

Abstract
A growing body of evidence demonstrates that susceptibility and progression of both acute and chronic central nervous system disease in the newborn is closely associated with an innate immune response that can manifest from either direct infection and/or infection-triggered damage. A common feature of many of these diseases is the systemic exposure of the neonate to bacterial infections that elicit brain inflammation. In recent years, the importance of innate immune receptors in newborn brain injury, the so-called Toll-like receptors, has been demonstrated. In this paper we will discuss how neonatal sepsis, with particular emphasis on Escherichia coli, coagulase-negative staphylococci, and group B streptococcal infections in preterm infants, and Toll-like receptor-mediated inflammation can increase the vulnerability of the newborn brain to injury.
1. Introduction
Perinatal brain injury represents a significant clinical problem [1]. A growing body of evidence demonstrates that susceptibility and progression of both acute and chronic central nervous system (CNS) disease is closely associated with an innate immune response that can manifest from either direct infection and/or infection-triggered damage [2]. A common feature of these diseases is the systemic activation of inflammatory mediators, which via the blood can disrupt the blood-brain barrier, affect the circumventricular organs in the brain (which lack a blood-brain barrier), or interact with the brain endothelium, thereby eliciting brain inflammation [3]. Furthermore, the presence of activated inflammatory cells derived from systemic circulation or from dormant brain resident populations is a key feature of many CNS diseases. More recently, the importance of innate immune receptors in CNS injury, the so-called Toll-like receptors (TLRs), has also been emphasized. In this paper we will focus on how neonatal sepsis and TLR-mediated inflammation increase the vulnerability of the newborn brain.
2. Neonatal Sepsis and Brain Injury
Infants with sepsis have an increased incidence of cerebral palsy [4] and white matter abnormalities [5–11]. In a large study of 6093 extremely low birth weight (<1000 g) infants, those who were infected (including early-onset sepsis, suspected sepsis (culture negative), and had necrotizing enterocolitis (NEC)) were more likely to have cerebral palsy than children who did not have a neonatal infection [12]. In another recent large sample-size study involving 1155 infants born at 23 to 27 weeks gestation, it was found that children who had both late bacteremia (positive blood culture result after the first postnatal week) and surgical NEC were at increased risk of diparetic cerebral palsy compared with children who had neither [13]. Moreover, by comparing outcomes of 150 infants with periventricular leukomalacia (PVL) with controls matched for gestational age, it was found that infants with bacterial sepsis were twice as likely to develop PVL, and those with meningitis were almost four times as likely to develop white matter disease [14]. Similar findings were noted in a smaller case-control study, where associations between cerebral palsy, clinical chorioamnionitis and sepsis were demonstrated [15]. Moreover, there was an increased incidence of Gram-negative bacterial and fungal infections in a very low birth weight population, and these infants were at significantly increased risk for moderate to severe cerebral palsy and neurodevelopmental impairment at 18 months of age [16].
2.1. Bacterial Pathogens in Neonatal Sepsis
Escherichia coli is one of the main pathogens causing early-onset infections in preterm neonates, accounting for up to 40% of the cases of bacteremia among very low birth weight preterm infants (<1,500 g) [17]. Cerebral white matter injury has been found by MRI following Escherichia coli meningitis in human newborn infants [18]. Furthermore, Escherichia coli induce brain damage in a number of antenatal rabbit and rodent models [19–26]. Also, in a recent study, white matter injury was demonstrated in an animal model of neonatal Escherichia coli sepsis in 5-day-old rat pups [27]. Experimental studies show that early-life Escherichia coli exposure can also have long-term effects, influencing the vulnerability to other factors in adulthood, for example, age-related cognitive decline [28] as well as attenuated glial and cytokine responses to amphetamine challenge [29].
In recent years, coagulase-negative staphylococci (CONS) have emerged as the most prevalent and important neonatal pathogens, responsible for approximately 50% of all episodes of late-onset neonatal sepsis in neonatal intensive care units around the world [30–33]. CONS cause significant morbidity, mortality, and healthcare costs worldwide in preterm newborns, especially in very low birth weight infants [34–38]. The vulnerability of preterm infants to CONS infection has been suggested to be due to the special characteristics of the premature infant's innate immunity [39]. Although there is no direct evidence of CONS causing perinatal brain injury, the presence of CONS in the chorioamnion space at delivery is associated with increased risk for the development of cerebral palsy in preterm infants [40, 41]. Further, in children with an established diagnosis of cerebral palsy, who are admitted to pediatric intensive care, there is a high rate of carriage of abnormal bacteria, including CONS [42].
In very low birth weight preterm infants with early onset neonatal sepsis, the rate of group B streptococcal (GBS) infections is relatively low in comparison with E. coli infections [17]. There is no direct evidence of GBS sepsis playing a role in cerebral palsy; however, nearly half of all infants who survive an episode of GBS meningitis suffer from long-term neurodevelopmental sequelae [43]. Further, extensive cortical neuronal injury was found in GBS-infected neonatal rats, which was mediated through reactive oxygen intermediates [44, 45].
3. Toll-Like Receptor-Mediated Vulnerability of the Immature Brain
3.1. Toll-Like Receptors
Toll-like receptors (TLRs) play a central role in primary recognition of infectious and viral pathogens. The presence of all 13 known TLRs has been demonstrated in the brain [46–48]. TLR4 mediates cellular activation in response to LPS derived from Escherichia coli [49], while CONS [39] and GBS infections [50] are, at least partly, believed to be mediated by TLR2. Interestingly, the role of TLRs in nonbacterial-induced brain injury has also recently been highlighted [51]. TLRs signal through the recruitment of intracellular adaptor proteins, followed by activation of protein kinases and transcription factors that induce the production of inflammatory mediators (Figure 1). The adaptor protein MyD88 is used by most TLRs, except TLR3, while the TRIF adaptor protein is used only by TLR3 and TLR4. LPS-induced activation of TLR4 elicits, via both MyD88 and TRIF, a broad inflammatory response in tissues, including the immature brain [52].
Figure 1.
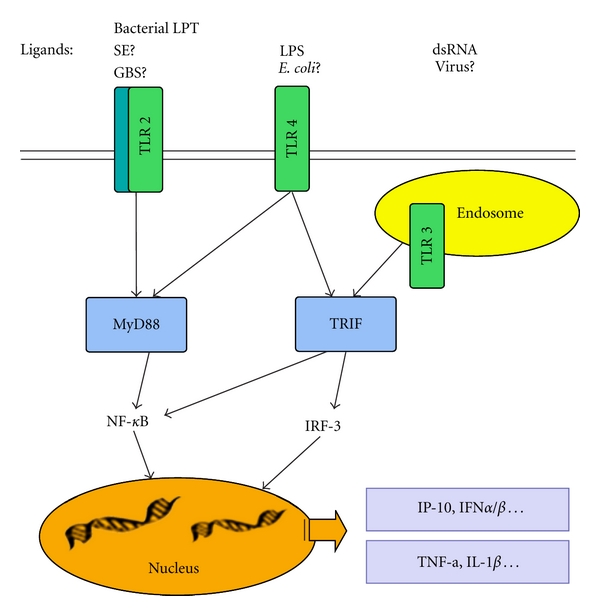
Diagram outlining infectious agents, TLRs, and major signaling pathways. Abbreviations: SE: S. epidermidis; GBS: group B streptococcus; LPT: lipopeptides. LPS: lipopolysaccharide; MyD88: myeloid differentiation primary response gene (88); TRIF: TIR domain-containing adaptor inducing interferon-β-mediated transcription factor; NF-κB: nuclear factor-KappaB; IRF: interferon regulatory factor; IP-10: interferon gamma-induced protein 10; IFN: interferon; TNF: tumor necrosis factor; IL-1: Interleukin -1.
3.2. TLR Expression during Brain Development
There is relatively little information regarding the expression of TLRs in the developing brain. During embryonic life, protein expression of both TLR-3 and -8 has been identified [53, 54], while TLR-2 expression is relatively low before birth and increases during the first two weeks of life [55]. We have shown that mRNA for TLR1-9 is expressed in the neonatal mouse brain [56]. It appears that some of the TLRs may play important roles during normal brain development, as TLR2 inhibits neural progenitor cell proliferation during the embryonic period, and TLR3 deficiency increases proliferation of neural progenitor cells, while TLR8 stimulation inhibits neurite outgrowth [53–55]. In support, TLR2 and TLR4 have been shown to regulate hippocampal neurogenesis in the adult brain [57].
3.3. LPS-Induced Brain Injury
We, and others, have shown that systemic administration of LPS results in brain injury in both fetal and newborn animals [58–60]. These injuries appear, both histologically and by MRI analysis, to be very similar to those found in preterm infants [61]. Furthermore, it is now well established that pre-exposure to LPS can increase the vulnerability of the immature brain to hypoxia-ischemia (HI), in both rats [62, 63] and mice [64]. These effects are TLR4 [65] and MyD88 dependent [64, 66]. In a recent study, it was also shown that a very low dose of LPS, specifically increased the vulnerability of the immature white matter [67]. Low-dose LPS (0.05 mg/kg) sensitized HI injury in P2 rat pups by selectively reducing myelin basic protein expression and the number of oligodendrocytes while increasing neuroinflammation and blood-brain barrier damage in the white matter. The neuroinflammatory responses to LPS/HI appears to be age dependent [68]. Rat pups subjected to LPS/HI at P1 responded with weak cytokine response, while there was a prominent upregulation of cytokines in P12 pups subjected to the same insult. Interestingly, IL-1β was upregulated at both ages; IL-1β injections sensitize the newborn brain to excitotoxicity [69] and repeated IL-1β exposure during the neonatal period induces preterm like brain injury in mice [70].
Although it has clearly been demonstrated that LPS can increase the vulnerability to HI, under certain circumstances LPS can also induce tolerance to brain injury. We have shown that the time interval between LPS exposure and the subsequent HI is imperative to the outcome [71, 72], where a 24 h interval seems to induce a tolerant state that makes the brain less vulnerable. This has been confirmed by others who have implicated several possible mechanisms, including upregulation of corticosterone [73], which is further supported by the fact that administration of dexamethasone prevents learning impairment following LPS/HI in neonatal rats [74]. Furthermore, Akt-mediated eNOS upregulation in neurons and vascular endothelial cells have been implicated in LPS-induced preconditioning [75].
The importance of the time interval between LPS and other insults seems to be a generalized phenomenon. We have recently demonstrated in an in vitro model that conditioned medium from LPS-activated microglia affects the antioxidant Nrf2 system and cell survival in astrocytes in a time-dependent manner. LPS-induced inflammation had dual, time-dependent, effects on the Nrf2 system in that sustained activation (72 h) of GSK3beta and p38 downregulated the Nrf2 system, possibly via the activation of histone deacetylases, changes that were not observed with a 24 h (tolerance) interval [76, 77]. These studies support our previous report demonstrating that reductions in antioxidants were more pronounced when HI was preceded by LPS injection in 8-day rats 3 days prior to the HI insult [78].
3.4. Other TLRs in Perinatal Brain Injury
Compared to TLR4, much less is known about other TLRs in perinatal brain injury. As mentioned above, TLR2, TLR3, and TLR8 can affect normal brain development [53–55]. Activation of TLR2 in neonatal mice decreases volume of cerebral gray matter, white matter in the forebrain, and cerebellar molecular layer [79]. Further, we have recently demonstrated the expression of both TLR1 and TLR2 in the neonatal mouse brain following HI. In these studies, TLR2 deficiency resulted in reduced infarct volume after HI, while TLR-1-deficient mice were not protected [56].
Maternal viral immune activation is believed to increase the risk of psychiatric disorders such as schizophrenia in offspring, and in order to examine this relationship, several authors have investigated the vulnerability of the fetal brain to synthetic double-stranded RNA, polyriboinosinic-polyribocytidilic acid (poly I:C), a TLR3 agonist. Maternal injection with poly I:C towards the end of gestation (≥G15) causes sensorimotor gating deficits in the adult offspring in mice [80] and increased sensitivity to the locomotor-stimulating effects of MK-801 [81]. The effects of Poly I:C appear to be gestational age dependent [82]. Maternal Poly I:C injection on GD9, but not GD17, significantly impaired sensorimotor gating and reduced prefrontal dopamine D1 receptors in adulthood, whereas prenatal immune activation in late gestation impaired working memory, potentiated the locomotor reaction to a NMDA-receptor antagonist, and reduced hippocampal NMDA-receptor subunit 1 expression. In particular, Poly I:C injections early during rodent pregnancy affect structural brain development, such as a transient decrease of myelin basic protein in the neonatal offspring [83] and cerebellar pathology [84].
4. Conclusion
E. coli infections are common in preterm neonates, and considerable evidence suggests that E. coli-induced inflammation play a role in the development of white matter damage in preterm infants. There is much less data available concerning the importance of two other common neonatal pathogens, CONS and GBS, in perinatal brain injury. Furthermore, it is becoming clear that TLRs have important roles during development and may be involved in both pathogen-induced damage as well as so called “sterile” HI-induced inflammation. In order to better understand the underlying causes of perinatal brain injury, the interaction between common neonatal pathogens and TLRs in the newborn brain deserves further investigation.
Acknowledgments
This work was supported by grants from the Swedish Research Council (VR 2009-2630, C. Mallard; VR K2009-54X-21119-01-4, X. Wang), ALFGBG-142881 (C. Mallard), Leducq Foundation (C. Mallard), the European Union (HEALTH-F2-2009-241778, Neurobid, C. Mallard), Åhléns stiftelsen (C. Mallard), Frimurare barnhusfonden (C. Mallard), Lundgrenska stiftelserna (C. Mallard, and X. Wang), and National Natural Science Foundation of China (30973240, X. Wang).
References
- 1.Marlow N, Wolke D, Bracewell MA, Samara M. Neurologic and developmental disability at six years of age after extremely preterm birth. The New England Journal of Medicine. 2005;352(1):9–19. doi: 10.1056/NEJMoa041367. [DOI] [PubMed] [Google Scholar]
- 2.Dammann O, Leviton A. Maternal intrauterine infection, cytokines, and brain damage in the preterm newborn. Pediatric Research. 1997;42(1):1–8. doi: 10.1203/00006450-199707000-00001. [DOI] [PubMed] [Google Scholar]
- 3.Hagberg H, Mallard C. Effect of inflammation on central nervous system development and vulnerability. Current Opinion in Neurology. 2005;18(2):117–123. doi: 10.1097/01.wco.0000162851.44897.8f. [DOI] [PubMed] [Google Scholar]
- 4.Leviton A, Gilles FH. An epidemiologic study of perinatal telencephalic leucoencephalopathy in an autopsy population. Journal of the Neurological Sciences. 1973;18(1):53–66. doi: 10.1016/0022-510x(73)90020-8. [DOI] [PubMed] [Google Scholar]
- 5.Faix RG, Donn SM. Association of septic shock caused by early-onset group B streptococcal sepsis and periventricular leukomalacia in the preterm infant. Pediatrics. 1985;76(3):415–419. [PubMed] [Google Scholar]
- 6.Murphy DJ, Hope PL, Johnson A. Neonatal risk factors for cerebral palsy in very preterm babies: case-control study. British Medical Journal. 1997;314(7078):404–408. doi: 10.1136/bmj.314.7078.404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Msall ME, Buck GM, Rogers BT, et al. Multivariate risks among extremely premature infants. Journal of Perinatology. 1994;14(1):41–47. [PubMed] [Google Scholar]
- 8.Wheater M, Rennie JM. Perinatal infection is an important risk factor for cerebral palsy in very-low-birthweight infants. Developmental Medicine and Child Neurology. 2000;42(6):364–367. doi: 10.1017/s0012162200000670. [DOI] [PubMed] [Google Scholar]
- 9.Shah DK, Doyle LW, Anderson PJ, et al. Adverse neurodevelopment in preterm infants with postnatal sepsis or necrotizing enterocolitis is mediated by white matter abnormalities on magnetic resonance imaging at term. Journal of Pediatrics. 2008;153(2):170–175. doi: 10.1016/j.jpeds.2008.02.033. [DOI] [PubMed] [Google Scholar]
- 10.Volpe JJ. Postnatal sepsis, necrotizing entercolitis, and the critical role of systemic inflammation in white matter injury in premature infants. Journal of Pediatrics. 2008;153(2):160–163. doi: 10.1016/j.jpeds.2008.04.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schmitz T, Heep A, Groenendaal F, et al. Interleukin-1β, interleukin-18, and interferon-γ expression in the cerebrospinal fluid of premature infants with posthemorrhagic hydrocephalus-markers of white matter damage? Pediatric Research. 2007;61(6):722–726. doi: 10.1203/pdr.0b013e31805341f1. [DOI] [PubMed] [Google Scholar]
- 12.Stoll BJ, Hansen NI, Adams-Chapman I, et al. Neurodevelopmental and growth impairment among extremely low-birth-weight infants with neonatal infection. JAMA. 2004;292(19):2357–2365. doi: 10.1001/jama.292.19.2357. [DOI] [PubMed] [Google Scholar]
- 13.Martin CR, Dammann O, Allred EN, et al. Neurodevelopment of extremely preterm infants who had necrotizing enterocolitis with or without late bacteremia. Journal of Pediatrics. 2010;157(5):751–756. doi: 10.1016/j.jpeds.2010.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Graham EM, Holcroft CJ, Rai KK, Donohue PK, Allen MC. Neonatal cerebral white matter injury in preterm infants is associated with culture positive infections and only rarely with metabolic acidosis. American Journal of Obstetrics and Gynecology. 2004;191(4):1305–1310. doi: 10.1016/j.ajog.2004.06.058. [DOI] [PubMed] [Google Scholar]
- 15.Michael O’Shea T, Klinepeter KL, Meis PJ, Dillard RG. Intrauterine infection and the risk of cerebral palsy in very low-birthweight infants. Paediatric and Perinatal Epidemiology. 1998;12(1):72–83. [PubMed] [Google Scholar]
- 16.Benjamin DK, Stoll BJ, Fanaroff AA, et al. Neonatal candidiasis among extremely low birth weight infants: risk factors, mortality rates, and neurodevelopmental outcomes at 18 to 22 months. Pediatrics. 2006;117(1):84–92. doi: 10.1542/peds.2004-2292. [DOI] [PubMed] [Google Scholar]
- 17.Stoll BJ, Hansen NI, Higgins RD, et al. Very low birth weight preterm infants with early onset neonatal sepsis: the predominance of Gram-negative infections continues in the National Institute of Child Health and Human Development Neonatal Research Network, 2002–2003. Pediatric Infectious Disease Journal. 2005;24(7):635–639. doi: 10.1097/01.inf.0000168749.82105.64. [DOI] [PubMed] [Google Scholar]
- 18.Shah DK, Daley AJ, Hunt RW, Volpe JJ, Inder TE. Cerebral white matter injury in the newborn following Escherichia coli meningitis. European Journal of Paediatric Neurology. 2005;9(1):13–17. doi: 10.1016/j.ejpn.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 19.Kannan S, Saadani-Makki F, Muzik O, et al. Microglial activation in perinatal rabbit brain induced by intrauterine inflammation: detection with 11C-(R)-PK11195 and small-animal PET. Journal of Nuclear Medicine. 2007;48(6):946–954. doi: 10.2967/jnumed.106.038539. [DOI] [PubMed] [Google Scholar]
- 20.Yuan TM, Yu HM, Gu WZ, Li JP. White matter damage and chemokine induction in developing rat brain after intrauterine infection. Journal of Perinatal Medicine. 2005;33(5):415–422. doi: 10.1515/JPM.2005.074. [DOI] [PubMed] [Google Scholar]
- 21.Debillon T, Gras-Leguen C, Leroy S, Caillon J, Rozé JC, Gressens P. Patterns of cerebral inflammatory response in a rabbit model of intrauterine infection-mediated brain lesion. Developmental Brain Research. 2003;145(1):39–48. doi: 10.1016/s0165-3806(03)00193-7. [DOI] [PubMed] [Google Scholar]
- 22.Davies JK, Shikes RH, Sze CI, et al. Histologic inflammation in the maternal and fetal compartments in a rabbit model of acute intra-amniotic infection. American Journal of Obstetrics and Gynecology. 2000;183(5):1088–1093. doi: 10.1067/mob.2000.108888. [DOI] [PubMed] [Google Scholar]
- 23.Bo Hyun Yoon, Chong Jai Kim, Romero R, et al. Experimentally induced intrauterine infection causes fetal brain white matter lesions in rabbits. American Journal of Obstetrics and Gynecology. 1997;177(4):797–802. doi: 10.1016/s0002-9378(97)70271-0. [DOI] [PubMed] [Google Scholar]
- 24.Pang Y, Rodts-Palenik S, Cai Z, Bennett WA, Rhodes PG. Suppression of glial activation is involved in the protection of IL-10 on maternal E. coli induced neonatal white matter injury. Developmental Brain Research. 2005;157(2):141–149. doi: 10.1016/j.devbrainres.2005.03.015. [DOI] [PubMed] [Google Scholar]
- 25.Rodts-Palenik S, Wyatt-Ashmead J, Pang Y, et al. Maternal infection-induced white matter injury is reduced by treatment with interleukin-10. American Journal of Obstetrics and Gynecology. 2004;191(4):1387–1392. doi: 10.1016/j.ajog.2004.06.093. [DOI] [PubMed] [Google Scholar]
- 26.Wallace KL, Lopez J, Shaffery JP, Wells A, Paul IA, Bennett WA. Interleukin-10/Ceftriaxone prevents E. coli-induced delays in sensorimotor task learning and spatial memory in neonatal and adult Sprague-Dawley rats. Brain Research Bulletin. 2010;81(1):141–148. doi: 10.1016/j.brainresbull.2009.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Loron G, Olivier P, See H, et al. Ciprofloxacin prevents myelination delay in neonatal rats subjected to E. coli sepsis. Annals of Neurology. 2011;69(2):341–351. doi: 10.1002/ana.22190. [DOI] [PubMed] [Google Scholar]
- 28.Bilbo SD. Early-life infection is a vulnerability factor for aging-related glial alterations and cognitive decline. Neurobiology of Learning and Memory. 2010;94(1):57–64. doi: 10.1016/j.nlm.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bland ST, Beckley JT, Watkins LR, Maier SF, Bilbo SD. Neonatal Escherichia coli infection alters glial, cytokine, and neuronal gene expression in response to acute amphetamine in adolescent rats. Neuroscience Letters. 2010;474(1):52–57. doi: 10.1016/j.neulet.2010.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stoll BJ, Hansen N, Fanaroff AA, et al. Changes in pathogens causing early-onset sepsis in very-low-birth-weight infants. The New England Journal of Medicine. 2002;347(4):240–247. doi: 10.1056/NEJMoa012657. [DOI] [PubMed] [Google Scholar]
- 31.Stoll BJ, Hansen N, Fanaroff AA, et al. Late-onset sepsis in very low birth weight neonates: the experience of the NICHD Neonatal Research Network. Pediatrics. 2002;110(2):285–291. doi: 10.1542/peds.110.2.285. [DOI] [PubMed] [Google Scholar]
- 32.Bizzarro MJ, Raskind C, Baltimore RS, Gallagher PG. Seventy-five years of neonatal sepsis at Yale: 1928–2003. Pediatrics. 2005;116(3):595–602. doi: 10.1542/peds.2005-0552. [DOI] [PubMed] [Google Scholar]
- 33.Makhoul IR, Sujov P, Smolkin T, Lusky A, Reichman B. Pathogen-specific early mortality in very low birth weight infants with late-onset sepsis: a national survey. Clinical Infectious Diseases. 2005;40(2):218–224. doi: 10.1086/426444. [DOI] [PubMed] [Google Scholar]
- 34.Stoll BJ, Hansen N. Infections in VLBW infants: studies from the NICHD Neonatal Research Network. Seminars in Perinatology. 2003;27(4):293–301. doi: 10.1016/s0146-0005(03)00046-6. [DOI] [PubMed] [Google Scholar]
- 35.Vuong C, Otto M. Staphylococcus epidermidis infections. Microbes and Infection. 2002;4(4):481–489. doi: 10.1016/s1286-4579(02)01563-0. [DOI] [PubMed] [Google Scholar]
- 36.Isaacs D. A ten year, multicentre study of coagulase negative staphylococcal infections in Australasian neonatal units. Archives of Disease in Childhood. 2003;88(2):F89–F93. doi: 10.1136/fn.88.2.F89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Healy CM, Hulten KG, Palazzi DL, Campbell JR, Baker CJ. Emergence of new strains of methicillin-resistant Staphylococcus aureus in a neonatal intensive care unit. Clinical Infectious Diseases. 2004;39(10):1460–1466. doi: 10.1086/425321. [DOI] [PubMed] [Google Scholar]
- 38.López Sastre JB, Coto Cotallo D, Fernández Colomer B, et al. Neonatal sepsis of nosocomial origin: an epidemiological study from the ‘Grupo de Hospitales Castrillo’. Journal of Perinatal Medicine. 2002;30(2):149–157. doi: 10.1515/JPM.2002.019. [DOI] [PubMed] [Google Scholar]
- 39.Strunk T, Richmond P, Simmer K, Currie A, Levy O, Burgner D. Neonatal immune responses to coagulase-negative staphylococci. Current Opinion in Infectious Diseases. 2007;20(4):370–375. doi: 10.1097/QCO.0b013e3281a7ec98. [DOI] [PubMed] [Google Scholar]
- 40.Mittendorf R, Covert R, Kohn J, Roizen N, Khoshnood B, Lee KS. The association of coagulase-negative staphylococci isolated from the chorioamnion at delivery and subsequent development of cerebral palsy. Journal of Perinatology. 2001;21(1):3–8. doi: 10.1038/sj.jp.7200474. [DOI] [PubMed] [Google Scholar]
- 41.Mittendorf R, Roizen N, Moawad A, Khoshnood B, Lee KS. Association between cerebral palsy and coagulase-negative staphylococci. The Lancet. 1999;354(9193):1875–1876. doi: 10.1016/S0140-6736(99)01111-3. [DOI] [PubMed] [Google Scholar]
- 42.Thorburn K, Jardine M, Taylor N, Reilly N, Sarginson RE, Van Saene HKF. Antibiotic-resistant bacteria and infection in children with cerebral palsy requiring mechanical ventilation. Pediatric Critical Care Medicine. 2009;10(2):222–226. doi: 10.1097/PCC.0b013e31819368ac. [DOI] [PubMed] [Google Scholar]
- 43.Edwards MS, Rench MA, Haffar AAM. Long-term sequelae of group B streptococcal meningitis in infants. Journal of Pediatrics. 1985;106(5):717–722. doi: 10.1016/s0022-3476(85)80342-5. [DOI] [PubMed] [Google Scholar]
- 44.Kim YS, Sheldon RA, Elliott BR, Liu Q, Ferriero DM, Tauber MG. Brain injury in experimental neonatal meningitis due to group B streptococci. Journal of Neuropathology and Experimental Neurology. 1995;54(4):531–539. doi: 10.1097/00005072-199507000-00007. [DOI] [PubMed] [Google Scholar]
- 45.Leib SL, Kim YS, Chow LL, Sheldon RA, Täuber MG. Reactive oxygen intermediates contribute to necrotic and apoptotic neuronal injury in an infant rat model of bacterial meningitis due to group B streptococci. The Journal of Clinical Investigation. 1996;98(11):2632–2639. doi: 10.1172/JCI119084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bsibsi M, Ravid R, Gveric D, Van Noort JM. Broad expression of Toll-like receptors in the human central nervous system. Journal of Neuropathology and Experimental Neurology. 2002;61(11):1013–1021. doi: 10.1093/jnen/61.11.1013. [DOI] [PubMed] [Google Scholar]
- 47.Mishra BB, Gundra UM, Teale JM. Expression and distribution of Toll-like receptors 11-13 in the brain during murine neurocysticercosis. Journal of Neuroinflammation. 2008;5, article 53 doi: 10.1186/1742-2094-5-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mishra BB, Mishra PK, Teale JM. Expression and distribution of Toll-like receptors in the brain during murine neurocysticercosis. Journal of Neuroimmunology. 2006;181(1-2):46–56. doi: 10.1016/j.jneuroim.2006.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tapping RI, Akashi S, Miyake K, Godowski PJ, Tobias PS. Toll-like receptor 4, but not Toll-like receptor 2, is a signaling receptor for Escherichia and Salmonella lipopolysaccharides. Journal of Immunology. 2000;165(10):5780–5787. doi: 10.4049/jimmunol.165.10.5780. [DOI] [PubMed] [Google Scholar]
- 50.Henneke P, Berner R. Interaction of neonatal phagocytes with group B streptococcus: recognition and Response. Infection and Immunity. 2006;74(6):3085–3095. doi: 10.1128/IAI.01551-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hanisch UK, Johnson TV, Kipnis J. Toll-like receptors: roles in neuroprotection? Trends in Neurosciences. 2008;31(4):176–182. doi: 10.1016/j.tins.2008.01.005. [DOI] [PubMed] [Google Scholar]
- 52.Eklind S, Hagberg H, Wang X, et al. Effect of lipopolysaccharide on global gene expression in the immature rat brain. Pediatric Research. 2006;60(2):161–168. doi: 10.1203/01.pdr.0000228323.32445.7d. [DOI] [PubMed] [Google Scholar]
- 53.Lathia JD, Okun E, Tang SC, et al. Toll-like receptor 3 is a negative regulator of embryonic neural progenitor cell proliferation. Journal of Neuroscience. 2008;28(51):13978–13984. doi: 10.1523/JNEUROSCI.2140-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ma Y, Li J, Chiu I, et al. Toll-like receptor 8 functions as a negative regulator of neurite outgrowth and inducer of neuronal apoptosis. Journal of Cell Biology. 2006;175(2):209–215. doi: 10.1083/jcb.200606016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Okun E, Griffioen KJ, Gen Son T, et al. TLR2 activation inhibits embryonic neural progenitor cell proliferation. Journal of Neurochemistry. 2010;114(2):462–474. doi: 10.1111/j.1471-4159.2010.06778.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stridh L, Smith PLP, Naylor AS, Wang X, Mallard C. Regulation of Toll-like receptor 1 and -2 in neonatal mice brains after hypoxia-ischemia. Journal of Neuroinflammation. 2011;8, article 85 doi: 10.1186/1742-2094-8-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rolls A, Shechter R, London A, et al. Toll-like receptors modulate adult hippocampal neurogenesis. Nature Cell Biology. 2007;9(9):1081–1088. doi: 10.1038/ncb1629. [DOI] [PubMed] [Google Scholar]
- 58.Duncan JR, Cock ML, Scheerlinck JPY, et al. White matter injury after repeated endotoxin exposure in the preterm ovine fetus. Pediatric Research. 2002;52(6):941–949. doi: 10.1203/00006450-200212000-00021. [DOI] [PubMed] [Google Scholar]
- 59.Mallard C, Welin AK, Peebles D, Hagberg H, Kjellmer I. White matter injury following systemic endotoxemia or asphyxia in the fetal sheep. Neurochemical Research. 2003;28(2):215–223. doi: 10.1023/a:1022368915400. [DOI] [PubMed] [Google Scholar]
- 60.Wang X, Hellgren G, Löfqvist C, et al. White matter damage after chronic subclinical inflammation in newborn mice. Journal of Child Neurology. 2009;24(9):1171–1178. doi: 10.1177/0883073809338068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dean J, van de Looij Y, Sizonenko SV, et al. Delayed cortical impairment following lipopolysaccharide exposure in preterm fetal sheep. doi: 10.1002/ana.22480. Annals of Neurology. In press. [DOI] [PubMed] [Google Scholar]
- 62.Eklind S, Mallard C, Leverin AL, et al. Bacterial endotoxin sensitizes the immature brain to hypoxic-ischaemic injury. European Journal of Neuroscience. 2001;13(6):1101–1106. doi: 10.1046/j.0953-816x.2001.01474.x. [DOI] [PubMed] [Google Scholar]
- 63.Yang L, Sameshima H, Ikeda T, Ikenoue T. Lipopolysaccharide administration enhances hypoxic-ischemic brain damage in newborn rats. Journal of Obstetrics and Gynaecology Research. 2004;30(2):142–147. doi: 10.1111/j.1447-0756.2003.00174.x. [DOI] [PubMed] [Google Scholar]
- 64.Wang X, Stridh L, Li W, et al. Lipopolysaccharide sensitizes neonatal hypoxic-ischemic brain injury in a MyD88-dependent manner. Journal of Immunology. 2009;183(11):7471–7477. doi: 10.4049/jimmunol.0900762. [DOI] [PubMed] [Google Scholar]
- 65.Lehnardt S, Massillon L, Follett P, et al. Activation of innate immunity in the CNS triggers neurodegeneration through a Toll-like receptor 4-dependent pathway. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(14):8514–8519. doi: 10.1073/pnas.1432609100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dean JM, Wang X, Kaindl AM, et al. Microglial MyD88 signaling regulates acute neuronal toxicity of LPS-stimulated microglia in vitro. Brain, Behavior, and Immunity. 2010;24(4):776–783. doi: 10.1016/j.bbi.2009.10.018. [DOI] [PubMed] [Google Scholar]
- 67.Wang LW, Chang YC, Lin CY, Hong JS, Huang CC. Low-dose lipopolysaccharide selectively sensitizes hypoxic ischemia-induced white matter injury in the immature brain. Pediatric Research. 2010;68(1):41–47. doi: 10.1203/PDR.0b013e3181df5f6b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Brochu M-E, Girard S, Lavoie K, Sébire G. Developmental regulation of the neuroinflammatory responses to LPS and/or hypoxia-ischemia between preterm and term neonates: an experimental study. Journal of Neuroinflammation. 2011;8, article 55 doi: 10.1186/1742-2094-8-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dommergues MA, Patkai J, Renauld JC, Evrard P, Gressens P. Proinflammatory cytokines and interleukin-9 exacerbate excitotoxic lesions of the newborn murine neopallium. Annals of Neurology. 2000;47(1):54–63. [PubMed] [Google Scholar]
- 70.Favrais G, Van De Looij Y, Fleiss B, et al. Systemic inflammation disrupts the developmental program of white matter. doi: 10.1002/ana.22489. Annals of Neurology. In press. [DOI] [PubMed] [Google Scholar]
- 71.Eklind S, Mallard C, Arvidsson P, Hagberg H. Lipopolysaccharide induces both a primary and a secondary phase of sensitization in the developing rat brain. Pediatric Research. 2005;58(1):112–116. doi: 10.1203/01.PDR.0000163513.03619.8D. [DOI] [PubMed] [Google Scholar]
- 72.Wang X, Hagberg H, Nie C, Zhu C, Ikeda T, Mallard C. Dual role of intrauterine immune challenge on neonatal and adult brain vulnerability to hypoxia-ischemia. Journal of Neuropathology and Experimental Neurology. 2007;66(6):552–561. doi: 10.1097/01.jnen.0000263870.91811.6f. [DOI] [PubMed] [Google Scholar]
- 73.Ikeda T, Yang L, Ikenoue T, Mallard C, Hagberg H. Endotoxin-induced hypoxic-ischemic tolerance is mediated by up-regulation of corticosterone in neonatal rat. Pediatric Research. 2006;59(1):56–60. doi: 10.1203/01.pdr.0000191140.87314.ce. [DOI] [PubMed] [Google Scholar]
- 74.Ikeda T, Mishima K, Aoo N, et al. Dexamethasone prevents long-lasting learning impairment following a combination of lipopolysaccharide and hypoxia-ischemia in neonatal rats. American Journal of Obstetrics and Gynecology. 2005;192(3):719–726. doi: 10.1016/j.ajog.2004.12.048. [DOI] [PubMed] [Google Scholar]
- 75.Lin HY, Wu CL, Huang CC. The akt-endothelial nitric oxide synthase pathway in lipopolysaccharide preconditioning-induced hypoxic-ischemic tolerance in the neonatal rat brain. Stroke. 2010;41(7):1543–1551. doi: 10.1161/STROKEAHA.109.574004. [DOI] [PubMed] [Google Scholar]
- 76.Correa F, Ljunggren E, Mallard C, Nilsson M, Weber SG, Sandberg M. The Nrf2-inducible antioxidant defense in astrocytes can be both up- and down-regulated by activated microglia:Involvement of p38 MAPK. Glia. 2011;59(5):785–799. doi: 10.1002/glia.21151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Correa F, Mallard C, Nilsson M, Sandberg M. Activated microglia decrease histone acetylation and Nrf2-inducible anti-oxidant defence in astrocytes: restoring effects of inhibitors of HDACs, p38 MAPK and GSK3β . Neurobiology of Disease. 2011;44(1):142–151. doi: 10.1016/j.nbd.2011.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wang X, Svedin P, Nie C, et al. N-Acetylcysteine reduces lipopolysaccharide-sensitized hypoxic-ischemic brain injury. Annals of Neurology. 2007;61(3):263–271. doi: 10.1002/ana.21066. [DOI] [PubMed] [Google Scholar]
- 79.Du X, Fleiss B, Li H, et al. Systemic stimulation of TLR2 impairs neonatal mouse brain development. PLoS One. 2011;6(5) doi: 10.1371/journal.pone.0019583. Article ID e19583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Shi L, Fatemi SH, Sidwell RW, Patterson PH. Maternal influenza infection causes marked behavioral and pharmacological changes in the offspring. Journal of Neuroscience. 2003;23(1):297–302. doi: 10.1523/JNEUROSCI.23-01-00297.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zuckerman L, Weiner I. Maternal immune activation leads to behavioral and pharmacological changes in the adult offspring. Journal of Psychiatric Research. 2005;39(3):311–323. doi: 10.1016/j.jpsychires.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 82.Meyer U, Nyffeler M, Yee BK, Knuesel I, Feldon J. Adult brain and behavioral pathological markers of prenatal immune challenge during early/middle and late fetal development in mice. Brain, Behavior, and Immunity. 2008;22(4):469–486. doi: 10.1016/j.bbi.2007.09.012. [DOI] [PubMed] [Google Scholar]
- 83.Makinodan M, Tatsumi K, Manabe T, et al. Maternal immune activation in mice delays myelination and axonal development in the hippocampus of the offspring. Journal of Neuroscience Research. 2008;86(10):2190–2200. doi: 10.1002/jnr.21673. [DOI] [PubMed] [Google Scholar]
- 84.Shi L, Smith SEP, Malkova N, Tse D, Su Y, Patterson PH. Activation of the maternal immune system alters cerebellar development in the offspring. Brain, Behavior, and Immunity. 2009;23(1):116–123. doi: 10.1016/j.bbi.2008.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]