

Abstract
Purpose of review
This review summarizes the recent data on the ‘Autoimmune Concept of Atherosclerosis’, according to which the first stage of this disease is due to an autoimmune reaction against arterial endothelial cells expressing heat shock protein 60 (HSP60) and adhesion molecules when stressed by classical atherosclerosis risk factors. Special emphasis is put on oxidized low-density lipoproteins as early endothelial stressors.
Recent findings
Plasma cholesterol and LDL levels considered ‘normal’ by the medical community are possibly too high from an evolutionary viewpoint. The proinflammatory milieu at sites of early atherosclerotic lesions could be conducive to oxidation of LDL in situ. LDL oxidation can also take place at nonvascular sites or in the circulation under general proinflammatory conditions explaining its proatherosclerotic role in ‘normocholesterolemic’ individuals.
Summary
We hypothesize that the plasma cholesterol and LDL levels currently considered normal are evolutionarily too high. Cholesterol and/or oxidized low-density lipoprotein, even as a mild HSP60-inducing endothelial stressor, function as a ubiquitous risk factor. If this hypothesis is true, most members of developed societies might be at risk to develop atherosclerotic plaques at anti-HSP60-immunity-triggered intimal inflammatory foci, irrespective of the primary risk-factor(s).
Keywords: atherosclerosis, cholesterol, classical atherosclerosis risk factors, heat shock protein, oxidized low-density lipoprotein, vascular-associated dendritic cells
Introduction
Inflammatory processes have been demonstrated pathohistologically in advanced atherosclerotic lesions (plaques). This review focuses on the incipient inflammatory stage of atherogenesis. In early lesions, activated T cells are the first mononuclear cells invading the arterial intima [1-3], which is already populated with a network of dendritic cells [4]. Next, monocytes (macrophages in situ), a few B cells, and finally vascular smooth muscle cells (SMCs) [1] immigrate into the intima. A depletion of T or B cells leads to an attenuation of atherosclerosis [5,6••,7]. However, T cells seem to be essential for atherosclerosis development, whereas B cells and antibodies play an accelerating and perpetuating role. In late lesions, macrophages, together with vascular SMCs, represent the most abundant subpopulation by far thus exceeding T-cell numbers [1,3]. Although intra-lesional fibroblasts and extracellular matrix (ECM) components, especially by formation of the fibrous lesional cap, contribute to plaque stability, the progressive proinflammatory conditions also lead to upregulation of matrix metalloproteinase (MMP) expression and concomitant downregulation of tissue inhibitors of metalloproteinases (TIMPs), culminating in plaque rupture [8-10]. These inflammatory processes are the basis of a vicious cycle mediated by the release of increasing quantities of proinflammatory cytokines and chemokines, promoting the influx of additional inflammatory cells into the intima. Quantitative and qualitative deficiency of T regulatory cells (Tregs) seems to significantly contribute to intralesional T effector (Teff) cells getting out of control [11,12•].
With respect to innate immunity, triggering by microbial components, for example, via binding to Toll-like receptors (TLRs), and activation of signaling via the MyD88–IRAK pathway, emerged as the most likely atherogenic mechanism [13,14,15••,16•]. In addition, TLR stimulation promotes the accumulation of lipids in macrophages and consequently foam cell formation [17-19]. With respect to adaptive immunity, biochemically altered oxidized low-density lipoproteins (oxLDLs), as well as heat shock protein 60 (HSP60) might be responsible for direct immune stimulation of Teffs or immune-complex deposition in atherosclerotic plaques [20].
The autoimmune concept of atherosclerosis
The ‘Autoimmune Hypothesis of Atherosclerosis’, according to which the earliest stage of atherosclerosis is due to an autoimmune reaction against HSP60, was first formulated in 1992 [21]. It was then just a hypothesis based on our experimental and clinical findings [21]. Today, based on a wealth of data supporting the hypothesis, one can rightly speak of the ‘Autoimmune Concept of Atherosclerosis’, recently discussed in great detail [22•• ]. The ‘Autoimmune Concept of Atherosclerosis’ is summarized in Fig. 1. The core of this concept is that an HSP60-induced inflammatory process initiates atherosclerosis, and all other events, including the formation of foam cells, which occur later.
Figure 1. The ‘autoimmune concept of atherosclerosis’ is based on the well proven fact that all healthy human beings develop protective immunity against microbial (bacterial and parasitic) heat shock protein 60 as well as bona fide physiological autoimmunity against biochemically modified autologous HSP60 produced by and released from stressed and/or disintegrated cells.

Heat shock protein 60 (HSP60) of various bacterial species displays over 95% sequence homology, and there is over 50% homology at the DNA and protein levels between prokaryotic and eukaryotic (including human) HSP60. When arterial endothelial cells are stressed by classical atherosclerosis risk factors, they simultaneously express HSP60 and adhesion molecules (ICAM-1, VCAM-1, and ELAM-1) on their surface, making them target cells for pre-existing beneficial cellular and humoral anti-HSP60 immunity. HSP60-reactive T cells invade the arterial intima and initiate the first inflammatory stage of atherosclerosis, which is followed by monocytes/macrophages and vascular smooth muscle cells. Anti-hHSP60 (auto)antibodies accelerate and perpetuate the disease. This first inflammatory stage of atherosclerosis is still reversible. However, in the continued presence of risk factors, severe advanced atherosclerosis (plaques) develops with deleterious consequences, such as myocardial infarction, stroke, and claudication. Thus, atherosclerosis is ‘the price we pay for pre-existing anti-HSP60 immunity’ if we maltreat our arteries by exposing them to classical risk factors.  , HSP60;
, HSP60;  , altered HSP60;
, altered HSP60;  , cell-specific proteins;
, cell-specific proteins;  , adhesion molecules;
, adhesion molecules;  , T cell;
, T cell;  , anti-hHSP60 antibody. Partly adapted from Servier Medical Art.
, anti-hHSP60 antibody. Partly adapted from Servier Medical Art.
According to in-vitro and in-vivo findings, the incipient inflammatory stage of atherosclerosis results from various classical atherosclerosis risk factors. All classical risk factors lead to the simultaneous surface expression of HSP60 and adhesion molecules on endothelial cells and thus are early endothelial stressors. Figure 2 is a schematic representation of the HSP60-inducing potential of various atherosclerosis risk factors expressed as approximately fold abundance of expression at RNA and protein levels. Arterial endothelial cells, lifelong ‘prestressed’ by the higher arterial blood pressure and flow conditions, are more susceptible to stress induced by different risk factors than venous endothelial cells [23,24]. Venous bypass conduits subjected to arterial blood pressure and flow conditions also develop atherosclerosis-like lesions and finally restenosis [23,24]. Recently, this also has been shown in an aortic valve regurgitation model [25]. Synthesis of proinflammatory cytokines, chemokines, and growth factors also contribute significantly to endothelial cell dysfunction. Human endothelial cells exposed to increased laminar shear stress display a specific change in behavior and modified gene expression, most probably entailing atheroprotective effects [26•].
Figure 2. All classical atherosclerosis risk factors studied in our laboratory so far act as endothelial cell stressors leading to the simultaneous surface expression of heat shock protein 60 and adhesion molecules (ICAM-1, VCAM-1, and ELAM-1).
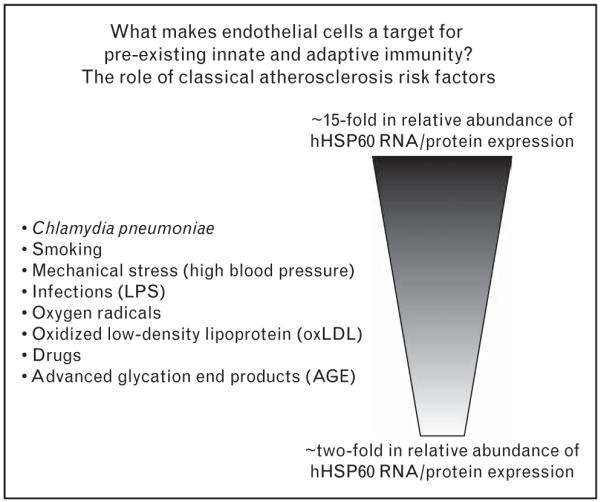
An approximate ranking of the respective stressor potential on protein and/or RNA levels is given. All data in the figure relate to the in-vitro treatment of human umbilical vein endothelial cells (HUVECS). The invivo effects of mechanical stress and bacterial lipopolysaccharide (LPS) have been shown in rats and rabbits, respectively. In several instances, comparative results were obtained with human arterial vs. venous endothelial cells showing a higher susceptibility of the former for the stressor effect of risk factors.
Vascular-associated dendritic cells
Antigen presentation is a key event in adaptive immune response. Dendritic cells, professional antigen-presenting cells (APCs), induce primary and secondary immune responses. Resident vascular-associated dendritic cells (VADCs) are present within the tertiary lymphoid structures in the aortic adventitia of atherosclerotic vessels [27•]. These might represent a unique subpopulation of dendritic cells, with distinct ultrastructural features including a unique tubulovesicular system. Ultrastructural investigations of normal arterial intima and early atherosclerotic lesions reveal two main populations of VADCs – type I and type II [28,29]. These become increasingly heterogeneous during lesion development [30]. In atherosclerotic lesions, dendritic cells often form clusters with T cells, suggesting that direct activation of dendritic cells might occur within the arterial wall [30]. Accumulation of dendritic cells in arterial lesions is associated with plaque growth and inflammation [31]. Dendritic cells are irregularly distributed in atherosclerotic plaques, with the highest frequency in areas of neovascularization in the shoulder of the plaque. Interestingly, not all foci of plaque neovascularization contain large numbers of lymphocytes but all areas of neovascularization consistently contain VADCs [30].
Newly arrived dendritic cells and dendritic cell precursors invading the aortic lesion might come not only from the circulation but also from the adventitia via vasa vasorum. Dendritic cells may easily migrate from vasa vasorum to capillaries [32,33], and ingrowth of vasa vasorum together with the lymphatics to advanced atherosclerotic plaques might facilitate the exchange of dendritic cell precursors and maturing dendritic cells between the arterial tissue and lymph [29,30]. We have shown earlier that the intima of healthy human babies, children, and young adults harbors an accumulation of mononuclear cells exactly at those sites subjected to hemodynamic stress (turbulent flow) that tend to develop atherosclerotic lesions later in life. In analogy to the mucosa-associated lymphoid tissue (MALT), we designated these accumulations as vascular-associated lymphoid tissue (VALT) [3,4,34,35]. VALT consists of Langerhans-like VADCs, T cells, resident macrophages, and scarce mast cells distributed throughout the subendothelial layer of the arterial intima of normocholesterolemic humans, rabbits, and wild type C57BL/6J mice [3,4,36,37]. In the arterial wall, VADCs may have a surveillance role, and by priming T cells and cross-presenting these antigens to T cells, they may facilitate tolerization against autoantigens [38-40]. Prolonged contact of blood with the arterial wall in areas exposed to turbulent flow compared to areas subjected to laminar shear stress enable VADCs to capture potentially harmful exogenous or autoantigens and present these to T cells. T cells entering the intima may also be tolerized against arterial antigens presented by VADCs [27•,41].
Scavenger receptors, expressed on dendritic cells, can mediate oxLDL uptake and induce a proinflammatory cytokine profile triggering dendritic cell maturation and differentiation [42]. Intimal lipids can also be taken up by resident intimal dendritic cells [43•• ]. Mice receiving malondialdehyde-modified LDL (MDA-LDL) pulsed myeloid dendritic cells show more extensive atherosclerotic lesions than controls, with increased inflammatory hallmarks and antigen-specific immune responses [44]. Deficiency of APC invariant chain (CD74) reduces atherosclerosis in mice and impairs adaptive immune response to ‘endogenous’ disease-specific antigens (MDA-LDL and HSP60) [45]. In the absence of CD11c+ cells, there was a striking 55% reduction in early lipid accumulation in the aortic wall [46]. Accumulated lipid was found only in extracellular spaces, apparently ignored by circulating monocytes. These data suggest that, under hypercholesterolemic conditions, VADCs can regulate the accumulation of lipid in the earliest stages of plaque formation and are central in the atherosclerosis process because of their direct effect on both cholesterol homeostasis and immune responses [47]. Whether VADCs can capture circulating native and/or oxidized LDL that may interact with the endothelium and thereby collecting much of the earliest cholesterol that accumulates in the arterial intima is presently unclear.
Tolerogenic dendritic cells have recently been shown to be useful in treating autoimmune disorders and improving allograft survival. Tolerogenic dendritic cells pulsed with native ApoB100 reduce the autoimmune response against LDL and attenuate atherosclerosis [48•• ]. Furthermore, vaccination using oxLDL-pulsed mature dendritic cells effectively reduces atherosclerotic lesion formation, inducing oxLDL-specific T cells, and enhancing the levels of anti-oxLDL IgG [49]. The therapeutic use of dendritic cell-pulsed antigen(s) to induce a specific tolerogenic response is limited, but encouraging.
Circulating levels of oxidized low-density lipoproteins
Many authors believe that LDL oxidation does not take place in the circulation, and therefore it must occur in the subendothelial space of the arterial wall. However, small quantities of lipoprotein with many of the characteristics of oxLDL have been found circulating in human plasma [50] with higher levels in atherosclerotic patients than healthy controls [51]. No correlation was found between clinical symptoms and plasma oxLDL levels, indicating its pathogenic role in the early stages of the disease. This is also supported by the fact that plasma oxLDL levels are elevated prior to atherosclerosis progression [52]. Furthermore, the reduction in plasma oxLDL levels coincided with lesion development and oxLDL accumulation in the atherosclerotic intima [53], suggesting that plasma oxLDL can migrate between the intimal regions and circulation. Increased levels of oxLDL are present in human gingival crevicular fluid compared to plasma of healthy individuals [54], indicating that oxLDL could be generated in inflamed extra-arterial tissues, transferred to the circulation, rapidly taken up into the arterial wall, and contribute to the perpetuation of atherosclerosis.
The presence of circulating antibodies against oxLDL suggests its availability as an antigen also outside the vascular system [55]. A variety of oxidized lipid products also have been identified in plasma, and degradation products of some of these have even been demonstrated in urine, with increased levels in atherosclerosis patients [56]. The source of these compounds is not clear. Cholesterol crystals, detected not only in the necrotic cores but also in the subendothelial areas in early atherosclerotic settings, trigger inflammasome activation both in humans and in mouse, leading to interleukin-1β secretion [57 ••,58]. Their possible source may be the circulation.
Oxidized LDL in early atherosclerotic lesions
LDL can be oxidized enzymatically, nonenzymatically by transition metal ions, and by other catalysts, triggering both innate and adaptive immunity. The process of LDL oxidation is assumed to occur in two main steps. Minimally oxLDL (mmLDL), which retains its affinity to the LDL receptor, activates antiapoptotic signaling and induces inflammatory changes. The recruitment of inflammatory cells may result in the production of a large variety of cytokines and chemokines that could continue the oxidation process of LDL [59]. With further LDL lipid oxidization, and LDL protein modification, loss of recognition by the LDL receptor and a shift to recognition by scavenger receptors occur, leading to foam cell formation, predominantly in anti-inflammatory (M2) macrophages. As a result, activated M2 macrophages shift to a proinflammatory profile [60•]. A variety of macrophage phenotypes have been found in plaques, which may all have different impacts on facets of plaque development [61]. The modified forms of LDL are more proatherogenic than native LDL. Mucosal immunizations with native LDL peptides show atheroprotective effects [62]. Previously, no T cells were believed to react with native LDL components. New data show that peripheral T cells in atherosclerotic mice recognize peptide motifs of native LDL particles and ApoB100 and, surprisingly, oxidation extinguishes rather than promotes LDL-dependent T-cell activation [63•• ]. oxLDL-reactive T cells can be localized in plaques, lymph nodes, and in the plasma of atherosclerosis patients and experimental animals. oxLDL has been demonstrated to precede accumulation of monocytes and formation of fatty streaks [64]. Several mechanisms for increased monocyte recruitment attributable to the presence of modified LDL in the arterial wall have been identified. Modified LDL can enhance monocyte adhesion by inducing the expression of adhesion molecules, chemotactic, and growth factors in endothelial cells, and also activate platelets, with increased platelet-monocyte aggregation, and monocyte adherence to the endothelium [65,66•,67]. The major pathways in the early lesion development are summarized in Fig. 3.
Figure 3. Compared with venous endothelial cells, arterial endothelial cells are exposed to higher mechanical stress because of the arterial blood pressure and flow conditions.

This is especially true at branching points subjected to an increased turbulent hemodynamic shear stress predisposing these sites for the development of atherosclerotic lesions. (1–2) Classical atherosclerosis risk factors first act as endothelial stressors leading to the simultaneous expression of heat shock protein 60 (HSP60) and adhesion molecules. (3) HSP60-reactive T cells adhere to stressed endothelial target cells and transmigrate into the intima with its pre-existing network of Langerhans-like dendritic cells that act as antigen-processing and antigen-presenting cells similar to the situation in the skin. Whether sensitization of T cells attracted to these sites occurs in situ or in the draining lymph nodes remains to be seen. (4–6) Whether LDL oxidation occurs already in the circulation or in the mononuclear cell infiltrate in the intima by oxidative stress is still a matter of debate. It is, however, well proven that oxLDL itself acts as an endothelial stressor leading to HSP60 expression. Furthermore, activated platelets can bind LDL in the circulation, adhere to endothelial cells, and mediate monocyte attachment. Monocytes (loaded or not with LDL) can then migrate into the primary T-cell-dominated inflammatory intima where they can differentiate into macrophages. In a further step, smooth muscle cells (SMCs) from the media are attracted into the intima. Macrophages, dendritic cells, and vascular SMCs possess scavenger receptors, which enable them to take up oxLDL and transform into foam cells. (7) T cells initiate the disease, and local immune reaction creates a microenvironment with an altered balance of pro-inflammatory and anti-inflammatory mediators. (8) (Auto)antibodies accelerate and perpetuate the disease. (9) Continuing presence of risk factors led to endothelial damage. Partly adapted from Servier Medical Art.
Thus, although there is substantial evidence that oxidized forms of LDL are produced in the arteries of mice as well as men, definite data to establish that the lipids that generate foam cells derive from these oxidized lipoproteins, as opposed to native, aggregated, or non-oxidatively modified forms of LDL, are not yet available.
Moreover, if an increased oxLDL level is such an important – if not the most important – risk factor for the initiation and progression of the disease, why do the majority of patients display normal cholesterol serum levels?
Why is cholesterol such an important atherosclerosis risk factor?
In more than 60% of patients with clinical symptoms of atherosclerosis, total blood cholesterol levels and LDL levels are within the range currently considered to be ‘normal’, namely, 200 mg/dl or less for total cholesterol and 100 mg/dl or less for LDL cholesterol. Concentrations of native LDL in extracts of human intimal samples typically exceed 100 mg/dl. There are notable examples in which the primary atherogenetic role of high LDL cholesterol levels is obvious, such as cases of human heterozygous or homozygous familial hypercholesterolemia due to mutations of the LDL receptor. However, this condition does not reflect the ‘poor man’s’ atherosclerosis that to a large extent afflicts normocholesterolemic patients.
An explanation for the accumulation of LDL in atherosclerotic lesions of normocholesterolemic patients is still lacking. Our hypothesis is that cholesterol levels of human beings considered ‘normocholesterolemic’ in terms of current medical criteria are still significantly hypercholesterolemic from an evolutionary viewpoint. We are equipped with a stone-age genome, but live in times where cultural evolution has far outpaced biological evolution. Populations following a more ‘Western’ life-style, for instance, show relatively high serum cholesterol/LDL concentrations and a significantly higher rate of atherosclerosis than those adhering to a Mediterranean lifestyle regarding nutrition and physical exercise [68,69]. Recent imaging studies of 4000-year-old Egyptian mummies revealed the presence of atherosclerotic lesions in some of these suggesting that atherosclerosis is not exclusively a modern disease [70-72]. However, embalming and thus preservation of bodies, organs, and vessels was a privilege of affluent segments of these societies and do not reflect the situation of the malnourished majority enduring hard physical labor and probably displaying very low cholesterol/LDL levels. Also, after World Wars I and II, prisoners returning home after years of severe hunger combined with exhausting labor suffered from many diseases, but not atherosclerosis [73]. Furthermore, statins can reduce the incidence of major cardiovascular events in healthy persons without hypercholesterolemia (LDL cholesterol levels of ≤130 mg/dl) but with elevated high-sensitive C-reactive protein levels. These effects were consistent in all sub-groups evaluated, including those customarily considered to be at low risk (LDL cholesterol levels of ≤100 mg/dl) [74]. In addition, both statins and aspirin have been shown to have anti-inflammatory properties [74-76].
We hypothesize that a plethora of different risk factors can initially act as endothelial cell stressors and lead to the first inflammatory-immunological stage of atherosclerosis. In persons currently considered ‘normocholesterolemic’, these create local conditions that foster the influx of native and/or oxLDL also from the circulation. Plasma cholesterol or modified LDL levels are emerging as major risk factors in practically all clinical studies, as they are too high from the evolutionary point of view, act as primary endothelial cell stressors alone or together with other risk factors, and make an essential contribution to lesion formation proceeding from early nonfoam cell-dominated inflammatory lesions to fatty streaks and atheromas in ‘normocholesterolemic’ persons.
Conclusion
Our hypothesis is that from an evolutionary viewpoint, plasma cholesterol levels currently considered ‘normal’ are far too high in the majority of humans. Cholesterol – even as a relatively mild adhesion molecule and HSP60-inducing endothelial cell stressor – is a ubiquitous risk factor. If true, irrespective of their primary risk factor(s), most members of developed societies are at risk of developing fatty streaks and even atherosclerotic plaques, because of the presence of intimal inflammatory foci arising from pre-existing anti-HSP60 immunity.
Key points.
All classical atherosclerosis risk factors can act as endothelial stressors provoking the simultaneous surface expression of HSP60 and adhesion molecules at known arterial predilection sites.
HSP60 can then act as a ‘danger signal’ for the immune system recognized by pre-existing innate and adaptive anti-HSP60 immunity.
T cells predominate over macrophages in early atherosclerotic lesions. An increased number of vascular-associated dendritic cells can be found in areas of hemodynamic stress and they may play a role in early tolerization against autoantigens.
oxLDL exert relatively low levels of stress on endothelial cell but are nevertheless the most common risk factor for atherosclerosis. oxLDL lead to foam cell formation at sites of primary inflammation/immunological reaction.
The importance of cholesterol and oxLDL in atherogenesis may be because of the fact that from an evolutionary viewpoint, people currently considered ‘normocholesterolemic’ are in fact hypercholesterolemic.
Acknowledgements
This work has been supported by the Austrian Research Fund (FWF; P19881-B05), the EU Framework Program 6 (MOLSTROKE, LSHM-CT-2004-005206, EVGN; LSHM-CT-2003-S03254), the EU Framework Program 7, Large Scale Integrated Project: Novel approaches to reconstitute normal immune function at old age (TOLERAGE Health research grant; HEALTH-F4-2008-202156), the ERANET PathoGenoMics Program (European Initiative to Fight Chlamydial Infections by Unibased Genomics – ECIBUG), and the Propter Homines Foundation, FL.
Footnotes
Conflicts of interest: There are no conflicts of interest.
References and recommended reading
Papers of particular interest, published within the annual period of review, have been highlighted as:
• of special interest
•• of outstanding interest
Additional references related to this topic can also be found in the Current World Literature section in this issue (p 417).
- 1.Xu QB, Oberhuber G, Gruschwitz M, Wick G. Immunology of atherosclerosis: cellular composition and major histocompatibility complex class II antigen expression in aortic intima, fatty streaks, and atherosclerotic plaques in young and aged human specimens. Clin Immunol Immunopathol. 1990;56:344–359. doi: 10.1016/0090-1229(90)90155-j. [DOI] [PubMed] [Google Scholar]
- 2.Kleindienst R, Xu Q, Willeit J, et al. Immunology of atherosclerosis. Demonstration of heat shock protein 60 expression and T lymphocytes bearing alpha/beta or gamma/delta receptor in human atherosclerotic lesions. Am J Pathol. 1993;142:1927–1937. [PMC free article] [PubMed] [Google Scholar]
- 3.Millonig G, Malcom GT, Wick G. Early inflammatory-immunological lesions in juvenile atherosclerosis from the Pathobiological Determinants of Atherosclerosis in Youth (PDAY)-study. Atherosclerosis. 2002;160:441–448. doi: 10.1016/s0021-9150(01)00596-2. [DOI] [PubMed] [Google Scholar]
- 4.Millonig G, Niederegger H, Rabl W, et al. Network of vascular-associated dendritic cells in intima of healthy young individuals. Arterioscler Thromb Vasc Biol. 2001;21:503–508. doi: 10.1161/01.atv.21.4.503. [DOI] [PubMed] [Google Scholar]
- 5.Song L, Leung C, Schindler C. Lymphocytes are important in early atherosclerosis. J Clin Invest. 2001;108:251–259. doi: 10.1172/JCI11380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6 ••.Ait-Oufella H, Herbin O, Bouaziz JD, et al. B cell depletion reduces the development of atherosclerosis in mice. J Exp Med. 2010;207:1579–1587. doi: 10.1084/jem.20100155. [In contrast to the current concepts, this study shows that B cell activation plays an overall protective role in atherogenesis. This was shown after mature B cell depletion using a CD20-specific monoclonal antibody, which induced a significant reduction of atherosclerosis in various mouse models of the disease.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kyaw T, Tay C, Khan A, et al. Conventional B2 B cell depletion ameliorates whereas its adoptive transfer aggravates atherosclerosis. J Immunol. 2010;185:4410–4419. doi: 10.4049/jimmunol.1000033. [DOI] [PubMed] [Google Scholar]
- 8.Newby AC, George SJ, Ismail Y, et al. Vulnerable atherosclerotic plaque metalloproteinases and foam cell phenotypes. Thromb Haemost. 2009;101:1006–1011. [PMC free article] [PubMed] [Google Scholar]
- 9.Gaubatz JW, Ballantyne CM, Wasserman BA, et al. Association of circulating matrix metalloproteinases with carotid artery characteristics: the Atherosclerosis Risk in Communities Carotid MRI Study. Arterioscler Thromb Vasc Biol. 2010;30:1034–1042. doi: 10.1161/ATVBAHA.109.195370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Musial K, Zwolinska D. Matrix metalloproteinases (MMP-2,9) and their tissue inhibitors (TIMP-1,2) as novel markers of stress response and atherogenesis in children with chronic kidney disease (CKD) on conservative treatment. Cell Stress Chaperones. 2011;16:97–103. doi: 10.1007/s12192-010-0214-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mallat Z, Taleb S, Ait-Oufella H, Tedgui A. The role of adaptive T cell immunity in atherosclerosis. J Lipid Res. 2009;50(Suppl):S364–S369. doi: 10.1194/jlr.R800092-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12 •.Taleb S, Tedgui A, Mallat Z. Adaptive T cell immune responses and atherogenesis. Curr Opin Pharmacol. 2010;10:197–202. doi: 10.1016/j.coph.2010.02.003. [This review summarizes the current knowledge on the role of effector and regulatory T-cell immunity in atherosclerosis and discusses the contribution of the Th17 cells to this disease.] [DOI] [PubMed] [Google Scholar]
- 13.Bjorkbacka H, Kunjathoor VV, Moore KJ, et al. Reduced atherosclerosis in MyD88-null mice links elevated serum cholesterol levels to activation of innate immunity signaling pathways. Nat Med. 2004;10:416–421. doi: 10.1038/nm1008. [DOI] [PubMed] [Google Scholar]
- 14.Michelsen KS, Wong MH, Shah PK, et al. Lack of Toll-like receptor 4 or myeloid differentiation factor 88 reduces atherosclerosis and alters plaque phenotype in mice deficient in apolipoprotein E. Proc Natl Acad Sci U S A. 2004;101:10679–10684. doi: 10.1073/pnas.0403249101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15 ••.West XZ, Malinin NL, Merkulova AA, et al. Oxidative stress induces angiogenesis by activating TLR2 with novel endogenous ligands. Nature. 2010;467:972–976. doi: 10.1038/nature09421. [This study shows that the end products of lipid oxidation are recognized by TLR2 in a MyD88-dependent fashion on endothelial cells. These are generated during inflammation and wound healing and can accumulate at high levels in aging tissues. Conclusively, these findings demonstrate a new function of TLR2 as a sensor of oxidation-associated molecular patterns, providing a key link connecting inflammation, oxidative stress, innate immunity, and angiogenesis.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16 •.Cole JE, Navin TJ, Cross AJ, et al. Unexpected protective role for Toll-like receptor 3 in the arterial wall. Proc Natl Acad Sci U S A. 2011;108:2372–2377. doi: 10.1073/pnas.1018515108. [In contrast to the current concepts, TLRs are not always detrimental in vascular disease but can be relevant in repair mechanisms within the vascular wall. The authors of this study show that neointimal formation in a perivascular collar-induced injury model was reduced by the systemic administration of a dsRNA analog in a TLR3-dependent manner. Genetic deletion of TLR3 enhanced the development of elastica lamina damage and TLR3 deficiency accelerates the onset of atherosclerosis in this model.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nicolaou G, Erridge C. Toll-like receptor-dependent lipid body formation in macrophage foam cell formation. Curr Opin Lipidol. 2010;21:427–433. doi: 10.1097/MOL.0b013e32833cacd5. [DOI] [PubMed] [Google Scholar]
- 18.Higashimori M, Tatro JB, Moore KJ, et al. Role of Toll-like receptor 4 in intimal foam cell accumulation in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2011;31:50–57. doi: 10.1161/ATVBAHA.110.210971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miller YI, Choi SH, Wiesner P, et al. Oxidation-specific epitopes are danger-associated molecular patterns recognized by pattern recognition receptors of innate immunity. Circ Res. 2011;108:235–248. doi: 10.1161/CIRCRESAHA.110.223875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hansson GK, Hermansson A. The immune system in atherosclerosis. Nat Immunol. 2011;12:204–212. doi: 10.1038/ni.2001. [DOI] [PubMed] [Google Scholar]
- 21.Wick G, Kleindienst R, Dietrich H, Xu Q. Is atherosclerosis an autoimmune disease? Trends Food Sci Technol. 1992;3:114–119. [Google Scholar]
- 22 ••.Grundtman C, Kreutmayer SB, Almanzar G, et al. Heat shock protein 60 and immune inflammatory responses in atherosclerosis. Arterioscler Thromb Vasc Biol. 2011;31:960–968. doi: 10.1161/ATVBAHA.110.217877. [This review summarizes the most recent publications about innate and adptive immune reactions to HSP60 in the earliest stages of atherogenesis. Both beneficial protective immunity and bona fide autoimmunity are discussed.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dietrich H, Hu Y, Zou Y, et al. Mouse model of transplant arteriosclerosis: role of intercellular adhesion molecule-1. Arterioscler Thromb Vasc Biol. 2000;20:343–352. doi: 10.1161/01.atv.20.2.343. [DOI] [PubMed] [Google Scholar]
- 24.Henderson B, Tagwerker A, Mayrl C, et al. Progression of arteriovenous bypass restenosis in mice exposed to a 50 Hz magnetic field. Cell Stress Chaperones. 2003;8:373–380. doi: 10.1379/1466-1268(2003)008<0373:poabri>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou YQ, Zhu SN, Foster FS, et al. Aortic regurgitation dramatically alters the distribution of atherosclerotic lesions and enhances atherogenesis in mice. Arterioscler Thromb Vasc Biol. 2010;30:1181–1188. doi: 10.1161/ATVBAHA.110.204198. [DOI] [PubMed] [Google Scholar]
- 26 •.White SJ, Hayes EM, Lehoux S, et al. Characterisation of the differential response of endothelial cells exposed to normal and elevated laminar shear stress. J Cell Physiol. 2011 doi: 10.1002/jcp.22629. doi: 10.1002/jcp.22629. [This study shows that an increased laminar shear stress can induce a specific change in behavior, gene expression, ROS reductions, altering MAP kinase signaling, and reducing cAMP levels. These data indicate that elevated shared stress function as atheroprotective.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27 •.Cybulsky MI, Jongstra-Bilen J. Resident intimal dendritic cells and the initiation of atherosclerosis. Curr Opin Lipidol. 2010;21:397–403. doi: 10.1097/MOL.0b013e32833ded96. [This review highlights the importance of resident intimal dendritic cells located in predilected sites of the aorta and their function in the initiation of atherosclerosis.] [DOI] [PubMed] [Google Scholar]
- 28.Bobryshev YV, Lord RS. Structural heterogeneity and contacting interactions of vascular dendritic cells in early atherosclerotic lesions of the human aorta. J Submicrosc Cytol Pathol. 1996;28:49–60. [PubMed] [Google Scholar]
- 29.Bobryshev YV. Dendritic cells and their role in atherogenesis. Lab Invest. 2010;90:970–984. doi: 10.1038/labinvest.2010.94. [DOI] [PubMed] [Google Scholar]
- 30.Bobryshev YV, Lord RS. Mapping of vascular dendritic cells in atherosclerotic arteries suggests their involvement in local immune-inflammatory reactions. Cardiovasc Res. 1998;37:799–810. doi: 10.1016/s0008-6363(97)00229-0. [DOI] [PubMed] [Google Scholar]
- 31.Liu P, Yu YR, Spencer JA, et al. CX3CR1 deficiency impairs dendritic cell accumulation in arterial intima and reduces atherosclerotic burden. Arterioscler Thromb Vasc Biol. 2008;28:243–250. doi: 10.1161/ATVBAHA.107.158675. [DOI] [PubMed] [Google Scholar]
- 32.Bobryshev YV, Lord RS. Vascular-associated lymphoid tissue (VALT) involvement in aortic aneurysm. Atherosclerosis. 2001;154:15–21. doi: 10.1016/s0021-9150(00)00441-x. [DOI] [PubMed] [Google Scholar]
- 33.Shen LH, Zhou L, Wang BY, et al. Oxidized low-density lipoprotein induces differentiation of RAW264.7 murine macrophage cell line into dendritic-like cells. Atherosclerosis. 2008;199:257–264. doi: 10.1016/j.atherosclerosis.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 34.Wick G, Romen M, Amberger A, et al. Atherosclerosis, autoimmunity, and vascular-associated lymphoid tissue. FASEB J. 1997;11:1199–1207. doi: 10.1096/fasebj.11.13.9367355. [DOI] [PubMed] [Google Scholar]
- 35.Waltner-Romen M, Falkensammer G, Rabl W, Wick G. A previously unrecognized site of local accumulation of mononuclear cells. The vascular-associated lymphoid tissue. J Histochem Cytochem. 1998;46:1347–1350. doi: 10.1177/002215549804601202. [DOI] [PubMed] [Google Scholar]
- 36.Millonig G, Niederegger H, Wick G. Analysis of the cellular composition of the arterial intima with modified en face techniques. Lab Invest. 2001;81:639–641. doi: 10.1038/labinvest.3780273. [DOI] [PubMed] [Google Scholar]
- 37.Jongstra-Bilen J, Haidari M, Zhu SN, et al. Low-grade chronic inflammation in regions of the normal mouse arterial intima predisposed to atherosclerosis. J Exp Med. 2006;203:2073–2083. doi: 10.1084/jem.20060245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Packard RR, Maganto-Garcia E, Gotsman I, et al. CD11c(+) dendritic cells maintain antigen processing, presentation capabilities, and CD4(+) T-cell priming efficacy under hypercholesterolemic conditions associated with atherosclerosis. Circ Res. 2008;103:965–973. doi: 10.1161/CIRCRESAHA.108.185793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Choi JH, Do Y, Cheong C, et al. Identification of antigen-presenting dendritic cells in mouse aorta and cardiac valves. J Exp Med. 2009;206:497–505. doi: 10.1084/jem.20082129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhu SN, Chen M, Jongstra-Bilen J, Cybulsky MI. GM-CSF regulates intimal cell proliferation in nascent atherosclerotic lesions. J Exp Med. 2009;206:2141–2149. doi: 10.1084/jem.20090866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Niessner A, Weyand CM. Dendritic cells in atherosclerotic disease. Clin Immunol. 2010;134:25–32. doi: 10.1016/j.clim.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nickel T, Schmauss D, Hanssen H, et al. oxLDL uptake by dendritic cells induces upregulation of scavenger-receptors, maturation and differentiation. Atherosclerosis. 2009;205:442–450. doi: 10.1016/j.atherosclerosis.2009.01.002. [DOI] [PubMed] [Google Scholar]
- 43 ••.Paulson KE, Zhu SN, Chen M, et al. Resident intimal dendritic cells accumulate lipid and contribute to the initiation of atherosclerosis. Circ Res. 2010;106:383–390. doi: 10.1161/CIRCRESAHA.109.210781. [The authors of this study shows that an induction of hypercholesterolemia in mice can trigger a rapid ingestion of lipid by resident intimal dendritic cells, which can lead to an initiation of nascent foam cell lesion formation. Furthermore, when intimal dendritic cells were depleted the surface area of lipid accumulation was strongly reduced.] [DOI] [PubMed] [Google Scholar]
- 44.Hjerpe C, Johansson D, Hermansson A, et al. Dendritic cells pulsed with malondialdehyde modified low density lipoprotein aggravate atherosclerosis in Apoe(−/−) mice. Atherosclerosis. 2010;209:436–441. doi: 10.1016/j.atherosclerosis.2009.10.003. [DOI] [PubMed] [Google Scholar]
- 45.Sun J, Hartvigsen K, Chou MY, et al. Deficiency of antigen-presenting cell invariant chain reduces atherosclerosis in mice. Circulation. 2010;122:808–820. doi: 10.1161/CIRCULATIONAHA.109.891887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jung S, Unutmaz D, Wong P, et al. In vivo depletion of CD11c+ dendritic cells abrogates priming of CD8+ T cells by exogenous cell-associated antigens. Immunity. 2002;17:211–220. doi: 10.1016/s1074-7613(02)00365-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gautier EL, Huby T, Saint-Charles F, et al. Conventional dendritic cells at the crossroads between immunity and cholesterol homeostasis in atherosclerosis. Circulation. 2009;119:2367–2375. doi: 10.1161/CIRCULATIONAHA.108.807537. [DOI] [PubMed] [Google Scholar]
- 48 ••.Hermansson A, Johansson DK, Ketelhuth DF, et al. Immunotherapy with tolerogenic apolipoprotein B-100-loaded dendritic cells attenuates atherosclerosis in hypercholesterolemic mice. Circulation. 2011;123:1083–1091. doi: 10.1161/CIRCULATIONAHA.110.973222. [In this study, the authors demonstrate that tolerogenic dendritic cells pulsed with ApoB100 reduced the autoimmune response against LDL, reduced atherosclerotic lesions in the aorta, decreased T-cell infiltration, and decreased signs of systemic inflammation. This treatment may therefore represent a novel possibility for treatment or prevention of atherosclerosis.] [DOI] [PubMed] [Google Scholar]
- 49.Habets KL, van Puijvelde GH, van Duivenvoorde LM, et al. Vaccination using oxidized low-density lipoprotein-pulsed dendritic cells reduces atherosclerosis in LDL receptor-deficient mice. Cardiovasc Res. 2010;85:622–630. doi: 10.1093/cvr/cvp338. [DOI] [PubMed] [Google Scholar]
- 50.Sevanian A, Hwang J, Hodis H, et al. Contribution of an in vivo oxidized LDL to LDL oxidation and its association with dense LDL subpopulations. Arterioscler Thromb Vasc Biol. 1996;16:784–793. doi: 10.1161/01.atv.16.6.784. [DOI] [PubMed] [Google Scholar]
- 51.Nishi K, Itabe H, Uno M, et al. Oxidized LDL in carotid plaques and plasma associates with plaque instability. Arterioscler Thromb Vasc Biol. 2002;22:1649–1654. doi: 10.1161/01.atv.0000033829.14012.18. [DOI] [PubMed] [Google Scholar]
- 52.Kato R, Mori C, Kitazato K, et al. Transient increase in plasma oxidized LDL during the progression of atherosclerosis in apolipoprotein E knockout mice. Arterioscler Thromb Vasc Biol. 2009;29:33–39. doi: 10.1161/ATVBAHA.108.164723. [DOI] [PubMed] [Google Scholar]
- 53.Itabe H, Obama T, Kato R. The dynamics of oxidized LDL during atherogenesis. J Lipids. 2011;2011:418313. doi: 10.1155/2011/418313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sakiyama Y, Kato R, Inoue S, et al. Detection of oxidized low-density lipoproteins in gingival crevicular fluid from dental patients. J Periodontal Res. 2010;45:216–222. doi: 10.1111/j.1600-0765.2009.01226.x. [DOI] [PubMed] [Google Scholar]
- 55.Binder CJ. Natural IgM antibodies against oxidation-specific epitopes. J Clin Immunol. 2010;30(Suppl 1):S56–S60. doi: 10.1007/s10875-010-9396-3. [DOI] [PubMed] [Google Scholar]
- 56.Cavalca V, Minardi F, Scurati S, et al. Simultaneous quantification of 8-iso-prostaglandin-F(2alpha) and 11-dehydro thromboxane B(2) in human urine by liquid chromatography-tandem mass spectrometry. Anal Biochem. 2010;397:168–174. doi: 10.1016/j.ab.2009.10.014. [DOI] [PubMed] [Google Scholar]
- 57 ••.Duewell P, Kono H, Rayner KJ, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464:1357–1361. doi: 10.1038/nature08938. [The results in this study demonstrate that crystalline cholesterol acts as an endogenous danger signal and its deposition in arteries or elsewhere is an early cause rather than a late consequence of inflammation. These findings provide new insights into the pathogenesis of atherosclerosis and indicate new potential molecular targets for the therapy of this disease.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rajamaki K, Lappalainen J, Oorni K, et al. Cholesterol crystals activate the NLRP3 inflammasome in human macrophages: a novel link between cholesterol metabolism and inflammation. PLoS One. 2010;5:e11765. doi: 10.1371/journal.pone.0011765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ait-Oufella H, Taleb S, Mallat Z, Tedgui A. Recent advances on the role of cytokines in atherosclerosis. Arterioscler Thromb Vasc Biol. 2011;31:969–979. doi: 10.1161/ATVBAHA.110.207415. [DOI] [PubMed] [Google Scholar]
- 60 •.Van Tits LJ, Stienstra R, van Lent PL, et al. Oxidized LDL enhances pro-inflammatory responses of alternatively activated M2 macrophages: a crucial role for Kruppel-like factor 2. Atherosclerosis. 2011;214:345–349. doi: 10.1016/j.atherosclerosis.2010.11.018. [This study demonstrates that foam cell formation predominantly occurs in anti-inflammatory M2 macrophages. As a result, the phenotype of alternatively activated M2 macrophages shifts to a proinflammatory profile, which may play an important role in the pathogenesis of atherosclerosis.] [DOI] [PubMed] [Google Scholar]
- 61.Johnson JL, Newby AC. Macrophage heterogeneity in atherosclerotic plaques. Curr Opin Lipidol. 2009;20:370–378. doi: 10.1097/MOL.0b013e3283309848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Klingenberg R, Lebens M, Hermansson A, et al. Intranasal immunization with an apolipoprotein B-100 fusion protein induces antigen-specific regulatory T cells and reduces atherosclerosis. Arterioscler Thromb Vasc Biol. 2010;30:946–952. doi: 10.1161/ATVBAHA.109.202671. [DOI] [PubMed] [Google Scholar]
- 63 ••.Hermansson A, Ketelhuth DF, Strodthoff D, et al. Inhibition of T cell response to native low-density lipoprotein reduces atherosclerosis. J Exp Med. 2010;207:1081–1093. doi: 10.1084/jem.20092243. [The authors show that peripheral T cells in atherosclerotic mice recognize peptide motifs of native LDL particles and ApoB100, the protein moiety of LDL. Oxidation extingushes rather than promotes LDL-dependent T-cell activation. This indicates that cellular immunity towards native LDL protein might have a pathogenetic role in atherosclerosis.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Napoli C, D’Armiento FP, Mancini FP, et al. Fatty streak formation occurs in human fetal aortas and is greatly enhanced by maternal hypercholesterolemia. Intimal accumulation of low density lipoprotein and its oxidation precede monocyte recruitment into early atherosclerotic lesions. J Clin Invest. 1997;100:2680–2690. doi: 10.1172/JCI119813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Siegel-Axel D, Daub K, Seizer P, et al. Platelet lipoprotein interplay: trigger of foam cell formation and driver of atherosclerosis. Cardiovasc Res. 2008;78:8–17. doi: 10.1093/cvr/cvn015. [DOI] [PubMed] [Google Scholar]
- 66 •.Daub K, Seizer P, Stellos K, et al. Oxidized LDL-activated platelets induce vascular inflammation. Semin Thromb Hemost. 2010;36:146–156. doi: 10.1055/s-0030-1251498. [This study demonstrates that activated platlets internalize oxLDL and that oxLDL-laden platelets activate endothelium, inhibit endothelial regeneration, and promote foam cell development. Platelet oxLDL contributes to vascular inflammation and is able to promote atherosclerosis.] [DOI] [PubMed] [Google Scholar]
- 67.Ghosh A, Murugesan G, Chen K, et al. Platelet CD36 surface expression levels affect functional responses to oxidized LDL and are associated with inheritance of specific genetic polymorphisms. Blood. 2011;117:6355–6366. doi: 10.1182/blood-2011-02-338582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Panagiotakos DB, Pitsavos C, Chrysohoou C, et al. Status and management of blood lipids in Greek adults and their relation to socio-demographic, lifestyle and dietary factors: the ATTICA Study. Blood lipids distribution in Greece. Atherosclerosis. 2004;173:353–361. doi: 10.1016/j.atherosclerosis.2003.12.031. [DOI] [PubMed] [Google Scholar]
- 69.Braun LT. Cholesterol and triglyceride management: ‘if I take my medication, can I eat what I want?’. J Cardiovasc Nurs. 2010;25:241–246. doi: 10.1097/JCN.0b013e3181cec6d1. [DOI] [PubMed] [Google Scholar]
- 70.Allam AH, Thompson RC, Wann LS, et al. Computed tomographic assessment of atherosclerosis in ancient Egyptian mummies. JAMA. 2009;302:2091–2094. doi: 10.1001/jama.2009.1641. [DOI] [PubMed] [Google Scholar]
- 71.Charlier P, Huynh I. Assessment of atherosclerosis in Egyptian mummies. JAMA. 2010;303:1149–1150. doi: 10.1001/jama.2010.326. author reply 1150. [DOI] [PubMed] [Google Scholar]
- 72.David AR, Kershaw A, Heagerty A. Atherosclerosis and diet in ancient Egypt. Lancet. 2010;375:718–719. doi: 10.1016/s0140-6736(10)60294-2. [DOI] [PubMed] [Google Scholar]
- 73.Schettler G. Atherosclerosis during periods of food deprivation following World Wars I and II. Prev Med. 1983;12:75–83. doi: 10.1016/0091-7435(83)90174-3. [DOI] [PubMed] [Google Scholar]
- 74.Ridker PM, Danielson E, Fonseca FA, et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med. 2008;359:2195–2207. doi: 10.1056/NEJMoa0807646. [DOI] [PubMed] [Google Scholar]
- 75.Baigent C, Blackwell L, Collins R, et al. Aspirin in the primary and secondary prevention of vascular disease: collaborative meta-analysis of individual participant data from randomised trials. Lancet. 2009;373:1849–1860. doi: 10.1016/S0140-6736(09)60503-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dinarello CA. Anti-inflammatory agents: present and future. Cell. 2010;140:935–950. doi: 10.1016/j.cell.2010.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]