

Abstract
Digital PCR enables the absolute quantitation of nucleic acids in a sample. The lack of scalable and practical technologies for digital PCR implementation has hampered the widespread adoption of this inherently powerful technique. Here we describe a high-throughput droplet digital PCR (ddPCR) system that enables processing of ∼2 million PCR reactions using conventional TaqMan assays with a 96-well plate workflow. Three applications demonstrate that the massive partitioning afforded by our ddPCR system provides orders of magnitude more precision and sensitivity than real-time PCR. First, we show the accurate measurement of germline copy number variation. Second, for rare alleles, we show sensitive detection of mutant DNA in a 100 000-fold excess of wildtype background. Third, we demonstrate absolute quantitation of circulating fetal and maternal DNA from cell-free plasma. We anticipate this ddPCR system will allow researchers to explore complex genetic landscapes, discover and validate new disease associations, and define a new era of molecular diagnostics.
Detection and quantitation of specific nucleic acid sequences using PCR is fundamental to a large body of research and a growing number of molecular diagnostic tests. The first generation of PCR users performed end-point analysis by gel electrophoresis to obtain qualitative results. The advent of real-time PCR spawned a second generation that enabled quantitation by monitoring the progression of amplification after each cycle using fluorescence probes. In real-time PCR, quantitative information is obtained from the cycle threshold (CT), a point on the analogue fluorescence curve where the signal increases above background. External calibrators or normalization to endogenous controls are required to estimate the concentration of an unknown. Imperfect amplification efficiencies affect CT values which in-turn limits the accuracy of this technique for absolute quantitation.
Early pioneers(1) recognized that the combination of limiting dilution, end-point PCR, and Poisson statistics could yield an absolute measure of nucleic acid concentration, an approach that later became known as digital PCR.(2) In digital PCR, target DNA molecules are distributed across multiple replicate reactions at a level where there are some reactions that have no template and others that have one or more template copies present. After amplification to the terminal plateau phase of PCR, reactions containing one or more templates yield positive end-points, whereas those without template remain negative. The number of target DNA molecules present can be calculated from the fraction of positive end-point reactions using Poisson statistics, according to eq 1,
where λ is the average number of target DNA molecules per replicate reaction and p is the fraction of positive end-point reactions. From λ, together with the volume of each replicate PCR and the total number of replicates analyzed, an estimate of the absolute target DNA concentration is calculated. In digital PCR, the number of replicates, or partitions, largely defines the dynamic range of target DNA quantitation, where an order of magnitude increase in the number of replicates yields approximately an order of magnitude increase in dynamic range. Increasing the number of partitions also improves precision and therefore enables resolution of small concentration differences between nucleic acid sequences in a sample.(4) This is analogous to the relationship between the number of pixels and the resolution of a digital image. As digital PCR relies on a binary end-point threshold to assign each replicate reaction as either positive or negative (one or zero, respectively), it can tolerate wide variations in amplification efficiencies without affecting DNA copy number estimation. Despite its low-throughput and limited dynamic range, digital PCR by limiting dilution in microwell plates is still used today.(3) A practical and low-cost embodiment will unlock the potential of digital PCR and establish a third generation of PCR users and applications.
Currently there are two approaches used by commercially available digital PCR systems. The first approach uses microwells(5) or microfluidic chambers6−8 to split the sample into hundreds of nanoliter partitions. Microfluidic chips simplify reaction setup but are challenging to scale to achieve high-throughput. The second approach, called BEAMing,9,10 is based on emulsion PCR, where templates are clonally amplified in the presence of beads. Post-PCR, the emulsion is broken to recover the beads, which are subsequently labeled with a fluorescent hybridization probe and read by conventional flow-cytometry. BEAMing requires specialized heterogeneous assay schemes that add complexity to the workflow thereby limiting its adoption to a few applications including rare allele detection and DNA methylation.11−13 Overall, high costs, limited throughput, and complicated workflows have hampered the adoption of digital PCR.
We have developed an approach that uses water-in-oil droplets14−16 as the enabling technology to realize high-throughput digital PCR in a low-cost and practical format. Our approach takes advantage of simple microfluidic circuits and surfactant chemistries to divide a 20 μL mixture of sample and reagents into ∼20 000 monodisperse droplets (i.e., partitions). These droplets support PCR amplification of single template molecules using homogeneous assay chemistries and workflows similar to those widely used for real-time PCR applications (i.e., TaqMan). An automated droplet flow-cytometer reads each set of droplets after PCR at a rate of 32 wells per hour.
Results and Discussion
The droplet digital PCR (ddPCR) workflow requires the following steps (Figure 1): Eight assembled PCR reactions, each comprising template, ddPCR Mastermix and TaqMan reagents, are loaded into individual wells of a single-use injection molded cartridge. Next, droplet generation oil containing stabilizing surfactants is loaded and the cartridge placed into the droplet generator. By application of vacuum to the outlet wells, sample and oil are drawn through a flow-focusing junction where monodisperse droplets are generated at a rate of ∼1 000 per second. The surfactant-stabilized droplets flow to a collection well where they quickly concentrate due to density differences between the oil and aqueous phases, forming a packed bed above the excess oil. The densely packed droplets are pipet transferred to a conventional 96-well PCR plate and thermal cycled to end-point. After thermal cycling, the plate is transferred to a droplet reader. Here, droplets from each well are aspirated and streamed toward the detector where, en route, the injection of a spacer fluid separates and aligns them for single-file simultaneous two-color detection. TaqMan assays provide specific duplexed detection of target and reference genes. All droplets are gated based on detector peak width to exclude rare outliers (e.g., doublets, triplets). Each droplet has an intrinsic fluorescence signal resulting from the imperfect quenching of the fluorogenic probes enabling detection of negative droplets. For droplets that contain template, specific cleavage of TaqMan probes generates a strong fluorescence signal. On the basis of fluorescence amplitude, a simple threshold assigns each droplet as positive or negative. As the droplet volume is known, the fraction of positive droplets is then used to calculate the absolute concentration of the target sequence. For 20 000 droplets, the dynamic range for absolute quantitation spans from a single copy up to ∼100 000 copies. For human genomic DNA, this equates to an input DNA mass ranging from 3.3 fg to 330 ng per 20 μL reaction. As templates are randomly distributed across the droplet partitions, a Poisson correction extends the dynamic range into the realm where on average there are multiple copies per droplet. Statistical models are applied to calculate confidence limits of the concentration estimates and their ratios.4,17
Figure 1.
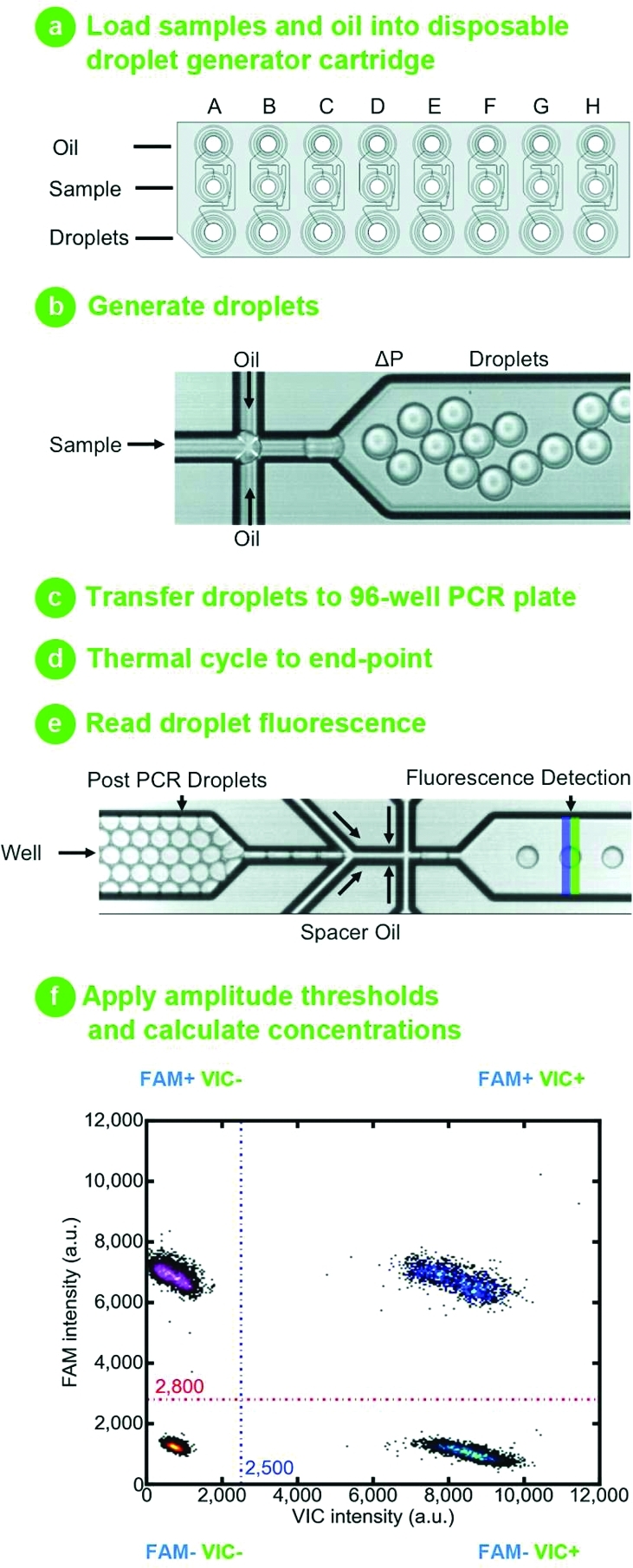
Droplet digital PCR workflow: (a) Samples and droplet generation oil are loaded into an eight-channel droplet generator cartridge. (b) A vacuum is applied to the droplet well, which draws sample and oil through a flow-focusing nozzle where monodisperse 1 nL droplets are formed. In <2 min, eight samples are converted into eight sets of 20 000 droplets. (c) The surfactant-stabilized droplets are pipet transferred to a 96-well PCR plate. (d) Droplet PCR amplification to end-point (35–45 cycles) is performed in a conventional thermal cycler. (e) The plate is loaded onto a reader which sips droplets from each well and streams them single-file past a two-color detector at the rate of ∼1 000 per second. (f) Droplets are assigned as positive or negative based on their fluorescence amplitude. The number of positive and negative droplets in each channel is used to calculate the concentration of the target and reference DNA sequences (see eq 1) and their Poisson-based 95% confidence intervals.
To demonstrate the immediate utility of this ddPCR system, we present data on three application areas of increasing interest to researchers: determination of copy number variation (CNV), detection of rare alleles and the absolute quantitation of circulating DNA in cell-free plasma. Each application was selected to highlight a distinct advantage that massive droplet partitioning affords to digital PCR. For CNV, the large number of replicates provides sufficient precision to accurately measure copy number states. For the detection of rare alleles, partitioning the target mutant DNA away from highly homologous wildtype DNA increases sensitivity. Finally, droplet partitioning enables accurate quantitation of nucleic acids from clinical samples over a wide dynamic range without external calibrators or endogenous controls.
CNVs are deletions and amplifications of genome segments ranging from hundreds to millions of base pairs in length that have been implicated in a broad spectrum of human disease.(18) Microarrays and the next-generation sequencing technologies have enabled and accelerated the discovery of new CNVs,(19) thereby further increasing the need for a high-throughput, low-cost approach to making precise CNV measurements with increased dynamic range for validation and follow-up studies. Although microarray technologies are valuable tools for CNV discovery,(20) they have limited dynamic range and are expensive to scale to large numbers of samples for population studies. Multiplex ligation-dependent probe amplification (MLPA)(21) is an assay that allows resolution of deletions or duplications for up to 40 targets but requires selection from a predefined test menu or extensive upfront assay optimization for new target panels. CNV investigators using methods based on real-time PCR have reported technical difficulty obtaining accurate copy number measurements.(22) Real-time PCR measurements are inherently imprecise, and copy number estimates can drift between cases and controls.
We measured the germline copy number variation of HapMap samples by ddPCR. Because increases in gene copy number are often the result of tandem gene duplications, we used restriction enzymes to predictably and efficiently separate linked copies of the target gene such that each sequence is encapsulated into its own droplet and counted separately. Restriction enzymes were selected to cut either side of the amplicon sequences avoiding known mutation sites(23) and methylation sensitivities. Physically shearing DNA using ultrasound or microfluidic devices is less attractive as it reduces the amount of target that can be amplified by PCR and can be challenging to implement in high-throughput without specialized equipment. Preamplification, an alternative strategy for separation of linked copies(24) has the potential to introduce bias between the target and reference genes.
Seven HapMap samples were screened for CNVs for three target genes. Each ddPCR reaction contained duplex TaqMan assay reagents for the target and reference genes. For MRGPRX1, the copy number states from 1 up to 6 were completely resolved from the results of a single well for each sample (Figure 2a). Lower CNV states for CYP2D6 and Chromosome X were also easily resolved, as shown. For 13 HapMap samples, our system estimated the copy number of CCL3L1, a gene associated with HIV-1/AIDS susceptibility(18) (Figure 2b). For DNA sample NA18507, next-generation sequencing estimated the CCL3L1 copy number to be 5.7(25) whereas our ddPCR system estimated 6.05. The estimate of 5.7 is likely due to under-sampling since the billions of reads of a next-generation sequencing run are distributed across the entire genome giving an average read-depth of only 30×. Thus, once target genes have been identified, greater precision can readily be achieved with ddPCR since the number of reads can be scaled almost arbitrarily. The current ddPCR system can achieve read depths of up to 20 000× for two genes from a single well. These data show that our ddPCR system is well suited for CNV population studies as it enables large numbers of samples to be tested against smaller sets of genes.
Figure 2.

Determination of copy number variation states by droplet digital PCR. (a) Measured copy number variation states in HapMap samples for MRGPRX1, Chromosome X, CYP2D6, and (b) CCL3L1. (c) Correlation of measured copy number alterations of GRB7 and ERBB2 in DNA extracted from normal and tumorous breast tissues. Each marker represents a CNV measurement from a single ddPCR well of ∼20 000 droplets. Error bars indicate the Poisson 95% confidence intervals for each copy number determination.
Sample heterogeneity can attenuate the measurement of copy number amplifications, which requires more precise measurements to discriminate smaller differences from normal. Somatic copy number alteration is the hallmark of many cancers. Without high-throughput technology for precise copy number quantitation, pathologists use fluorescent in situ hybridization (FISH) for diagnosing amplifications and deletions as this technique affords single-cell resolution. FISH and related techniques are expensive, laborious, and subject to large losses in performance due to other analytical factors.(26) Specific amplifications define tumor subtypes and guide therapy. For example, Her2 positive breast tumors respond to Trastuzumab (Herceptin). For a set of normal and tumor breast tissue samples, the measured copy numbers of ERBB2 and GRB7 correlated with the exception of two samples that showed lower GRB7 amplification (Figure 2c). These results were expected as the GRB7 gene is part of the HER2 amplicon and is coamplified in almost all breast tumors with 17q11-21 amplification.27,28 This ddPCR method provides the ability to scale the number of partitions by combining replicate wells to resolve fine copy number differences in heterogeneous mixtures and could foreseeably form the basis of more efficient diagnostic tests.
The second application demonstrates improved detection of rare mutant alleles by drastically reducing competitive PCR processes that occur in the presence of a highly homologous wild-type DNA background. With careful optimization, real-time PCR assays can detect down to the 1% mutant fraction. With the same assays, ddPCR partitions the competing background away from the mutant, effectively increasing the average mutant-to-wild-type ratio by 20 000 times. On average, the effective enrichment of the mutant molecules per PCR reaction is proportional to the number of sample partitions used. For a duplex TaqMan assay targeting the BRAF V600E mutation,(29) we show droplet partitioning detects 0.001% mutant fraction, 1 000 times lower than real-time PCR (Figure 3 and Supplementary Table 1 and Supplementary Figure 1 in the Supporting Information). With dependence on the amount recovered from clinical samples, more DNA can be loaded into the ddPCR system to push the detection limits down to even lower levels. This approach enables researchers to measure extremely low levels of mutant that could in turn lead to the improved detection of minimal residual disease and less invasive diagnostics.
Figure 3.

Detection of the BRAF V600E rare mutant allele in the presence of homologous wildtype DNA by droplet digital PCR. Serial dilutions of the mutant cell line DNA were prepared in a constant background of wildtype human genomic DNA. Droplet partitioning reduces competitive amplification effects allowing detection down to 0.001% mutant fraction, 1 000 times lower than real-time PCR. The mutant cell line contains 35% BRAF V600E, as measured by ddPCR.
We next evaluated the ability of this ddPCR system to quantitate DNA in clinical samples. Circulating DNA in cell-free plasma(30) has received increasing levels of attention as a sample type for developing noninvasive prenatal(31) and oncology(32) diagnostics. The cell-free DNA in plasma is highly fragmented(33) and present at low levels, which present challenges for quantitation. We enumerated fetal and total DNA in maternal cell-free plasma. For 19 maternal plasma samples taken between 10 and 20 weeks gestational age, the level of fetal (Figure 4a) and total DNA (Figure 4b) were measured for both male and female fetuses. A selective methylation-sensitive digest enabled the low-levels of hypermethylated RASSF1 fetal DNA(34) to be accurately quantified using our ddPCR system. With an absolute measure of SRY, RASSF1, and total DNA concentrations, the fetal load for each sample was calculated (Figure 4c). For male fetuses, a correlation of 93.7% between the hypermethylated RASSF1 fetal DNA and SRY fetal loads provided confidence in the estimates for female fetuses. On the basis of RASSF1 alone, fetal loads ranged from 2.1 to 11.9% and were in general agreement with those data collected by next-generation sequencing(35) that is currently limited to estimating fetal loads from male fetuses. This application demonstrates the capability of absolute quantitation of highly fragmented cell-free DNA in clinical samples.
Figure 4.
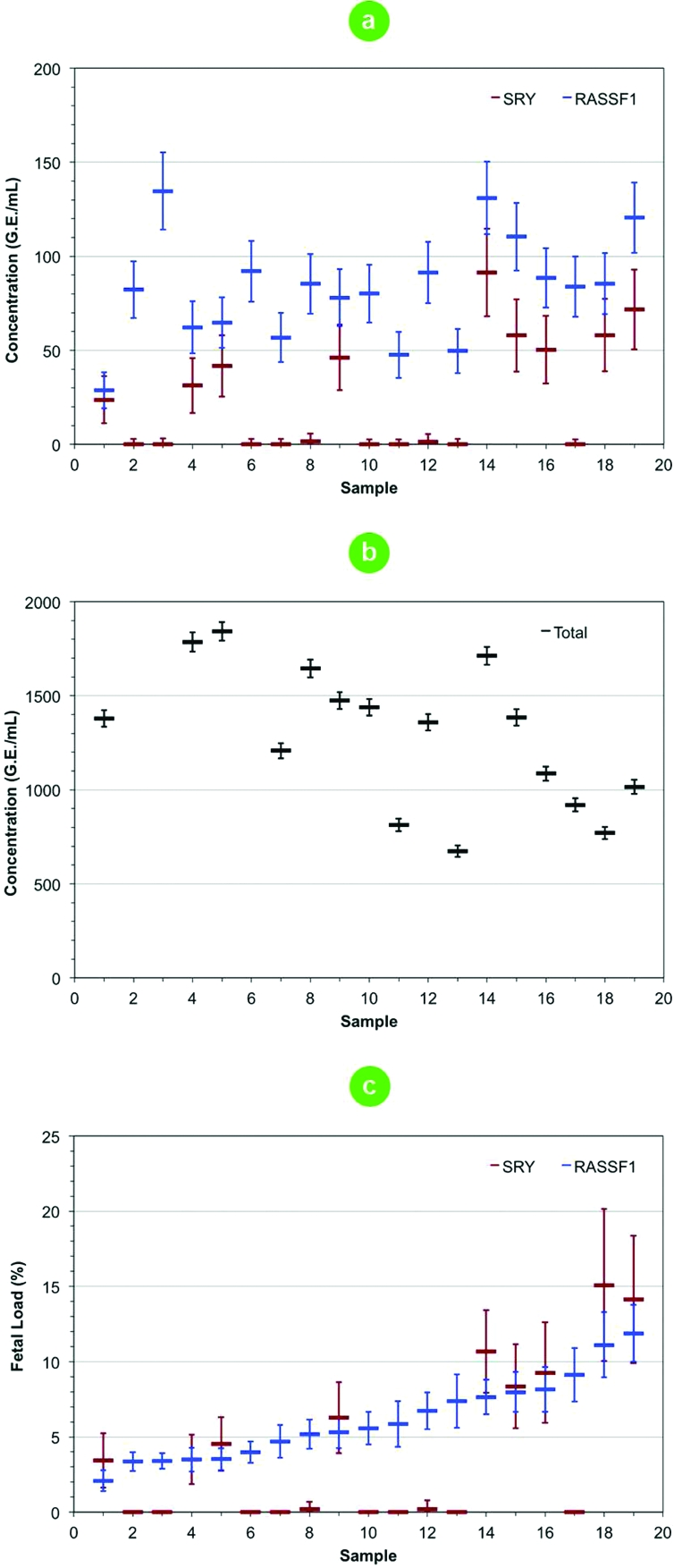
Absolute quantitation of circulating fetal and maternal DNA from cell-free plasma for male and female fetuses. (a) Quantitation of fetal DNA concentration using SRY (red bar) and hypermethylated RASSF1 (blue bar). The RASSF1 gene of circulating fetal DNA is hypermethylated whereas maternal DNA is hypomethylated. Methylation sensitive restriction enzymes selectively digested away the hypomethylated fraction, leaving the hypermethylated fetal DNA that was quantified. (b) Quantitation of total DNA concentration (black bar) represented as the weighted average from six independent assay measurements including undigested RASSF1 and β-actin as well as RNaseP and TERT. (c) Fetal loads as determined from the ratio of SRY to total (male fetuses only) and RASSF1 to total (male and female fetuses). For male fetuses, the Pearson’s correlation coefficient between SRY and RASSF1 fetal loads was 97.3%. Fetal DNA is not completely hypermethylated; therefore, the RASSF1 fetal loads measured for some samples are lower than those determined using SRY. Error bars represent the Poisson 95% confidence intervals of the concentration or the ratio in the case of fetal load estimates.
Overall, these data show that ddPCR offers a practical solution to realize precise estimates of DNA copy number with high-throughput. We anticipate this system will unlock the inherent power of digital PCR to more researchers for many applications.
Experimental Section
Droplet Digital PCR Workflow
The ddPCR workflow was described in Figure 1. The TaqMan PCR reaction mixture was assembled from a 2× ddPCR Mastermix (Bio-Rad), 20× primer, and probes (final concentrations of 900 and 250 nM, respectively) and template (variable volume) in a final volume of 20 μL. Each assembled ddPCR reaction mixture was then loaded into the sample well of an eight-channel disposable droplet generator cartridge (Bio-Rad). A volume of 60 μL of droplet generation oil (Bio-Rad) was loaded into the oil well for each channel. The cartridge was placed into the droplet generator (Bio-Rad). The cartridge was removed from the droplet generator, where the droplets that collected in the droplet well were then manually transferred with a multichannel pipet to a 96-well PCR plate. The plate was heat-sealed with a foil seal and then placed on a conventional thermal cycler and amplified to the end-point (40–55 cycles). After PCR, the 96-well PCR plate was loaded on the droplet reader (Bio-Rad), which automatically reads the droplets from each well of the plate (32 wells/h). Analysis of the ddPCR data was performed with QuantaSoft analysis software (Bio-Rad) that accompanied the droplet reader.
Determination of Copy Number Variation in HapMap Samples
For MRGPRX1, ChromosomeX, and CYP2D6, 4.4 μg of each purified human genomic DNA sample (Coriell) was digested with 10 units of RsaI (NEB) in 50 μL for 1 h at 37 °C. The digest was diluted 8-fold to 400 μL with TE buffer (pH 8.0) then 33 ng (3 μL) was assayed per 20 μL ddPCR reaction. For CCL3L1, 815 ng of each purified human genomic DNA sample (Coriell) was digested with 7.5 units of MseI (NEB) in 10 μL for 1 h at 37 °C. The digest was diluted 3.5-fold to 35 μL with TE buffer and then 69 ng (3 μL) was assayed per 20 μL ddPCR reaction. MRGPRX1 assay sequences(36) were (forward primer) 5′-TTAAGCTTCATCAGTATCCCCCA-3′, (reverse primer) 5′-CAAAGTAGGAAAACATCATCACAGGA-3′, and (probe) 6FAM-ACCATCTCTAAAATCCT-MGBNFQ. Chromosome X assay sequences(37) were (forward primer) 5′-GATGAGGAAGGCAATGATCC-3′, (reverse primer) 5′-TTGGCTTTTACCAAATAGGG-3′, and (probe) 5′-FAM-TGTTTCTCTCTGCCTGCACTGG-BHQ1-3′ (Integrated DNA Technologies). The CYPD2D6 (Hs00010001_cn) was purchased as a 20× premix of primers and FAM-MGBNFQ probe (Applied Biosystems). Modified CCL3L1 assay sequences(19) were (forward primer) 5′-GGGTCCAGAAATACGTCAGT-3′, (reverse primer) 5′-CATGTTCCCAAGGCTCAG-3′, and (probe) 6FAM-TTCGAGGCCCAGCGACCTCA-MGBNFQ. All CNV assays were duplexed with an RPP30 reference assay (forward primer) 5′-GATTTGGACCTGCGAGCG-3′, (reverse primer) 5′-GCGGCTGTCTCCACAAGT-3′, and (probe) VIC-CTGACCTGAAGGCTCT-MGBNFQ. Thermal cycling conditions were 95 °C × 10 min (1 cycle), 94 °C × 30 s and 60 °C × 60 s (40 cycles), 98 °C × 10 min (1 cycle), and 12 °C hold.
Determination of GRB7 and ERBB2 Copy Number Alterations
Purified DNA (20 ng) from each normal and tumorous breast tissue sample (D8235086-1, Biochain) was digested with 0.2 units of NlaIII in 10 μL for 1 h at 37 °C. The restricted DNA was added directly to ddPCR Mastermix at 8.8 ng (4.4 μL) per 20 μL of ddPCR reaction. ERBB2 (Hs02803918_cn) and GRB7 (Hs02139994_cn) assays were purchased as 20× premixes of primers and FAM-MGBNFQ probe (Applied Biosystems) and duplexed with the RPP30 reference assay described above. Thermal cycling conditions were 95 °C × 10 min (1 cycle), 94 °C × 30 s and 60 °C × 60 s (40 cycles), 98 °C × 10 min (1 cycle), and 12 °C hold.
Rare Allele Detection
A dilution series of BRAF V600E mutant DNA (HTB-38D) from a HT-29 cell line (ATCC) was prepared in a high, constant background (5 000 copies/μL) of wildtype DNA (NA19205, Coriell). For ddPCR, when the concentration of intact human genomic DNA is >66 ng/20 μL reaction, the accompanying increase in viscosity can cause the average droplet volume to change, which in turn could affect the accuracy of DNA quantitation. Therefore, for samples of this nature, restriction enzyme digestion is recommended to fragment the DNA and reduce solution viscosity. In our experience, once fragmented, the human genomic DNA concentration can exceed 1 μg/20 μL reaction without affecting the average droplet volume. Therefore, prior to ddPCR, each sample of the dilution series was digested with 40 U of HaeIII (NEB) in 100 μL containing 1× NEB buffer 4 and BSA. The BRAF V600E/wildtype duplex TaqMan assay used common primers (forward) 5′-CTACTGTTTTCCTTTACTTACTACACCTCAGA-3′, (reverse) 5′-ATCCAGACAACTGTTCAAACTGATG-3′, and specific probes (BRAF V600E) 6FAM-TAGCTACAGAGAAATC-MGBNFQ and (wildtype) VIC-CTAGCTACAGTGAAATC-MGBNFQ. Eight ddPCR wells were used for each sample of the dilution series. Thermal cycling conditions were 95 °C × 10 min (1 cycle), 94 °C × 30 s and 62.7 °C × 60 s (55 cycles), and 12 °C hold.
Quantitation of Cell-Free Fetal and Total DNA in Maternal Plasma
Whole blood (3 × 10 mL) was collected (ProMedDx) from healthy pregnant donors, between 10 and 20 weeks of gestational age, by venipuncture into cell-free DNA BCT tubes (Streck) according to the manufacturer’s instructions. Fetus gender was determined by ultrasound within 6 weeks of sample collection. The tubes were stored for up to 48 h at room temperature then shipped overnight at 4 °C to Bio-Rad where they were processed upon receipt. The whole blood was centrifuged for 10 min at 1 600g, the supernatant removed and transferred to a new tube, centrifuged for 10 min at 16 000g, the supernatant removed, and transferred to a new tube, then the cell-free plasma was stored at −80 °C. Cell-free plasma (5 mL) was thawed and cell-free DNA isolated using the QIAmp Circulating Nucleic Acid Kit (Qiagen) according to the manufactuerʼs protocol and eluted in AVE buffer (150 μL). A portion of the eluate (99 μL) was subjected to a single-tube digest containing HhaI (30 U), HpaII (60 U), and BstUI (30 U) in 1× NEB buffer 4 in a total volume of 120 μL. A second portion of the eluate (33 μL) was used in a no-digest control mixture where restriction enzymes were substituted for water. The mixtures were incubated for 37 °C for 2 h, 60 °C for 2 h, then 65 °C for 20 min. The restriction enzyme digested mixture was split and subjected to three ddPCR duplexed assays of SRY/TERT, RASSF1/RNaseP, and RASSF1/β-actin. The restriction enzyme mixture cuts unmethylated RASSF1 and β-actin TaqMan templates but not SRY, RNaseP, or TERT. The no-digest control mixture was split and subjected to two ddPCR duplexed assays of RASSF1/RNaseP and RASSF1/β-actin. β-Actin is hypomethylated in both fetal and maternal DNA and is completely digested by the enzyme cocktail.
RASSF1(34) and SRY(37) assays were reported previously. RNaseP and TERT copy number reference assays were purchased commercially (Applied Biosystems). The β-actin assay was modified from Chan et al. (forward primer) 5′- GCAAAGGCGAGGCTCTGT-3′, (reverse primer) 5′-CGTTCCGAAAGTTGCCTTTTATGG-3′, and (probe) VIC-ACCGCCGAGACCGCGTC-MGBNFQ. For RASSF1/RNaseP and RASSF1/β-actin duplexes, 1× GC-Rich Solution (Roche) was used as a component of the assembled ddPCR reaction mixtures. Thermal cycling conditions were 95 °C × 10 min (1 cycle), 95 °C × 30 s and 60 °C × 60 s (45 cycles), and 4 °C hold.
For each sample, six independent assay measurements of total DNA concentration (G.E/mL) were made from one TERT, one β-actin, two RASSF1, and two RNaseP assays. Each assay measurement comprised data from seven replicate ddPCR wells. We combined the droplet counts (positive and negative) from all seven replicate wells to yield a single “metawell”. The concentration and confidence intervals for each of the 6 measurement metawells were computed.(4) The appropriate dilution factors were applied to yield total cell-free DNA concentration (G.E./mL) and the confidence interval is scaled accordingly. The weighted mean of the six total measurements was calculated, where weights are inverses of confidence interval variances of these measurements. For digested RASSF1, there are two independent assay measurements, which are also combined in the same manner. For SRY, there is one measurement that was used directly, with scaling by a factor of 2 to account for haploidy. Fetal load is then computed as a ratio with the associated Poisson 95% confidence intervals.
Acknowledgments
The project described was supported by Grant Number R01EB010106 from the National Institute of Biomedical Imaging and Bioengineering. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Biomedical Imaging and Bioengineering or the National Institutes of Health.
Supporting Information Available
Additional information as noted in text. This material is available free of charge via the Internet at http://pubs.acs.org.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Sykes P. J.; Neoh S. H.; Brisco M. J.; Hughes E.; Condon J.; Morley A. A. Biotechniques 1992, 13, 444–449. [PubMed] [Google Scholar]
- Vogelstein B.; Kinzler K. W. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 9236–9241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCaughan F.; Dear P. H. J. Pathol. 2010, 220, 297–306. [DOI] [PubMed] [Google Scholar]
- Dube S.; Qin J.; Ramakrishnan R. PLoS One 2008, 3, e2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison T.; Hurley J.; Garcia J.; Yoder K.; Katz A.; Roberts D.; Cho J.; Kanigan T.; Ilyin S. E.; Horowitz D.; Dixon J. M.; Brenan C. J. Nucleic Acids Res. 2006, 34, e123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren L.; Bryder D.; Weissman I. L.; Quake S. R. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 17807–17812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottesen E. A.; Hong J. W.; Quake S. R.; Leadbetter J. R. Science 2006, 314, 1464–1467. [DOI] [PubMed] [Google Scholar]
- Fan H. C.; Quake S. R. Anal. Chem. 2007, 79, 7576–7579. [DOI] [PubMed] [Google Scholar]
- Dressman D.; Yan H.; Traverso G.; Kinzler K. W.; Vogelstein B. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 8817–8822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diehl F.; Li M.; He Y.; Kinzler K. W.; Vogelstein B.; Dressman D. Nat. Methods 2006, 3, 551–559. [DOI] [PubMed] [Google Scholar]
- Diehl F.; Diaz L. A. Jr. Curr. Opin. Oncol. 2007, 19, 36–42. [DOI] [PubMed] [Google Scholar]
- Li M.; Diehl F.; Dressman D.; Vogelstein B.; Kinzler K. W. Nat. Methods 2006, 3, 95–97. [DOI] [PubMed] [Google Scholar]
- Li M.; Chen W. D.; Papadopoulos N.; Goodman S. N.; Bjerregaard N. C.; Laurberg S.; Levin B.; Juhl H.; Arber N.; Moinova H.; Durkee K.; Schmidt K.; He Y.; Diehl F.; Velculescu V. E.; Zhou S.; Diaz L. A. Jr.; Kinzler K. W.; Markowitz S. D.; Vogelstein B.; Nat. Biotechnol. 2009, 27(9), 858−863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beer N. R.; Hindson B. J.; Wheeler E. K.; Hall S. B.; Rose K. A.; Kennedy I. M.; Colston B. W. Anal. Chem. 2007, 79, 8471–8475. [DOI] [PubMed] [Google Scholar]
- Beer N. R.; Wheeler E. K.; Lee-Houghton L.; Watkins N.; Nasarabadi S.; Hebert N.; Leung P.; Arnold D. W.; Bailey C. G.; Colston B. W. Anal. Chem. 2008, 80, 1854–1858. [DOI] [PubMed] [Google Scholar]
- Kiss M. M.; Ortoleva-Donnelly L.; Beer N. R.; Warner J.; Bailey C. G.; Colston B. W.; Rothberg J. M.; Link D. R.; Leamon J. H. Anal. Chem. 2008, 80, 8975–8981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver S.; Dube S.; Mir A.; Qin J.; Sun G.; Ramakrishnan R.; Jones R. C.; Livak K. J. Methods 2010, 50, 271–276. [DOI] [PubMed] [Google Scholar]
- Gonzalez E.; Kulkarni H.; Bolivar H.; Mangano A.; Sanchez R.; Catano G.; Nibbs R. J.; Freedman B. I.; Quinones M. P.; Bamshad M. J.; Murthy K. K.; Rovin B. H.; Bradley W.; Clark R. A.; Anderson S. A.; O’Connell R J.; Agan B. K.; Ahuja S. S.; Bologna R.; Sen L.; Dolan M. J.; Ahuja S. K. Science 2005, 307, 1434–1440. [DOI] [PubMed] [Google Scholar]
- Sudmant P. H.; Kitzman J. O.; Antonacci F.; Alkan C.; Malig M.; Tsalenko A.; Sampas N.; Bruhn L.; Shendure J.; Eichler E. E. Science 2010, 330, 641–646.21030649 [Google Scholar]
- Carter N. P. Nat. Genet. 2007, 39, S16–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schouten J. P.; McElgunn C. J.; Waaijer R.; Zwijnenburg D.; Diepvens F.; Pals G. Nucleic Acids Res. 2002, 30, e57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldhous M. C.; Abu Bakar S.; Prescott N. J.; Palla R.; Soo K.; Mansfield J. C.; Mathew C. G.; Satsangi J.; Armour J. A. Hum. Mol. Genet. 2010, 19, 4930–4938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherry S. T.; Ward M. H.; Kholodov M.; Baker J.; Phan L.; Smigielski E. M.; Sirotkin K. Nucleic Acids Res. 2001, 29, 308–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin J.; Jones R. C.; Ramakrishnan R. Nucleic Acids Res. 2008, 36, e116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkan C.; Kidd J. M.; Marques-Bonet T.; Aksay G.; Antonacci F.; Hormozdiari F.; Kitzman J. O.; Baker C.; Malig M.; Mutlu O.; Sahinalp S. C.; Gibbs R. A.; Eichler E. E. Nat. Genet. 2009, 41, 1061–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartlett J. M.; Ibrahim M.; Jasani B.; Morgan J. M.; Ellis I.; Kay E.; Connolly Y.; Campbell F.; O’Grady A.; Barnett S.; Miller K. Amer. J. Clin. Pathol. 2009, 131, 106–111. [DOI] [PubMed] [Google Scholar]
- Kauraniemi P.; Kuukasjarvi T.; Sauter G.; Kallioniemi A. Am. J. Pathol. 2003, 163, 1979–1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luoh S. W. Cancer Genet. Cytogenet. 2002, 136, 43–47. [DOI] [PubMed] [Google Scholar]
- Benlloch S.; Paya A.; Alenda C.; Bessa X.; Andreu M.; Jover R.; Castells A.; Llor X.; Aranda F. I.; Massuti B. J. Mol. Diagn. 2006, 8, 540–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo Y. M.; Corbetta N.; Chamberlain P. F.; Rai V.; Sargent I. L.; Redman C. W.; Wainscoat J. S. Lancet 1997, 350, 485–487. [DOI] [PubMed] [Google Scholar]
- Wright C. F.; Burton H. Hum. Reprod. Update 2009, 15, 139–151. [DOI] [PubMed] [Google Scholar]
- Pathak A. K.; Bhutani M.; Kumar S.; Mohan A.; Guleria R. Clin. Chem. 2006, 52, 1833–1842. [DOI] [PubMed] [Google Scholar]
- Fan H. C.; Blumenfeld Y. J.; Chitkara U.; Hudgins L.; Quake S. R. Clin. Chem. 2010, 56, 1279–1286. [DOI] [PubMed] [Google Scholar]
- Tong Y. K.; Jin S.; Chiu R. W.; Ding C.; Chan K. C.; Leung T. Y.; Yu L.; Lau T. K.; Lo Y. M. Clin. Chem. 2010, 56, 90–98. [DOI] [PubMed] [Google Scholar]
- Fan H. C.; Blumenfeld Y. J.; Chitkara U.; Hudgins L.; Quake S. R. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 16266–16271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosono N.; Kubo M.; Tsuchiya Y.; Sato H.; Kitamoto T.; Saito S.; Ohnishi Y.; Nakamura Y. Hum. Mutat. 2008, 29, 182–189. [DOI] [PubMed] [Google Scholar]
- Fan H. C.; Blumenfeld Y. J.; El-Sayed Y. Y.; Chueh J.; Quake S. R. Am. J. Obstet. Gynecol. 2009, 200 (543), e541–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.