

Abstract
Scaffolding, the problem of ordering and orienting contigs, typically using paired-end reads, is a crucial step in the assembly of high-quality draft genomes. Even as sequencing technologies and mate-pair protocols have improved significantly, scaffolding programs still rely on heuristics, with no guarantees on the quality of the solution. In this work, we explored the feasibility of an exact solution for scaffolding and present a first tractable solution for this problem (Opera). We also describe a graph contraction procedure that allows the solution to scale to large scaffolding problems and demonstrate this by scaffolding several large real and synthetic datasets. In comparisons with existing scaffolders, Opera simultaneously produced longer and more accurate scaffolds demonstrating the utility of an exact approach. Opera also incorporates an exact quadratic programming formulation to precisely compute gap sizes (Availability: http://sourceforge.net/projects/operasf/).
Key words: parametric complexity, genome assembly, quadratic programming, scaffolding
1. Introduction
With the advent of second-generation sequencing technologies, while the cost of sequencing has decreased dramatically, the challenge of reconstructing genomes from the large volumes of fragmentary read data has remained daunting. Newly developed protocols for second-generation sequencing can generate paired-end reads (reads from the ends of a fragment of known approximate length) for a range of library sizes (Ng et al., 2006) and third-generation strobe sequencing protocols (Eid et al., 2009) provide linking information that, in principle, can be valuable for assembling a genome. In recent work, the importance of paired reads has been further highlighted, with some authors even questioning the need for long reads in the presence of libraries with large insert lengths (Chaisson et al., 2009; Zerbino et al., 2009).
Scaffolding, the problem of using the connectivity information from paired reads to order and orient partially reconstructed contig sequences in the genome, has been well studied in the assembly literature with several algorithms proposed in recent years (Myers et al., 2000; Kent and Haussler, 2001; Pevzner and Tang, 2001; Huson et al., 2002; Pop et al., 2004; Zerbino et al., 2009; Dayarian et al., 2010). In Huson et al. (2002), the authors presented a natural formulation of this problem (in terms of finding an ordering of sequences that minimizes paired read violations) and showed that its decision version is NP-complete. Several related problems are also known to be NP-complete (Kent and Haussler, 2001; Pop et al., 2004) and hence, to maintain efficiency and scalability, existing algorithms have relied on various heuristic solutions. For instance, in Huson et al. (2002), the authors proposed a greedy solution that iteratively merges scaffolds connected by the most paired reads. Similarly, the algorithm proposed in the Phusion assembler (Mullikin and Ning, 2003) relies on a greedy heuristic based on the distance contraints imposed by the paired reads. Other approaches, used in assemblers such as ARACHNE and JAZZ (Jaffe et al., 2003; Aparicio et al., 2002), also employ error-correction steps to minimize the potential impact of misjoins from heuristic searches.
In addition to paired reads, similarity to a reference genome (Pop et al., 2004; Richter et al., 2007; Husemann and Stoye, 2010) and restriction-map based approaches (Kent and Haussler, 2001; Nagarajan et al., 2008) have been used to order contigs, partly because they can lead to a more computationally tractable problem. However, while reference-guided assembly uses potentially misleading synteny information, restriction-map based approaches can produce an ambiguous order and find it hard to place small contigs (Nagarajan et al., 2008). Paired-end reads therefore remain the most general source of information for generating high-quality scaffolds.
In this work, we focus on the problem of scaffolding of a set of contigs using paired-end reads, though similar ideas could be extended to multi-contig constraints from sources such as strobe sequencing and restriction maps (Eid et al., 2009). Unlike existing solutions which use heuristics, we provide a combinatorial algorithm that is guaranteed to find the optimal scaffold under a natural criterion similar to that in Huson et al., (2002). By exploiting the tractability of the problem and a contraction step that leverages the structure of the graph, our scaffolder (Opera) effectively constructs scaffolds for large genomic datasets. The fundamental advantages of this approach are twofold: Firstly, the algorithm provides a solution that explains/uses as much of the paired read data as possible (as we show, this also translates into a more complete and reliable scaffold in practice). Secondly, the algorithm provides a clear guarantee on the quality of the assembly and avoids overly aggressive assembly heuristics that can produce large scaffolds at the expense of assembly errors.
While libraries from new sequencing technologies generate a vast amount of paired-end reads that provide detailed connectivity information, assembly and mapping errors from shorter read lengths and an abundance of chimeric mate-pairs in some protocols (Ng et al., 2006) can complicate the scaffolding effort. We show how these sources of error can be handled in our optimization framework in a robust fashion. We also employ a quadratic programming formulation (and an efficient solver) to compute gap sizes that best agree with the mate-pair library derived constraints. Our experiments with several large real and synthetic datasets suggest that these theoretical advantages in Opera do translate to larger, more reliable and well-defined scaffolds when compared to existing programs.
2. Methods
2.1. Definitions
In a typical whole-genome shotgun sequencing project, randomly sheared fragments of DNA are sequenced, using one or more of the several sequencing technologies that are now available. The resulting reads are then assembled in silico to produce longer contig sequences (Pop, 2004). In addition, the reads are often generated from the ends of long fragments (of known approximate sizes and from one or more libraries), and this information is used to link together contigs and order and orient them (Fig. 1).
FIG. 1.

Paired-read and scaffold graph. (a) Paired-read constraints on order and orientation of contigs (pointed boxes). (b) A set of paired-reads and contigs. (c) The resulting scaffold graph. (d) A scaffold for the graph in (c).
Consider a set of contigs 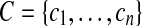 . For every
. For every 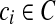 , we denote the two possible orientations as ci and −ci. A scaffold is then given by a signed permutation of the contigs as well as a list of gap sizes between adjacent contigs (Fig. 1d). Given two contigs ci and cj linked by a paired-read (i.e. one end falls on ci and the other end on cj), the relative orientation of the contigs suggested by the paired-read can be encoded as a bidirected edge in a graph (Fig. 1a, c). We then say that a paired-read is concordant in a scaffold if the suggested orientation is satisfied and the distance between the reads is less then a specified maximum1 library size τ.
, we denote the two possible orientations as ci and −ci. A scaffold is then given by a signed permutation of the contigs as well as a list of gap sizes between adjacent contigs (Fig. 1d). Given two contigs ci and cj linked by a paired-read (i.e. one end falls on ci and the other end on cj), the relative orientation of the contigs suggested by the paired-read can be encoded as a bidirected edge in a graph (Fig. 1a, c). We then say that a paired-read is concordant in a scaffold if the suggested orientation is satisfied and the distance between the reads is less then a specified maximum1 library size τ.
2.2. Scaffold graph
Given a set of contigs and a mapping of paired-reads to contigs, we use an “edge bundling” step as described in Huson et al. (2002) to construct a scaffold graph (actually bidirected multi-graph) where contigs are nodes and are connected by scaffold edges representing multiple paired-reads that suggest a similar distance and orientation for the contigs (Fig. 1b, c). After the bundling step, existing scaffolders typically filter edges with reads less than an arbitrary (sometimes user-specified) threshold. This is done to reduce the number of incorrect edges in the graph from chimeric paired-reads. Instead of setting this threshold arbitrarily and independent of genome size or sequence coverage, we use the following simulation to determine an appropriate threshold: we simulate chimeric reads by selecting paired-reads at random and exchanging their partners. This is then repeated till a significant proportion of the reads (say 10%) are chimeric. We then bundle the chimeric reads as before and repeat the simulation a 100 times to determine the scaffold edge with most chimeric reads supporting it (say d) and set the threshold to be one more than that (i.e., d + 1). This then effectively removes the “stochastic noise” introduced by chimeric constructs and allows the main scaffolding algorithm to focus on systematic assembly and mapping errors that lead to incorrect scaffold edges.
Extrapolating the notion of concordance to scaffold edges we get the following natural formulation of the Scaffolding Problem:
Definition 1 (Scaffolding Problem)
Given a scaffold graph G, find a scaffold S of the contigs that maximizes the number of concordant edges in the graph.
As this problem is analagous to that in Huson et al. (2002) (where the optimality criterion is the number of concordant paired-reads), it is easy to modify their proof to show that the decision version of the scaffolding problem is NP-complete.
2.3. Computational tractability
The scaffolding problem that we defined (as well as the one in Huson et al. [2002]) does not specifically delineate a structure for the scaffold graph. In practice, however, the scaffold graph is constrained by the fact that paired-read libraries have an upper-bound τ and contigs have a minimum length, lmin. This defines an upper-bound on the number of contigs that can be spanned by a paired-read, i.e., the width of the library (or w, where 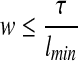 . Here we show that considering width as a fixed parameter, we can indeed construct an algorithm that is polynomial in the size of the graph. This is similar to the work in Saxe (1980), where the focus is on a bounded version of the graph bandwith problem. The scaffolding problem can be seen as a generalization of a bounded version of the graph bandwith problem where nodes and edges in the graph have orientations and lengths and not all edge constraints have to be satisfied.
. Here we show that considering width as a fixed parameter, we can indeed construct an algorithm that is polynomial in the size of the graph. This is similar to the work in Saxe (1980), where the focus is on a bounded version of the graph bandwith problem. The scaffolding problem can be seen as a generalization of a bounded version of the graph bandwith problem where nodes and edges in the graph have orientations and lengths and not all edge constraints have to be satisfied.
For ease of exposition we first consider the special case where the optimal scaffold in a scaffold graph has no discordant edges (i.e., a bounded-width graph). We consider the case of discordant edges in Section 2.4. Also, without loss of generality, we assume that the graph is connected (otherwise, we can compute optimal scaffolds for each component independently). Finally, it is easy to see that we can limit our search to scaffolds where all gap sizes are 0 (we show how more appropriate gap sizes can be computed in Section 2.7). We begin with a few definitions: For a scaffold graph G = (V, E), a partial scaffold S′ is a scaffold on a subset of the contigs and the dangling set, D(S′), is the set of edges from S′ to V − S′. The active region A(S′) is then the shortest suffix of S′ such that all dangling edges are adjacent to a contig in A(S′). A partial scaffold S′ is said to be valid if all edges in the induced subgraph are concordant.
We now describe a dynamic programming based search over the space of scaffolds to find the optimal scaffold. Note that a naive search for an optimal scaffold would enumerate over all possible signed permutations of the contigs and count the number of concordant scaffold edges. Since there are 2|V ||V |! possible signed permutations, this approach is clearly not feasible. Instead, we can limit our search over an equivalence class of partial scaffolds as shown in the following lemma.
Lemma 1
Consider two valid partial scaffolds 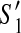 and
and 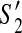 of the scaffold graph G. If
of the scaffold graph G. If 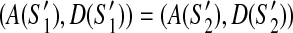 then (1)
then (1)  and
and 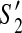 contain the same set of contigs; and (2) both or neither of them can be extended to a solution.
contain the same set of contigs; and (2) both or neither of them can be extended to a solution.
Proof
For (1), suppose there exists a contig c which appears in 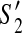 but not in
but not in 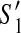 . Since G = (V, E) is a connected graph, there exists a path (in an undirected sense)
. Since G = (V, E) is a connected graph, there exists a path (in an undirected sense) 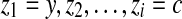 in G = (V, E) where y is the first contig in the active region of both
in G = (V, E) where y is the first contig in the active region of both 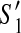 and
and 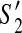 while
while 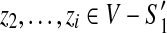 . For
. For 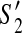 , since y is the first contig which appears in the active region of
, since y is the first contig which appears in the active region of 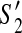 , we have
, we have 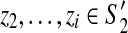 . Hence, (z1, z2) is a dangling edge of
. Hence, (z1, z2) is a dangling edge of 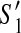 but not a dangling edge of
but not a dangling edge of 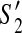 which gives us a contradiction.
which gives us a contradiction.
For (2), let S″ be any scaffold of 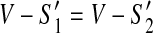 . Since
. Since 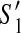 and
and  have the same active region,
have the same active region, 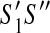 has no discordant edges if and only if
has no discordant edges if and only if 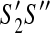 has no discordant edges.
has no discordant edges.
Based on the above lemma, the algorithm in Figure 2 starts from an empty scaffold S = ∅, extends it a contig at a time to search over the equivalence class of partial scaffolds, and finds a scaffold with no discordant edges (if it exists). The proof of correctness of the algorithm follows directly from Lemma 1.
FIG. 2.

An algorithm for generating a scaffold for a bounded-width scaffold graph.
We prove the runtime complexity of the algorithm in the following theorem.
Theorem 1
Given a scaffold graph G = (V, E) and an empty scaffold, the algorithm Scaffold-Bounded-Width runs in O(|E||V|w) time.
Proof
The number of contigs in an active region is bounded by w and each contig has two possible orientations. Hence, the set of possible active regions is O((2|V|)w). Every contig in an active region has ≤w dangling edges. Thus, a given active region has at most 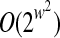 possible dangling sets. The number of equivalence classes is therefore bounded by
possible dangling sets. The number of equivalence classes is therefore bounded by 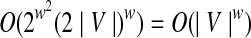 (treating w as a constant). For each equivalence class, updating the active region and the dangling set in steps 6 and 7 takes O(|E|) time.
(treating w as a constant). For each equivalence class, updating the active region and the dangling set in steps 6 and 7 takes O(|E|) time.
The runtime analysis presented here is clearly a coarse-grained analysis and with some more work, tighter bounds can be proven (e.g., since we extend the scaffold in only one direction, we do not need to keep track of the dangling set). However, the main point here is that, for a fixed w, the worst-case runtime of the algorithm is polynomial in the size of the graph; i.e., we have a tractable algorithm for the problem. In the next section, we discuss how this analysis can be extended to the case where not all edges in the optimal scaffold are concordant.
2.4. Minimizing discordant edges
Treating the width parameter as a fixed constant is a special case of the scaffolding problem and correspondingly the NP-completeness result discussed in Section 2.2 does not hold. In the following theorem (with proof in the Appendix) we show that the decision version of the scaffolding problem (allowing for discordant edges) is NP-complete even when the width of the paired-end library is treated as a constant.
Theorem 2
Given a scaffold graph G and treating the library width w as a constant, the problem of deciding if there exists a scaffold S with less than p discordant edges is NP-complete.
Theorem 2 suggests that we cannot hope to design an algorithm that is polynomial in p, the number of discordant edges. However, treating p as a constant as well, we can extend the algorithm in Section 2.3 and still maintain a runtime polynomial in the size of the graph. The basic idea here is that we need to extend the notion of equivalence class by keeping track of discordant edges from the partial scaffold (denoted by X(S′) for a partial scaffold S′). Also, we redefine the dangling set to contain only concordant edges and note that as the scaffold is only extended in one direction, the dangling set is completely determined by the active region and the set of discordant edges. Then the following lemma is a straightforward extension of Lemma 1:
Lemma 2
Consider two partial scaffolds  and
and 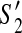 of G with less than p discordant edges. If
of G with less than p discordant edges. If 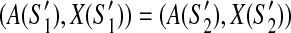 then (1)
then (1) 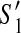 and
and 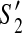 contain the same set of contigs; and (2) both or neither of them can be extended to a solution.
contain the same set of contigs; and (2) both or neither of them can be extended to a solution.
Based on this lemma, an extension of the algorithm in Figure 2 that handles discordant edges is presented in Figure 3. In addition, we extend the runtime analysis in the following lemma:
FIG. 3.

An algorithm for generating a scaffold with at most p discordant edges.
Lemma 3
Consider a scaffold graph G = (V, E) and let p be the maximum allowed number of discordant edges. The algorithm Scaffold runs in O(|V |w|E|p+1) time.
Proof
As before, the set of possible active regions is O(|V|w). Also, there are at most O(|E|p) possible sets of discordant edges. Finally, for each equivalence class, updating the active region and the set of discordant edges in steps 6 and 7 takes O(|E|) time.
To convert this algorithm into one that optimizes over p, we can rely on a branch-and-bound approach where (1) a quick heuristic search is used to find a good solution and define an upper-bound on p and (2) the upper-bound is refined as better solutions are found and the search is not terminated till all extensions have been explored in step 4. We implemented such an approach but found that in some cases our heuristic search would return a poor upper-bound and thus affect the runtime of the algorithm. To get around this, our current implementation tries each value of p (starting from 0) and stops when a scaffold can be constructed (the total runtime is still O(|V|w|E|p+1)).
Note that while the worst-case runtime bound suggests that if p increases by one, runtime would increase by a factor proportional to the size of the graph, in practice, we observe only a constant factor increase (i.e., runtime growth is Cp where C ≤ 5). For real datasets, we can further exploit the structure of the graph, and one idea that improves runtimes significantly is detailed below.
2.5. Graph contraction
Contigs assembled from whole-genome shotgun sequencing data come in a range of sizes and often a successful assembly produces several contigs longer than the paired-read library thresholds (τ). For a particular library size, we label such contigs as border contigs and note the fact that a scaffold derived from such a scaffold graph will not have concordant library edges spanning a border contig. For a scaffold graph G = (V, E), we then define G′ = (V′, E′) as a fenced subgraph of G if edges in E from V − V′ to V′ are always adjacent to a border contig. For example, Figure 4b shows a fenced subgraph of the scaffold graph in Figure 4a.
FIG. 4.

Contracting the Scaffold Graph. (a) Original scaffold graph G. (b) A fenced subgraph of G (with optimal scaffolds ″3 4 − 5″ and ″8 − 9 10″). (c) The new graph after contraction of optimal scaffolds for the subgraph in (b).
We now prove a lemma on the relationship between optimal scaffolds of G′ and G.
Lemma 4
Given a scaffold graph G = (V, E), let G′ = (V′, E′) be a fenced subgraph of G. Suppose 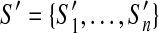 forms the optimal scaffold set of G′ (disconnecting scaffolds connected by discordant edges). There exists an optimal scaffold set S of G where every
forms the optimal scaffold set of G′ (disconnecting scaffolds connected by discordant edges). There exists an optimal scaffold set S of G where every 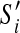 is a subpath of some scaffold of S.
is a subpath of some scaffold of S.
Proof
Let S be an optimal scaffold set of G that does not contain S′ as subpaths. We construct a new scaffold set that does, by first removing all contigs in V′. For each remaining partial scaffold whose end was adjacent to a border contig b, we append that end to the corresponding scaffold 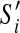 (with b on its end and in the right orientation). This new scaffold is at least as optimal as S. To see this, note that the number of concordant edges between nodes in V − V′ as well as those between nodes in V and V − V′ has remained the same. Also, since S′ is optimal for G′ the number of concordant edges in V′ could only have gone up.
(with b on its end and in the right orientation). This new scaffold is at least as optimal as S. To see this, note that the number of concordant edges between nodes in V − V′ as well as those between nodes in V and V − V′ has remained the same. Also, since S′ is optimal for G′ the number of concordant edges in V′ could only have gone up.
Based on the lemma, we devise a recursive, graph contraction based algorithm to compute the optimal scaffold, and this is outlined in Figure 5 and illustrated by an example in Figure 4.
FIG. 5.

A recursive graph contraction based algorithm to compute an optimal scaffold for a scaffold graph G.
2.6. Handling of repeat contigs
Repeat regions in the genome are often assembled (especially by short-read assemblers) as a single contig and in the scaffolding stage, information from paired-reads could help place such contigs in multiple scaffold locations. The optimization algorithm described here can be naturally extended to handle such cases but due to space constraints we do not explore this extension here and instead filter such contigs (based on read coverage greater than 1.5 times the genomic mean) before scaffolding with Opera.
2.7. Determination of gap sizes
After the order and the orientation of contigs in a scaffold have been computed, the constraints imposed by the paired-reads can also be used to determine the sizes of intervening gaps between contigs. This then serves as an important guide for genome finishing efforts as well as downstream analysis. Since scaffold edges can span multiple gaps and impose competing constraints on their sizes, we adopt a maximum likelihood approach to compute gap sizes:
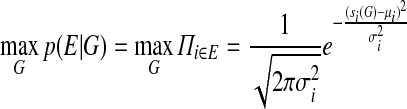 |
(1) |
where E is the set of scaffold edges (whose sizes follow normal distribution with parameters μi, σi), G is the set of gap sizes, and si(G) is the observed separation for scaffold edge i determined from the gap sizes. If ci is the total length of contig sequences spanned by a scaffold edge and Gi is the set of gaps spanned, then we can reformulate this as the minimization of the following quadratic function:
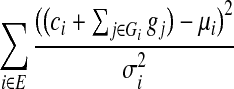 |
(2) |
where gj are the gap sizes. The resulting quadratic program (with gap sizes bounded by τ) can be shown to have a positive definite Q matrix with a unique solution that can be found by the Goldfarb-Idnani active-set dual method in polynomial time (Goldfarb and Idnani, 1983). This procedure thus efficiently computes gap sizes that optimize a clear likelihood function while taking all scaffold edges into account. As we show below, this also leads to improved estimates for gap sizes in practice.
3. Experimental Results
3.1. Datasets
To evaluate Opera, we compared it against existing programs—Velvet (Zerbino et al., 2009), Bambus (Pop et al., 2004) and SOPRA (Dayarian et al., 2010)—on a dataset for B. pseudomallei (Nandi et al., 2010) as well as synthetic datasets for E. coli, S. cerevisiae, and D. melanogaster (chromosome X). The synthetic datasets were generated using Metasim (Richter et al., 2008) (with default Illumina error models), and the reference genome in each case was downloaded from the NCBI website. Similar to the real dataset, for the synthetic sets, we simulated a high-coverage read library as well as a low-coverage paired-read library. Detailed information about the datasets can be found in Table 1.
Table 1.
Test Datasets and Sequencing Statistics
| E. coli | B. pseudomallei | S. cerevisiae | D. melanogaster | |
|---|---|---|---|---|
| Size (Mbp) | 4.6 | 7 | 12.1 | 22.4 |
| Chromosomes | 1 | 2 | 16 | 1 |
| Reads (length, insert size, coverage) | 80 bp, 300±30 bp 40× | 100 bp (454 reads) 20× | 80 bp, 300±30 bp 40× | 80 bp, 300 ± 30 bp 40× |
| Paired-reads (length, insert size, coverage) | 50 bp, 10±1 Kbp 2× | 20 bp, 10±1.5 Kbp 2.8× | 50 bp, 10±1 Kbp 2× | 50 bp, 10 ± 1 Kbp 2× |
Note that, for a library where insert size is μ ± σ, τ was set to μ + 6σ and in all cases lmin was set to 500 bp. For the synthetic datasets, 10% of the large-insert library reads were made chimeric by exchanging read-ends at random.
In all cases (except for B. pseudomallei), the reads were assembled and scaffolded using Velvet (with default parameters and k = 31). For Bambus and Opera, contigs assembled by Velvet were provided as input and scaffolded with the aid of the paired-read library. For SOPRA, we used the combined Velvet-SOPRA pipeline as described in Dayarian et al., (2010). In the case of B. pseudomallei, the 454 reads were assembled using Newbler (available at http://www.454.com) and scaffolded using Bambus and Opera (Velvet and SOPRA cannot directly take contigs as input).
3.2. Scaffold contiguity
For each dataset and for each method, we assessed the contiguity of the reported set of scaffolds, by the N50 size (the length 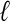 of the longest scaffold such that at least half of the genome is covered by scaffolds longer than
of the longest scaffold such that at least half of the genome is covered by scaffolds longer than 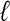 ) and the length of the longest scaffold. We also report the total number of scaffolds as well as the number of scaffolds with more than one contig (Table 2). As can be seen from Table 2, Opera consistently produces the smallest number of scaffolds, the largest N50 sizes and the largest single scaffold.
) and the length of the longest scaffold. We also report the total number of scaffolds as well as the number of scaffolds with more than one contig (Table 2). As can be seen from Table 2, Opera consistently produces the smallest number of scaffolds, the largest N50 sizes and the largest single scaffold.
Table 2.
A Comparison of Scaffold Contiguity for Different Methods
| E. coli | B. pseudomallei | S. cerevisiae | D. melanogaster | |
|---|---|---|---|---|
| Scaffolds (non-singletons) | ||||
| Velvet | 241 (2) | — | 1131 (26) | 2148 (23) |
| Bambus | 200 (9) | 183 (62) | 1085 (39) | 2062 (42) |
| SOPRA | 545 (90) | — | 2171 (308) | 4927 (149) |
| Opera | 3 (2) | 3 (2) | 31 (22) | 36 (15) |
| N50 (Mbp) | ||||
| Velvet | 3.02 | — | 0.55 | 1.88 |
| Bambus | 0.73 | 0.25 | 0.36 | 1.05 |
| SOPRA | 0.05 | — | 0.04 | 0.03 |
| Opera | 3.02 | 3.81 | 0.65 | 3.18 |
| Maximum length (Mbp) | ||||
| Velvet | 3.02 | — | 0.96 | 4.31 |
| Bambus | 1.35 | 0.47 | 0.72 | 2.33 |
| SOPRA | 0.14 | — | 0.15 | 0.82 |
| Opera | 3.02 | 3.81 | 1.04 | 7.69 |
3.3. Scaffold correctness
To check the correctness of the reported scaffolds, we aligned the corresponding contigs to the reference genome using MUMmer (Kurtz et al., 2004). Consecutive contigs in a scaffold that do not have the same order and orientation in the reference genome were then counted as breakpoints in the scaffold (Table 3). In all datasets, Opera reports scaffolds with fewer breakpoints and therefore with greater agreement with the reference genome. Table 3 also reports the number of discordant edges seen in the scaffolds for the various methods (SOPRA is not compared as it uses a different set of contigs), and as expected, Opera produces the best results under this criteria.
Table 3.
Comparison of Scaffold Correctness for Different Methods
| E. coli | B. pseudomallei | S. cerevisiae | D. melanogaster | |
|---|---|---|---|---|
| No. of breakpoints | ||||
| Velvet | 3 | — | 6 | 11 |
| Bambus | 31 | — | 57 | 107 |
| SOPRA | 1 | — | 67 | 10 |
| Opera | 0 | — | 1 | 4 |
| No. of discordant edges | ||||
| Velvet | 4 | — | 7 | 16 |
| Bambus | 19 | 673 | 55 | 423 |
| Opera | 1 | 19 | 3 | 4 |
Breakpoints were not assessed for B. pseudomallei due to the lack of a finished reference for the sequenced strain.
3.4. Running time and gaps
The current implementation of Opera is in JAVA (for ease of programming) and has not been optimized for runtime or memory (the datasets tested typically used a few MB of memory). However, despite this Opera had favorable runtimes on all datasets (Table 4). This is likely due to the fact that it can effectively contract the scaffold graph while it searches for the optimal scaffold. We also compared the gap sizes estimated by the scaffolders, and in general, Velvet and Opera had the most consistent scaffolds and gap sizes. For gaps (≤1 Kbp) shared by their scaffolds, both Velvet and Opera produced accurate gap size estimates for S. cerevisiae, but Velvet had more gaps with relative error >10% (13 compared to 7 for Opera). For D. melanogaster, both scaffolders had many more gaps with relative error >10%, but Opera was slightly better (31 versus 36 for Velvet).
Table 4.
Runtime Comparison
| E. coli | B. pseudomallei | S. cerevisiae | D. melanogaster | |
|---|---|---|---|---|
| Time | ||||
| Bambus | 50 s | 16 min | 2 min | 3 min |
| SOPRA | 49 min | — | 2 h | 5 h |
| Opera | 4 s | 7 min | 11 s | 30 s |
Note that we do not report results for Velvet, as it does not have a stand-alone scaffolding module.
4. Discussion
In this article, we explored a formal approach to the problem of scaffolding of a set of contigs using a paired-read library. As we describe in the methods, despite the computational complexity of the problem, we can devise a tractable algorithm for scaffolding. Furthermore, by exploiting the structure of the scaffold graph (using a graph contraction procedure), this method can scaffold large graphs and long paired-read libraries (e.g., the B. pseudomallei graph has more than 900 contigs).
Our experimental results, while limited, do suggest that Opera can more fully utilize the connectivity information provided by paired-reads. When compared with existing heuristic approaches, Opera simultaneously produces longer scaffolds and with fewer errors. This highlights the utility of minimizing the number of discordant edges in the scaffold graph and suggests that good approximation algorithms for this problem could achieve similar results with better scalability.
We plan to explore several promising extensions to Opera including the use of strobe-sequencing reads and information from overlaping contigs to improve the scaffolds. Another extension is to incorporate additional quality metrics (such as a lower-bound for the library size) to help differentiate between solutions that are equally good under the current optimality criterion. A C++ version of Opera (handling repeat contigs as well) will be publicly available soon.
5. Appendix
Proof of Theorem 2
Proof
Given a scaffold, it is easy to see that it can be checked in polynomial time, and hence the problem is in NP.
We now show a reduction from the (1, 2)-traveling salesperson problem. Given a complete graph H = (V, E) whose edges are of weight either 1 or 2, the (1, 2)-traveling salesperson problem asks if a weight k path exists that visits all vertices.
To construct a scaffold graph G = (V′, E′), we set V′ = V and E′ to a subset of E in which all edges with weight 2 are discarded (for every pair of such nodes 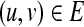 , there are actually two bidirected edges in E′ corresponding to the permutations uv and −u − v). Note that the graph G can be constructed from H in polynomial time and while the reduction is for the case w = 0 it extends in a straightforward fashion for other values (by inserting a path of w contig nodes between every pair of nodes from V).
, there are actually two bidirected edges in E′ corresponding to the permutations uv and −u − v). Note that the graph G can be constructed from H in polynomial time and while the reduction is for the case w = 0 it extends in a straightforward fashion for other values (by inserting a path of w contig nodes between every pair of nodes from V).
We now show that H has a path of weigth L + 2(|V|−1 − L) if and only if G has a solution which omits |E′| − L edges, where L is the number of weight-1 edges in a scaffold of G.
Suppose H has a path of length L + 2(|V| − 1 − L), i.e., the path has L edges of weight 1 and (|V| − 1 − L) edges of weight 2. Then, in G, we can construct a scaffold S which consists of these L edges of weight 1 (by choosing the appropriate bidirected edge). S is a valid scaffold which omits |E′| − L edges in G.
Suppose G has a scaffold which omits |E′| − L edges (for multiple independent scaffolds, consider them in any order). Since the weights of all edges in G are 1, all edges in the solution connect two adjacent nodes. As H is a clique, if there is no edge in a pair of adjacent nodes in the solution, there must be an edge of weight 2 in H. Then, a travelling-salesperson path of length L + 2(|V| − 1 − L) can be constructed by selecting all such missing edges from H.
Footnotes
In principle, a lower-bound can also be determined and used in defining concordant edges.
Acknowledgments
We would like to thank Mihai Pop for stimulating interest in this topic and Pramila Nuwantha Ariyaratne for providing advice on datasets. This work was supported in part by the Biomedical Research Council/Science and Engineering Research Council of A*STAR (Agency for Science, Technology and Research) Singapore and by the MOEs AcRF Tier 2 funding (R grant-252-000-444-112). S.G. is supported by a NUS graduate scholarship.
Disclosure Statement
No competing financial interests exist.
References
- Aparicio S. Chapma J. Stupka E., et al. Whole-genome shotgun assembly and analysis of the genome of Fugu rubripes. Science. 2002;297:1301–1310. doi: 10.1126/science.1072104. [DOI] [PubMed] [Google Scholar]
- Chaisson M.J. Brinza D. Pevzner P.A. De novo fragment assembly with short mate-paired reads: does the read length matter? Genome Res. 2009;19:336–346. doi: 10.1101/gr.079053.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dayarian A. Michael T.P. Sengupta A.M. SOPRA: scaffolding algorithm for paired reads via statistical optimization. BMC Bioinform. 2010;11:345. doi: 10.1186/1471-2105-11-345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eid J. Fehr A. Gray J., et al. Real-time DNA sequencing from single polymerase molecules. Science. 2009;323:133–138. doi: 10.1126/science.1162986. [DOI] [PubMed] [Google Scholar]
- Husemann P. Stoye J. Phylogenetic comparative assembly. Alg. Mol. Biol. 2010;5:3. doi: 10.1186/1748-7188-5-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldfarb D. Idnani A. A numerically stable dual method for solving strictly convex quadratic programs. Math. Program. 1983:27. [Google Scholar]
- Huson D.H. Reinert K. Myers E.W. The greedy path-merging algorithm for contig scaffolding. J ACM. 2002;49:603–615. [Google Scholar]
- Jaffe D.B. Butler J. Gnerre S., et al. Whole-genome sequence assembly for mammalian genomes: Arachne 2. Genome Res. 2003;13:91–96. doi: 10.1101/gr.828403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent W.J. Haussler D. Assembly of the working draft of the human genome with GigAssembler. Genome Res. 2001;11:1541–1548. doi: 10.1101/gr.183201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtz S.A. Phillippy A. Delcher A.L., et al. Versatile and open software for comparing large genomes. Genome Biol. 2004;5:R12. doi: 10.1186/gb-2004-5-2-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacCallum I. Przybylksi D. Gnerre S., et al. ALLPATHS2: small genomes assembled accurately and with high continuity from short paired reads. Genome Biol. 2009;10:R103. doi: 10.1186/gb-2009-10-10-r103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullikin J.C. Ning Z. The phusion assembler. Genome Res. 2003;13:81–90. doi: 10.1101/gr.731003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers E.W. Sutton G.G. Delcher A.L., et al. A whole-genome assembly of Drosophila. Science. 2000;287:2196–2204. doi: 10.1126/science.287.5461.2196. [DOI] [PubMed] [Google Scholar]
- Nagarajan N. Read T.D. Pop M. Scaffolding and validation of bacterial genome assemblies using optical restriction maps. Bioinformatics. 2008;24:1229–1235. doi: 10.1093/bioinformatics/btn102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nandi T. Ong C. Singh A.P., et al. A genomic survey of positive selection in Burkholderia pseudomallei provides insights into the evolution of accidental virulence. PLoS Pathogens. 2010;6:4. doi: 10.1371/journal.ppat.1000845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng P. Tan J.J. Ooi H.S., et al. Multiplex sequencing of paired-end ditags (MS-PET): a strategy for the ultra-high-throughput analysis of transcriptomes and genomes. Nucleic Acids Res. 2006;34:e84. doi: 10.1093/nar/gkl444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pevzner P.A. Tang H. Fragment assembly with double-barreled data. Bioinformatics. 2001;17:225–233. doi: 10.1093/bioinformatics/17.suppl_1.s225. [DOI] [PubMed] [Google Scholar]
- Pop M. Kosack S.D. Salzberg S.L. Hierarchical scaffolding with bambus. Genome Res. 2004;14:149–159. doi: 10.1101/gr.1536204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pop M. Phillipy A. Delcher A.L., et al. Comparative genome assembly. Brief. Bioinform. 2004;5:237–248. doi: 10.1093/bib/5.3.237. [DOI] [PubMed] [Google Scholar]
- Pop M. Shotgun sequence assembly. Adv. Computers. 2004;60:1. [Google Scholar]
- Richter D.C. Schuster S.C. Huson D.H. OSLay: optimal syntenic layout of unfinished assemblies. Bioinformatics. 2007;23:1573–1579. doi: 10.1093/bioinformatics/btm153. [DOI] [PubMed] [Google Scholar]
- Richter D.C. Ott F. Schmid R., et al. Metasim: a sequencing simulator for genomics and metagenomics. PloS One. 2008;3:10. doi: 10.1371/journal.pone.0003373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxe J. Dynamic programming algorithms for recognizing small-bandwidth graphs in polynomial time. SIAM J. Algebraic Discr. Methods. 1980;1:363–369. [Google Scholar]
- Zerbino D.R. McEwen G.K. Marguiles E.H., et al. Pebble and rock band: heuristic resolution of repeats and scaffolding in the velvet short-read de novo assembler. PLoS ONE. 2009;4:12. doi: 10.1371/journal.pone.0008407. [DOI] [PMC free article] [PubMed] [Google Scholar]