, binary species tree under parsimony, it is natural to seek the refinement of the gene tree that results in the minimum number of extra lineages over all possible refinements (Fig. 5). In this case, the MDC problem can be formulated as follows: given a set 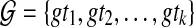 in which each gti may only be partially resolved, we seek a species tree ST and binary refinements
in which each gti may only be partially resolved, we seek a species tree ST and binary refinements 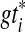 of gti so that
of gti so that 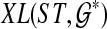 is minimized, where
is minimized, where 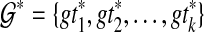 . This problem is at least as hard as the MDC problem, which is conjectured to be NP-hard.
. This problem is at least as hard as the MDC problem, which is conjectured to be NP-hard.
FIG. 5.

Illustration of optimal and non-optimal reconciliations of a rooted, non-binary gene tree gt with a rooted, binary species tree ST, which yield 0 and 3 extra lineages, respectively.
A Quadratic Algorithm for Optimal Refinement of Gene Trees Under MDC
We begin with the problem of finding an optimal refinement of a given gene tree gt with respect to a given species tree ST, with both trees rooted:
Definition 1
(Optimal tree refinement w.r.t. MDC (OTRMDC))
Input: Species tree ST and gene tree gt, both rooted and leaf-labelled by set
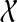 of taxa.
of taxa.Output: Binary rooted tree gt* refining gt that minimizes XL(ST, t) over all refinements t of gt. We denote gt* by OTRMDC(ST, gt).
We show that OTRMDC(ST, gt) can be solved in O(n2) time, where n is the number of leaves in either tree. For 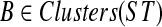 and gene tree gt, we define FB(gt) to be the number of nodes in gt that have at least one child whose cluster is B-maximal. We will show that for a given rooted gene tree gt and rooted binary species tree ST, the optimal refinement t* of gt will satisfy
and gene tree gt, we define FB(gt) to be the number of nodes in gt that have at least one child whose cluster is B-maximal. We will show that for a given rooted gene tree gt and rooted binary species tree ST, the optimal refinement t* of gt will satisfy  . Therefore, to compute the score of the optimal refinement of one gene tree gt, it suffices to compute FB(gt) for every
. Therefore, to compute the score of the optimal refinement of one gene tree gt, it suffices to compute FB(gt) for every 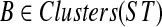 .
.
The algorithm to compute the score of the optimal refinement of gt first computes the set of B-maximal clusters, which takes O(n) time by Lemma 1. It then computes FB(gt), for each B; this requires an additional O(n) time per B, for a total cost of O(n2) time:
Algorithm for OTRMDC(ST, gt): To compute the optimal refinement, we have a slightly more complicated algorithm.
Step 1: Preprocessing. We begin by computing HST(v) for every node
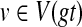 , as described above; this takes O(n) time overall.
, as described above; this takes O(n) time overall.Step 2: Refine at every high degree node. We then visit each internal node v of gt that has more than two children, and we modify the tree gt locally at v by replacing the rooted star tree at v by a tree defined by the topology induced in ST by the images under the mapping HST of v and v's children. The order in which we visit the nodes is irrelevant.
We now make precise how this modification of gt at node v is performed. We denote by Tree(ST, gt, v) the tree formed as follows. First, we compute the subtree of ST induced by the images of v and its children under the HST mapping. If a child y of v is mapped to an internal node of the induced subtree, we add a leaf ly and make it a child of HST(y); in this way, the tree we obtain has all the nodes in Children(v) identified with distinct leaves in Tree(ST, gt, v). (Although ST is assumed to be binary, Tree(ST, gt, v) may not be binary.) After we compute Tree(ST, gt, v), we modify gt by replacing the subtree of gt induced by v and its children with Tree(ST, gt, v). The subtree within the refinement that is isomorphic to Tree(ST, gt, v) is referred to as the local subtree at v.
Step 3: Completely refine if necessary. Finally, after the refinement at every node is complete, if the tree is not binary, we complete the refinement with an arbitrary refinement at v.
Theorem 2
Algorithm OTRMDC(ST, gt) takes O(n2) time, where ST and gt each have n leaves.
Proof
The first step (preprocessing the tree for constant time MRCA-queries, and computing HST(v) for every node 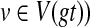 takes O(n) time. For a given node
takes O(n) time. For a given node 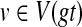 with m children, computing Tree(ST, gt, v) costs at most O(m2) time; this follows from Kannan et al. (1996) that shows that computing trees with m leaves using MRCA-queries requires only O(m2) MRCA queries and other operations. Therefore, modifying gt by refining around a given node v takes O(m2) time, where v has m children. Since the total number of edges in gt is bounded by n, the total cost of computing t* is no more than O(n2) time. ▪
with m children, computing Tree(ST, gt, v) costs at most O(m2) time; this follows from Kannan et al. (1996) that shows that computing trees with m leaves using MRCA-queries requires only O(m2) MRCA queries and other operations. Therefore, modifying gt by refining around a given node v takes O(m2) time, where v has m children. Since the total number of edges in gt is bounded by n, the total cost of computing t* is no more than O(n2) time. ▪
It is clear that the algorithm is well-defined, so that the order in which we visit the nodes in V (gt) does not impact the output.
Observation 1
Let gt be an arbitrary rooted gene tree, gt′ a refinement of gt, and ST an arbitrary rooted binary species tree. Then kB(gt′) ≥ FB(gt) for all clusters B of ST.
Proof
For a given cluster B of ST, we define an equivalence relation on the set of B-maximal clusters in the tree gt, by A1 ∼ A2 if A1 and A2 are siblings (i.e., the roots of the clades for these two clusters are siblings). It is then easy to see that the number of equivalence classes is FB(gt), and that 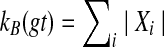 , where Xi is the ith equivalence class. For gt′ a refinement of gt, the number of B-maximal clusters can be reduced only when a new node is added to the tree and made the parent of a set of B-maximal clusters (and not parent to any clusters that are not B-maximal). Such an action reduces the number of B-maximal clusters, and also therefore the constitution of the equivalence classes. However, the number of equivalence classes cannot reduce, since the action only affects sibling sets of B-maximal clusters. (Note that it is possible for the number of equivalence classes to increase!) Therefore, kB(gt′) ≥ FB(gt′) ≥ FB(gt), since each equivalence class contributes at least one B-maximal cluster. ▪
, where Xi is the ith equivalence class. For gt′ a refinement of gt, the number of B-maximal clusters can be reduced only when a new node is added to the tree and made the parent of a set of B-maximal clusters (and not parent to any clusters that are not B-maximal). Such an action reduces the number of B-maximal clusters, and also therefore the constitution of the equivalence classes. However, the number of equivalence classes cannot reduce, since the action only affects sibling sets of B-maximal clusters. (Note that it is possible for the number of equivalence classes to increase!) Therefore, kB(gt′) ≥ FB(gt′) ≥ FB(gt), since each equivalence class contributes at least one B-maximal cluster. ▪
Theorem 3
Let gt be an arbitrary rooted gene tree, ST an arbitrary rooted binary species tree, t the result of the first two steps of OTRMDC(ST, gt), and t* an arbitrary refinement of t (thus t* = OTRMDC(ST, gt)). Then for all 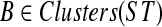 , FB(gt) = FB(t*) and no node in t or t* has more than one B-maximal child.
, FB(gt) = FB(t*) and no node in t or t* has more than one B-maximal child.
Proof
Step 2 of OTRMDC(ST, gt) can be seen as a sequence of refinements that begins with gt and ends with t, in which each refinement is obtained by refining around a particular node in gt. The tree t* = OTRMDC(ST, gt) is then obtained by refining t arbitrarily into a binary tree, if t is not fully resolved. Let the internal nodes of gt with at least three children be  . Thus,
. Thus, 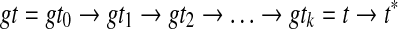 , where gti → gti+1 is the act of refining at node vi+1, and t → t* is an arbitrary refinement.
, where gti → gti+1 is the act of refining at node vi+1, and t → t* is an arbitrary refinement.
We begin by showing that FB(gti) = FB(gti+1), for 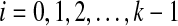 . When we refine at node vi, we modify the tree gti−1 by replacing the subtree immediately below node vi by Tree(ST, gt, vi), producing the local subtree below vi. Fix a cluster
. When we refine at node vi, we modify the tree gti−1 by replacing the subtree immediately below node vi by Tree(ST, gt, vi), producing the local subtree below vi. Fix a cluster 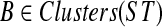 . If the cluster for vi in gti−1 does not have any B-maximal children, then refining at vi will not change FB, and hence FB(gti−1) = FB(gti). Otherwise, vi has at least one B-maximal child in gti−1. Since vi is not B-maximal within gti−1,vi also has at least one child in gti−1 that is not B-maximal. Hence, the tree gti produced by refining gti−1 at vi (using Tree(ST, gti, vi)) contains a node y that is an ancestor of all the B-maximal children of vi within gti−1 and not the ancestor of any other children of vi in gti−1. Therefore, the cluster for y is B-maximal within gti, and no other node that is introduced during this refinement is B-maximal within gti. Therefore within the local subtree at vi in gti there is exactly one node that defines a B-maximal cluster, and exactly one node that is the parent of at least one B-maximal cluster. As a result, FB(gti−1) = FB(gti).
. If the cluster for vi in gti−1 does not have any B-maximal children, then refining at vi will not change FB, and hence FB(gti−1) = FB(gti). Otherwise, vi has at least one B-maximal child in gti−1. Since vi is not B-maximal within gti−1,vi also has at least one child in gti−1 that is not B-maximal. Hence, the tree gti produced by refining gti−1 at vi (using Tree(ST, gti, vi)) contains a node y that is an ancestor of all the B-maximal children of vi within gti−1 and not the ancestor of any other children of vi in gti−1. Therefore, the cluster for y is B-maximal within gti, and no other node that is introduced during this refinement is B-maximal within gti. Therefore within the local subtree at vi in gti there is exactly one node that defines a B-maximal cluster, and exactly one node that is the parent of at least one B-maximal cluster. As a result, FB(gti−1) = FB(gti).
This argument also shows that any node in the local subtree at vi that is the parent of at least one B-maximal cluster is the parent of exactly one B-maximal cluster. On the other hand, if vi does not have any B-maximal child in gti−1, then there is no node in vi's local subtree that has any B-maximal children. In other words, after refining at node vi, any node within the local subtree at vi that has one or more B-maximal children has exactly one such child. As a result, at the end of Step 2 of OTRMDC(ST, gt), every node has at most one B-maximal child, for all 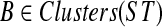 .
.
The last step of the OTRMDC algorithm produces an arbitrary refinement of t = gtk, if it is not fully resolved. But since no node in gtk can have more than one B-maximal child, if t* is a refinement of t = gtk then FB(t) = FB(t*). ▪
Theorem 4
Let gt be a rooted gene tree, ST a rooted binary species tree, both on set 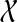 , t the result of the first two steps of OTRMDC(ST, gt), and t* any refinement of t. Then
, t the result of the first two steps of OTRMDC(ST, gt), and t* any refinement of t. Then  , and t* is a binary refinement of gt that minimizes XL(ST, t′) over all binary refinements t′ of gt.
, and t* is a binary refinement of gt that minimizes XL(ST, t′) over all binary refinements t′ of gt.
Proof
Let B be an arbitrary cluster in ST. By Theorem 3, FB(t*) = FB(gt). Also by Theorem 3, no node in t has more than one B-maximal child, and so kB(t) = FB(t). Since t* is an arbitrary refinement of t, it follows that kB(t*) = FB(t*), and so kB(t*) = FB(gt). By Observation 1, for all refinements t′ of gt, kB(t′) ≥ FB(gt). Hence kB(t′) ≥ kB(t*) for all refinements t′ of gt. Since this statement holds for an arbitrary cluster B in ST, it follows that XL(ST, t′) ≥ XL(ST, t*) for all refinements t′ of gt, establishing the optimality of t*. ▪
Corollary 3
Let ST be a species tree and 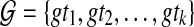 be a set of gene trees that may not be resolved. Let
be a set of gene trees that may not be resolved. Let 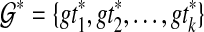 be a set of binary gene trees such that
be a set of binary gene trees such that 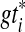 refines gti for each
refines gti for each 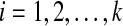 , and which minimizes
, and which minimizes 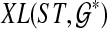 . Then
. Then 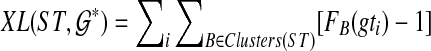 . Furthermore, the optimal resolution of each gene tree and its score can be computed in O(n2k) time.
. Furthermore, the optimal resolution of each gene tree and its score can be computed in O(n2k) time.
Solving MDC given rooted, non-binary gene trees
Corollary 3 allows us to compute the score of any species tree with respect to a set of rooted but unresolved gene trees. We can use this to find optimal species trees from rooted, non-binary gene trees, as we now show. We begin by formulating the MDC criterion for rooted, non-binary (RNB) gene trees, as follows:
Definition 2
(The MDC-RNB Problem)
Input: Set
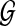 of rooted gene trees that are not necessarily binary.
of rooted gene trees that are not necessarily binary.Output: Rooted, binary species tree ST* and set
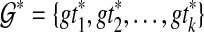 with
with 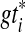 a binary tree refining gti, such that
a binary tree refining gti, such that 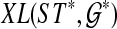 is minimum over all possible rooted binary species trees and sets
is minimum over all possible rooted binary species trees and sets 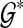 .
.
By Corollary 3, we can formulate the problem as a minimum-weight clique problem. The graph has one vertex for every subset of 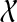 , and we set the weight of the vertex corresponding to subset B to be
, and we set the weight of the vertex corresponding to subset B to be 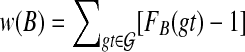 . We have edges between vertices if the two vertices are compatible (can both be contained in a tree). The solution is therefore a minimum weight clique with n − 2 vertices. And, as before, we can describe this as a maximum weight clique problem by having the weight be w′(B) = Q − w(B), for some large enough Q.
. We have edges between vertices if the two vertices are compatible (can both be contained in a tree). The solution is therefore a minimum weight clique with n − 2 vertices. And, as before, we can describe this as a maximum weight clique problem by having the weight be w′(B) = Q − w(B), for some large enough Q.
However, we can also address this problem using dynamic programming, as before. Let 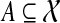 and
and 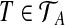 . Let
. Let 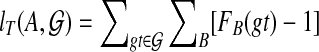 , as B ranges over the clusters of T. Let
, as B ranges over the clusters of T. Let 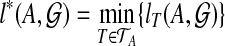 . Then
. Then 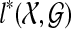 is the solution to the problem of inferring a species tree from rooted, non-binary gene trees.
is the solution to the problem of inferring a species tree from rooted, non-binary gene trees.
We set base cases 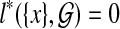 for all
for all 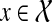 . We order the subproblems by the size of A, and compute
. We order the subproblems by the size of A, and compute 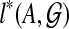 only after every
only after every 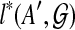 is computed for A′⊂ A. The DP formulation is
is computed for A′⊂ A. The DP formulation is
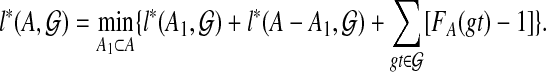 |
3.3. Unrooted, non-binary gene trees
When reconciling an unrooted and incompletely resolved gene tree with a rooted, binary species tree under parsimony, it is natural to seek the rooting and refinement of the gene tree that results in the minimum number of extra lineages over all possible rootings and refinements; see the illustration in Fig. 6. In this case, the MDC problem can be formulated as follows: given a set 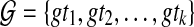 , with each gti a tree on
, with each gti a tree on 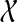 , but not necessarily rooted nor fully resolved, we seek a rooted, binary species tree ST and set
, but not necessarily rooted nor fully resolved, we seek a rooted, binary species tree ST and set 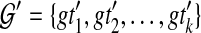 such that each
such that each 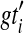 is a binary rooted tree that can be obtained by rooting and refining gti, so as to minimize
is a binary rooted tree that can be obtained by rooting and refining gti, so as to minimize 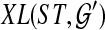 over all such
over all such 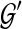 . As before, the computational complexity of this problem is unknown, but conjectured to be NP-hard.
. As before, the computational complexity of this problem is unknown, but conjectured to be NP-hard.
FIG. 6.

Illustration of optimal and non-optimal reconciliations of an unrooted, non-binary gene tree gt with a rooted, binary species tree ST, which yield 0 and 3 extra lineages, respectively.
Observation 2
For any gene tree gt and species tree ST, and t* the optimal refined rooted version of gt that minimizes XL(ST, t*) can be obtained by first rooting gt at some node, and then refining the resultant rooted tree. Thus, to find t*, it suffices to find a node 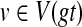 at which to root the tree t, thus producing a tree t′, so as to minimize
at which to root the tree t, thus producing a tree t′, so as to minimize 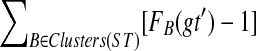 .
.
We now show how we will find an optimal root x.
Notation
Let x be a node in gt, and let gt(x) denote the tree obtained by rooting gt at x. Let gt be a given gene tree, ST a given species tree, and X a cluster of gt. Let r′(X) be the far endpoint of the parent edge of X within gt (i.e., the parent edge of X is (r, r′), where r is the root of X in gt). For a given cluster B of ST, we will say that two clusters A and A′ of gt are B-siblings if A and A′ are B-maximal clusters and their roots share a common neighbor. For a fixed cluster 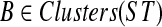 , we will refer to a maximal set of B-maximal clusters in gt that are pairwise siblings as a family of B-maximal clusters in gt. A family of B-maximal clusters will also be referred to as a family of ST-maximal clusters. We denote by gt(A) the subtree of gt induced by cluster A.
, we will refer to a maximal set of B-maximal clusters in gt that are pairwise siblings as a family of B-maximal clusters in gt. A family of B-maximal clusters will also be referred to as a family of ST-maximal clusters. We denote by gt(A) the subtree of gt induced by cluster A.
Lemma 5
Let A be a largest ST-maximal cluster in gt, and a = r′(A). Then a is not internal to any ST-maximal clade in gt, nor is a the root of any ST-maximal clade that is in a family of size at least two.
Proof
Let A be a largest ST-maximal cluster in gt, and suppose A is X-maximal for 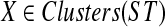 with a = r′(A). Suppose a is inside a B-maximal cluster Y, with Y produced by edge
with a = r′(A). Suppose a is inside a B-maximal cluster Y, with Y produced by edge 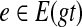 . If e is inside A, then Y ∩ A ≠ 0̸, A − Y ≠ 0̸, and
. If e is inside A, then Y ∩ A ≠ 0̸, A − Y ≠ 0̸, and 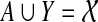 . Since A ⊆ X and Y ⊆ B, it follows that
. Since A ⊆ X and Y ⊆ B, it follows that 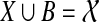 , and also that X ∩ B ≠ 0̸. Since X and B are both clusters in ST they must be compatible, and so one must contain the other. But then one must be the entire set
, and also that X ∩ B ≠ 0̸. Since X and B are both clusters in ST they must be compatible, and so one must contain the other. But then one must be the entire set 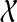 , which is a contradiction.
, which is a contradiction.
Suppose instead that a is the root of a B-maximal cluster Z of gt, with Z defined by edge e, and that Z has a sibling Z′. Thus, Z′ is also B-maximal, and the roots of Z and Z′ share a common neighbor. We consider first the case where the edge e defining cluster Z is the parent edge of A. In this case, the edge e in gt splits the tree into clusters A and Z. Since Z is B-maximal and A is X-maximal, and B and X are both clusters in ST, it follows that 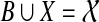 . Thus, the two subtrees off the root of ST are on leafset B and and on leafset X. But then Z = B and A = X. Since Z and Z′ are disjoint, it follows that Z′ ⊆ A, contradicting that Z′ ⊆ B.
. Thus, the two subtrees off the root of ST are on leafset B and and on leafset X. But then Z = B and A = X. Since Z and Z′ are disjoint, it follows that Z′ ⊆ A, contradicting that Z′ ⊆ B.
We now consider the case where the edge e defining Z is some other edge incident with a. But then since a is the root of Z, it follows that A is a proper subset of Z, contradicting that A is the largest ST-maximal cluster in gt.
Hence, for a the far endpoint of the parent edge of a largest ST-maximal cluster in gt, a is not the root of any ST-maximal cluster that is in a family of size two or more, nor is a internal to any ST-maximal cluster.
Theorem 5
Let A be a largest ST-maximal cluster in gt, and a = r′(A). Then FB(gt(a)) ≤ FB(gt(r)) for all clusters 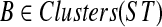 and for all nodes
and for all nodes 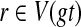 .
.
Proof
By Lemma 5, a is not internal to any ST-maximal cluster of gt, and a is not the root of any cluster that is in a family of size at least two. Let B be a cluster in ST. Since a is not internal to any B-maximal cluster and not the parent of any B-maximal cluster that has a sibling, it follows that the set of B-maximal clusters of gt(a) is identical to the set of B-maximal clusters of gt. Hence, the number of families of B-maximal clusters of gt(a) is identical to the number of families of B-maximal clusters of gt, and so equal to FB(gt(a)). Furthermore, it is easy to see that FB(gt(r)) is at least the number of families of B-maximal clusters of gt, no matter what r is. Hence, FB(gt(r)) ≥ FB(gt(a)) for all vertices r. ▪
The following two corollaries follow directly from Theorem 5:
Corollary 4
Let gt be an unrooted, not necessarily binary gene tree on 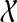 , and let ST be a rooted species tree on
, and let ST be a rooted species tree on 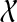 . Let
. Let 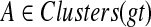 be a largest ST-maximal cluster, and a = r′(A) the far endpoint of the parent edge for A. If we root gt at a, then the resultant tree gt(a) minimizes
be a largest ST-maximal cluster, and a = r′(A) the far endpoint of the parent edge for A. If we root gt at a, then the resultant tree gt(a) minimizes 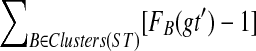 over all rooted versions gt′ of gt.
over all rooted versions gt′ of gt.
Corollary 5
Let 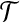 be a set of gene trees that are unrooted and not necessarily binary. For
be a set of gene trees that are unrooted and not necessarily binary. For 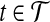 and
and 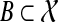 , define tB to be the rooted version of t formed by rooting t at r′(B). Then, the species tree ST that minimizes
, define tB to be the rooted version of t formed by rooting t at r′(B). Then, the species tree ST that minimizes 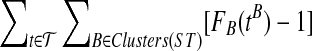 is an optimal species tree for
is an optimal species tree for 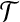 .
.
As a result, we can solve the problem using the clique and DP formulations as in the other versions of the MDC problem.
4. MDC: Multiple-Allele Cases
Thus far, we have assumed that exactly a single allele is sampled per species in the analysis. However, sequences of multiple individuals per species are becoming increasingly available. Therefore, it is necessary to develop methods that infer species trees from data sets that contain zero or more alleles per species for the different loci. Than and Nakhleh (2010) described how to extend the algorithms from Than and Nakhleh (2009) to the case of multiple alleles in a straightforward manner. To illustrate this case, consider the scenario in Figure 7. Here, the numbers of alleles sampled from the species A, B, C, D, and E, for the given locus are 3, 1, 2, 0, and 2, respectively. Under the MDC criterion, a cluster of alleles in the gene tree coalesce at their MRCA in the species tree, where the MRCA is taken over the set of species to which the alleles belong. For example, the cluster {A1, A2} coalescence on the parent edge of species A, whereas the cluster {A1, A2, B1} coalesces at the parent edge of cluster {A, B}. We now formalize the MDC criterion for this case.
FIG. 7.

Illustration of ILS and the MDC criterion in the case of multiple alleles. We denote by Xi an allele of species X. In particular, species D has no alleles sampled for the given locus.
Let ST be a rooted, binary species tree on set 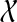 of taxa. Let gt be a gene tree injectively leaf-labeled by the elements of
of taxa. Let gt be a gene tree injectively leaf-labeled by the elements of 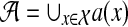 , where a(x) is a set of alleles for species x. In other words, every leaf in gt is labeled uniquely by an element in
, where a(x) is a set of alleles for species x. In other words, every leaf in gt is labeled uniquely by an element in 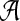 , but there may be labels in
, but there may be labels in 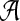 to which no leaf in gt is mapped. In Figure 7, we have a(A) = {A1, A2, A3}, a(B) = {B1}, a(C) = {C1, C2}, a(D) = 0̸, and a(E) = {E1, E2}.
to which no leaf in gt is mapped. In Figure 7, we have a(A) = {A1, A2, A3}, a(B) = {B1}, a(C) = {C1, C2}, a(D) = 0̸, and a(E) = {E1, E2}.
For every set of alleles W of a given locus, we denote by α(W) the set of all species that have alleles in W. In Figure 7, for the set W = {A1, A2, B1, C1, C2, A3}, we have α(W) = {A, B, C}. Using the α mapping, we can now define the MRCA mapping for the multiple-allele case.
Let v be a node in gt and, as before, denote by Cluster(v) its cluster. Then, the MRCA mapping H : V (gt) → V (ST) is defined by HST(v) = MRCAST(α(Clustergt(v))). Notice that under this mapping, if Cluster(v) contains only alleles of a single species (e.g., Cluster(v) ⊆ a(x) for some 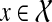 ), then HST(v) = x. Given a cluster A in gt and a cluster B in ST, we say that A is B-maximal if (1) α(A) ⊆ B, and (2) for all
), then HST(v) = x. Given a cluster A in gt and a cluster B in ST, we say that A is B-maximal if (1) α(A) ⊆ B, and (2) for all 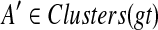 , if A ⊆ A′, then α(A′) ⊈ B. We set kB(gt) as before to be the number of B-maximal clusters within gt. Further, we say that cluster A in gt is ST-maximal if there is a cluster
, if A ⊆ A′, then α(A′) ⊈ B. We set kB(gt) as before to be the number of B-maximal clusters within gt. Further, we say that cluster A in gt is ST-maximal if there is a cluster 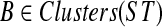 such that
such that 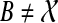 and A is B-maximal.
and A is B-maximal.
Now that we have established the definitions, Theorem 1 applies directly. (We show it here for the case where each gene is sampled in at least one individual per species.)
Theorem 6
Let ST be a rooted, binary species tree on set 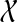 of taxa, and gt be a rooted, binary gene tree leaf-labeled by set
of taxa, and gt be a rooted, binary gene tree leaf-labeled by set 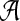 of alleles of
of alleles of 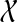 . Let B be a cluster in ST and let e be the parent edge of B in ST. Then kB(gt) is equal to the number of lineages on e in an optimal valid coalescent history. Therefore, XL(e, gt) = kB(gt) − 1, and
. Let B be a cluster in ST and let e be the parent edge of B in ST. Then kB(gt) is equal to the number of lineages on e in an optimal valid coalescent history. Therefore, XL(e, gt) = kB(gt) − 1, and 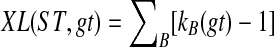 , where B ranges over the clusters of ST. Furthermore, a valid coalescent history f that achieves this total number of extra lineages can be produced by setting f (v) = HST(v) (i.e., f (v) = MRCAST(α(Clustergt(v)))) for all v.
, where B ranges over the clusters of ST. Furthermore, a valid coalescent history f that achieves this total number of extra lineages can be produced by setting f (v) = HST(v) (i.e., f (v) = MRCAST(α(Clustergt(v)))) for all v.
Proof
Let B = ClusterST(v) and e be the parent edge of node v. We prove that kB(gt) is equal to the number of lineages on e in an optimal valid coalescent history by induction on the height of node v (as before, the height of a node is the longest distance from the node to a leaf under it). In particular, as above, we show that kB(gt) = kC(gt) + kD(gt) − numcoal(B), where C and D are the children clusters of B in the species tree, and numcoal(B) is the number of coalescence events that occur on the parent edge of B in an optimal valid coalescent history.
For the base case consider node v of height 0, that is, v is a leaf. Further, assume v is labeled by taxon 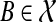 (here, the cluster B has a single element, therefore, we used B as the element of the cluster itself ). In this case, B has no children clusters, and the B-maximal clusters in g are exactly all maximal subtrees
(here, the cluster B has a single element, therefore, we used B as the element of the cluster itself ). In this case, B has no children clusters, and the B-maximal clusters in g are exactly all maximal subtrees 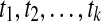 of gt, where L(ti) ⊆ a(B) for 1 ≤ i ≤ k. An optimal valid coalescent history will have all leaves of ti, for each i, coalesce within the parent edge of B. In other words, the number of lineages in the parent edge of B under such a valid coalescent history is k, which is equal to the number of B-maximal clusters within gt. Thus, the result holds for all nodes of height 0.
of gt, where L(ti) ⊆ a(B) for 1 ≤ i ≤ k. An optimal valid coalescent history will have all leaves of ti, for each i, coalesce within the parent edge of B. In other words, the number of lineages in the parent edge of B under such a valid coalescent history is k, which is equal to the number of B-maximal clusters within gt. Thus, the result holds for all nodes of height 0.
For the induction hypothesis, we assume the result holds for all nodes 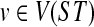 of height p, and prove the result for nodes of height p + 1. Let
of height p, and prove the result for nodes of height p + 1. Let 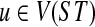 be a node such that v, w are its children, and their height is p. Further, let B = ClusterST(u) whose parent edge is e, C = ClusterST(v) and D = ClusterST(w). By the induction hypothesis, we have that kC(gt) and kD(gt) equal the number of extra lineages in an optimal valid coalescent history on the parent edges of clusters C and D, respectively. The number of lineages that “enter” edge e from below (i.e., from its endpoint that is closer to the leaves) in an optimal valid coalescent history is kC(gt) + kD(gt). If numcoal(B) is the number of coalescence events that take place on edge e in an optimal valid coalescent history, and given that each coalescence event decreases the number of lineages by 1, then the number of edges that “exit” e from above (i.e., from its endpoint that is closer to the root) is kC(gt) + kD(gt) − numcoal(B), which is, by definition, kB(gt). This completes the proof. ▪
be a node such that v, w are its children, and their height is p. Further, let B = ClusterST(u) whose parent edge is e, C = ClusterST(v) and D = ClusterST(w). By the induction hypothesis, we have that kC(gt) and kD(gt) equal the number of extra lineages in an optimal valid coalescent history on the parent edges of clusters C and D, respectively. The number of lineages that “enter” edge e from below (i.e., from its endpoint that is closer to the leaves) in an optimal valid coalescent history is kC(gt) + kD(gt). If numcoal(B) is the number of coalescence events that take place on edge e in an optimal valid coalescent history, and given that each coalescence event decreases the number of lineages by 1, then the number of edges that “exit” e from above (i.e., from its endpoint that is closer to the root) is kC(gt) + kD(gt) − numcoal(B), which is, by definition, kB(gt). This completes the proof. ▪
5. Experimental Evaluation
5.1. Methods
Simulated data
We generated species trees using the “Uniform Speciation” (Yule) module in the program Mesquite (Maddison and Maddison, 2004). Two sets of species trees were generated: one for 8 taxa plus an outgroup, and one for 16 taxa plus an outgroup. Each data set had 500 species trees. All of them have a total branch length of 800,000 generations excluding the outgroup. Within the branch of each species tree, 1, 2, 4, 8, 16, or 32 gene trees were simulated using the “Coalescence Contained Within Current Tree” module in Mesquite with the effective population size Ne equal 100,000. For each gene tree, 1, 2, or 4 alleles were sampled per species. We used the program Seq-gen (Rambaut and Grassly, 1997) to simulate the evolution of DNA sequences of length 2000 under the Jukes-Cantor model (Jukes and Cantor, 1969) down each of the gene trees (these settings are similar to those used in Maddison and Knowles [2006]).
Estimated gene trees
We estimated gene trees from these sequence alignments using default PAUP* heuristic maximum parsimony (MP) methods, returning the strict consensus of all optimal MP trees. We also tried the programs GARLI (Zwickl, 2006) and RAxML (Stamatakis, 2006) for maximum likelihood reconstruction of the gene trees, but ML trees produced very similar results, yet took more time than MP. We rooted each estimated tree at the outgroup in order to produce rooted estimated trees.
Estimated species trees
The “heuristic” version of our method uses only the clusters of the input gene trees, and the “exact” version uses all possible clusters on the taxon set. For some analyses using the heuristic MDC algorithms, the estimated species tree is not fully resolved. In this case, we followed this initial analysis with a search through the set of binary resolutions of the initial estimated species tree for a fully resolved tree that optimized the number of extra lineages. This additional step was limited to 5 minutes of analysis. The only cases where this additional search was not applied were when the polytomy (unresolved node) in the species tree was present in all gene trees; in these cases, any resolution is arbitrary and is as good (under the MDC) criterion as any other resolution.
For the 8-taxon data sets, we used both the exact and heuristic versions of all four algorithms. For the 16-taxon data sets, we used only the heuristic versions.
Measurements
We report the degree of resolution of each estimated gene tree, which is the number of internal branches in t divided by n − 3, where t has n leaves. We also report the Robinson-Foulds (RF) error (Robinson and Foulds, 1981) of estimated trees to the true trees, where the RF error is the total number of edges in the two trees that define bipartitions that are not shared by the other tree, divided by 2n − 6. A value 0 of the RF distance indicates the two trees are identical, and a value of 1 indicates the two trees are completely different (they disagree on every branch).
5.2. Results
The degree of resolution of the estimated gene trees varied between 0.6 for single-allele trees to 0.4 for 4-allele trees in the case of 8-taxon data sets, and between 0.5 for single-allele trees to 0.3 for 4-allele trees in the case of 16-taxon data sets (Fig. 8).
FIG. 8.

Degree of resolution of estimated gene trees. (Left) 8-taxon data sets. (Right) 16-taxon data sets.
With respect to topological accuracy of the estimated gene trees, we found that for 8 taxa, the RF distance is around 0.21. However, 98% of the estimated gene trees have no false positives; thus, all but 2% of the estimated gene trees can be resolved to match the true gene tree. Similarly, the RF distance for the 16-taxon data sets between true gene trees and reconstructed gene trees is around 0.27, but 96% have 0 false positive values.
We now discuss topological accuracy of the species trees estimated using our algorithms for solving the MDC problem. In Figure 9, we show results on running the exact and heuristic versions of the algorithms on 8-taxon data sets. We can see that the heuristic version of our algorithm is almost as accurate as the exact version for both single-allele gene trees and multiple-allele gene trees especially when the number of gene trees sampled are more than four. We also see that increasing the number of gene trees improves the accuracy of the estimated species tree, and that very good accuracy is obtainable from a small number of gene trees. And increasing the number of alleles sampled per species in the gene trees does not help improve the accuracy of the estimated species tree much. In the bottom row of Figure 9, we show results on running algorithms on 8-taxon estimated gene trees. We can see that knowing the true root instead of estimating the root is helpful when the number of gene trees is very small, but that otherwise our algorithm is able to produce comparable results even on unrooted gene trees. The results for 16-taxon data sets are shown in Figure 10, from which we also observe the same trends.
FIG. 9.

Performance of MDC on the 8-taxon data sets. (Top row) True gene trees: exact versus heuristic. (Bottom row) Estimated gene trees: rooted versus unrooted and exact versus heuristic. Columns from left to right correspond to gene trees having 1, 2, and 4 alleles sampled per species, respectively.
FIG. 10.

Performance of MDC on the 16-taxon data sets. (Top row) True gene trees: exact versus heuristic. (Bottom row) Estimated gene trees: rooted versus unrooted. Columns from left to right correspond to gene trees having 1, 2, and 4 alleles sampled per species, respectively.
Acknowledgments
We would also like to thank Carlo Baldassi for pointing out an error in an earlier draft.
This work was supported in part by NSF grant CCF-0622037, grant R01LM009494 from the National Library of Medicine, an Alfred P. Sloan Research Fellowship to L.N., a Guggenheim Fellowship to T.W., and by Microsoft Research New England support to T.W. The contents are solely the responsibility of the authors and do not necessarily represent the official views of the NSF, National Library of Medicine, the National Institutes of Health, the Alfred P. Sloan Foundation, the Guggenheim Foundation, or Microsoft Research New England.
Disclosure Statement
No competing financial interests exist.
References
- Bender M. Farach-Colton M. The LCA problem revisited. Latin Am. Theor. Inform. 2000:88–94. [Google Scholar]
- Dawkins R. The Ancestor's Tale. Houghton Mifflin; New York: 2004. [Google Scholar]
- Degnan J. Rosenberg N. Gene tree discordance, phylogenetic inference and the multispecies coalescent. Trends Ecol. Evol. 2009a;24:332–340. doi: 10.1016/j.tree.2009.01.009. [DOI] [PubMed] [Google Scholar]
- Degnan J.H. DeGiorgio M. Bryant D., et al. Properties of consensus methods for inferring species trees from gene trees. Syst. Biol. 2009;58:35–54. doi: 10.1093/sysbio/syp008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degnan J.H. Rosenberg N.A. Discordance of species trees with their most likely gene trees. PLoS Genet. 2006;2:762–768. doi: 10.1371/journal.pgen.0020068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degnan J.H. Rosenberg N.A. Gene tree discordance, phylogenetic inference and the multispecies coalescent. Trends Ecol. Evol. 2009b;24:332–340. doi: 10.1016/j.tree.2009.01.009. [DOI] [PubMed] [Google Scholar]
- Degnan J.H. Salter L.A. Gene tree distributions under the coalescent process. Evolution. 2005;59:24–37. [PubMed] [Google Scholar]
- Driskell A.C. Ané C. Burleigh J.G., et al. Prospects for building the tree of life from large sequence dstabases. Science. 2004;306:1172–1174. doi: 10.1126/science.1102036. [DOI] [PubMed] [Google Scholar]
- Edwards S.V. Liu L. Pearl D.K. High-resolution species trees without concatenation. Proc. Natl. Acad. Sci. USA. 2007;104:5936–5941. doi: 10.1073/pnas.0607004104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harel D. Tarjan R. Fast algorithms for finding nearest common ancestors. SIAM J. Comput. 1984;13:338–355. [Google Scholar]
- Hudson R.R. Testing the constant-rate neutral allele model with protein sequence data. Evolution. 1983;37:203–217. doi: 10.1111/j.1558-5646.1983.tb05528.x. [DOI] [PubMed] [Google Scholar]
- Jukes T.H. Cantor C.R. Evolution of protein molecules, 21–132. In: Munro H.N., editor. Mammalian Protein Metabolism. Academic Press; New York: 1969. [Google Scholar]
- Kannan S. Lawler E. Warnow T. Determining the evolutionary tree. J. Algorithms. 1996;21:26–50. [Google Scholar]
- Kubatko L. Carstens B. Knowles L. STEM: species tree estimation using maximum likelihood for gene trees under coalescence. Bioinformatics. 2009;25:971–973. doi: 10.1093/bioinformatics/btp079. [DOI] [PubMed] [Google Scholar]
- Kuo C.-H. Wares J.P. Kissinger J.C. The Apicomplexan whole-genome phylogeny: an analysis of incongruence among gene trees. Mol. Biol. Evol. 2008;25:2689–2698. doi: 10.1093/molbev/msn213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L. Pearl D.K. Species trees from gene trees: reconstructing Bayesian posterior distributions of a species phylogeny using estimated gene tree distributions. Syst. Biol. 2007;56:504–514. doi: 10.1080/10635150701429982. [DOI] [PubMed] [Google Scholar]
- Maddison W. Knowles L. Inferring phylogeny despite incomplete lineage sorting. Syst. Biol. 2006;55:21–30. doi: 10.1080/10635150500354928. [DOI] [PubMed] [Google Scholar]
- Maddison W. Maddison D. Mesquite: a modular system for evolutionary analysis. Version 1.01. 2004. http://mesquiteproject.org. [Sep 15;2011 ]. http://mesquiteproject.org
- Maddison W.P. Gene trees in species trees. Syst. Biol. 1997;46:523–536. [Google Scholar]
- Nei M. Stochastic errors in DNA evolution and molecular phylogeny, 133–147. In: Gershowitz H., editor; Rucknagel D.L., editor; Tashian R.E., editor. Evolutionary Perspectives and the New Genetics. Alan R. Liss; New York: 1986. [PubMed] [Google Scholar]
- Nei M. Molecular Evolutionary Genetics. Columbia University Press; New York: 1987. [Google Scholar]
- Pamilo P. Nei M. Relationships between gene trees and species trees. Mol. Biol. Evol. 1988;5:568–583. doi: 10.1093/oxfordjournals.molbev.a040517. [DOI] [PubMed] [Google Scholar]
- Pollard D.A. Iyer V.N. Moses A.M., et al. Widespread discordance of gene trees with species tree in Drosophila: evidence for incomplete lineage sorting. PLoS Genet. 2006;2:1634–1647. doi: 10.1371/journal.pgen.0020173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambaut A. Grassly N.C. Seq-gen: an application for the Monte Carlo simulation of DNA sequence evolution along phylogenetic trees. Comp. Appl. Biosci. 1997;13:235–238. doi: 10.1093/bioinformatics/13.3.235. [DOI] [PubMed] [Google Scholar]
- Robinson D. Foulds L. Comparison of phylogenetic trees. Math. Biosci. 1981;53:131–147. [Google Scholar]
- Rokas A. Williams B. King N., et al. Genome-scale approaches to resolving incongruence in molecular phylogenies. Nature. 2003;425:798–804. doi: 10.1038/nature02053. [DOI] [PubMed] [Google Scholar]
- Rosenberg N.A. The probability of topological concordance of gene trees and species trees. Theor. Pop. Biol. 2002;61:225–247. doi: 10.1006/tpbi.2001.1568. [DOI] [PubMed] [Google Scholar]
- Semple C. Steel M. Phylogenetics. Oxford University Press; Oxford, UK: 2003. [Google Scholar]
- Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22:2688–2690. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- Syring J. Willyard A. Cronn R., et al. Evolutionary relationships among Pinus (Pinaceae) subsections inferred from multiple low-copy nuclear loci. Am. J. Botany. 2005;92:2086–2100. doi: 10.3732/ajb.92.12.2086. [DOI] [PubMed] [Google Scholar]
- Tajima F. Evolutionary relationship of DNA sequences in finite populations. Genetics. 1983;105:437–460. doi: 10.1093/genetics/105.2.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahata N. Gene genealogy in three related populations: consistency probability between gene and population trees. Genetics. 1989;122:957–966. doi: 10.1093/genetics/122.4.957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Than C. Nakhleh L. Species tree inference by minimizing deep coalescences. PLoS Comput. Biol. 2009;5:e1000501. doi: 10.1371/journal.pcbi.1000501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Than C. Nakhleh L. Inference of parsimonious species phylogenies from multi-locus data by minimizing deep coalescences, 79–98. In: Knowles L., editor; Kubatko L., editor. Estimating Species Trees: Practical and Theoretical Aspects. Wiley-VCH; New York: 2010. [Google Scholar]
- Than C. Ruths D. Nakhleh L. PhyloNet: a software package for analyzing and reconstructing reticulate evolutionary relationships. BMC Bioinform. 2008a;9:322. doi: 10.1186/1471-2105-9-322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Than C. Sugino R. Innan H., et al. Efficient inference of bacterial strain trees from genome-scale multi-locus data. Bioinformatics. 2008b;24:i123–i131. doi: 10.1093/bioinformatics/btn149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C.-I. Inferences of species phylogeny in relation to segregation of ancient polymorphisms. Genetics. 1991;127:429–435. doi: 10.1093/genetics/127.2.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C.-I. Reply to Richard R. Hudson. Genetics. 1992;131:513. [Google Scholar]
- Zwickl D. University of Texas; Austin: 2006. Genetic algorithm approaches for the phylogenetic analysis of large biological sequence datasets under the maximum likelihood criterion. [Ph.D. dissertation]. [Google Scholar]