

Abstract
Gastrointestinal oncology is one of the foremost causes of death: the gastric cancer accounts for 10.4% of cancer deaths worldwide, the pancreatic cancer for 6%, and finally, the colorectal cancer for 9% of all cancer-related deaths. For all these gastrointestinal cancers, surgical tumor resection remains the primary curative treatment, but the overall 5-year survival rate remains poor, ranging between 20–25%; the addition of combined modality strategies (pre- or postoperative chemoradiotherapy or perioperative chemotherapy) results in 5-year survival rates of only 30–35%. Therefore, many investigators believe that the potential for making significant progress lies on understanding and exploiting the molecular biology of gastrointestinal tumors to investigate new therapeutic strategies such as specific immunotherapy. In this paper we will focus on recent knowledge concerning the role of T cells and the use of T adoptive immunotherapy in the treatment of gastrointestinal cancers.
1. Introduction
Gastrointestinal oncology is one of the foremost causes of death; regarding the gastric cancer (GC) the American Cancer Society estimated one million new cases, nearly 70% of them in developing countries, and about 800,000 deaths [1]; instead the pancreatic cancer (PC) is the fourth leading cause of cancer deaths among men and women, being responsible for 6% of all cancer-related deaths [2], and finally, the colorectal cancer (CRC) accounted for 9% of all cancer deaths (49, 920) in 2009 [3].
For all these gastrointestinal cancers, surgical tumor resection remains the primary curative treatment but the overall 5-year survival rate remains poor, ranging between 20–25% [4–6]. The addition of combined modality strategies (pre- or postoperative chemoradiotherapy or perioperative chemotherapy) results in 5-year survival rates of only 30–35% [7–9].
Therefore, many investigators believe that the potential for making significant progress lie on understanding and exploiting the molecular biology of gastrointestinal tumors to investigate new therapeutic strategies such as gene therapy [10] and especially specific immunotherapy [11–13].
Evidence from different analysis suggests a key role of the immune system in counterattack of cancer progression: tumors are 100 times more likely to occur in people who take immunosuppressive medications than in people with normal immune function [14], and, in opposition, heightened anti-tumor activity of the immune system has been suggested in many reports of spontaneous cancer regression [15]. Also, a positive correlation between tumor-infiltrating lymphocytes and patients' survival has been observed [16]; moreover tumor-specific T-cell responses have been found in patients with a variety type of tumors [17].
Immune defence against tumor is mediated through antigen-specific and nonspecific immune mechanisms (macrophage and NK cell lineage and soluble factors such as cytokines). The operational, instead, of the antigen-specific immune system is based on a division of tasks between T cells and B cells (Figure 1).
Figure 1.
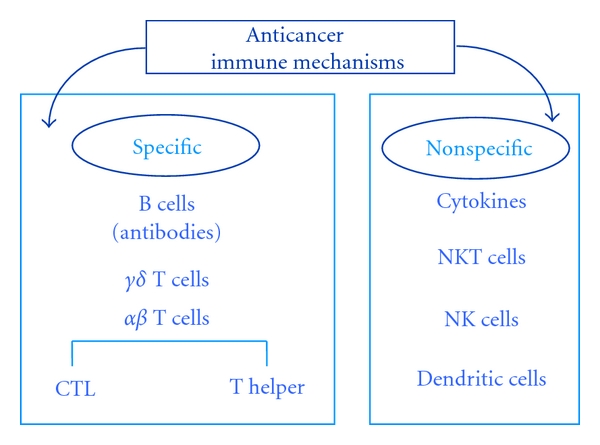
Innate and adaptive immune defence against cancer cells.
Various reagents (vaccines, infusion of T cells, or cytokines) can stimulate the immune system essentially through two mechanisms: (1) stimulation of the antitumor response, either by increasing the number of effector cells or by producing soluble mediators (e.g., cytokines); (2) alteration of tumor cells to increase their immunogenicity and susceptibility to immunological defences. However, the cancer cells have developed a number of different strategies to escape immune surveillance such as loss of tumor antigen expression, MHC downregulation, expression of Fas-L that can induce apoptosis in activated T cells, secretion of cytokines such as IL-10 (Interleukin-10) or TGF-β (Tumor grow factor-β), or generation of regulatory T (Treg) cells [18].
The requirement for an immune-based strategy in opposition to cancer is the induction of an effective tumor-specific immunity in order to break immunological tolerance to the tumor and generate antitumor immunity. To achieve this goal, several strategies as in preclinical models as in clinical trials are currently being investigated.
In this paper we will focus on recent knowledge concerning the role of T cells and the use of T adoptive immunotherapy in the treatment of gastrointestinal cancers.
2. Pancreatic Cancer
2.1. In Human and Animal Model T-Cell Response
Over the past 30 years, a large body of data has been accumulated showing that cancer patients generate B and T cells specific to antigens expressed on autologous pancreatic tumor cells [19–25]. PC expresses a variety of cancer-associated antigens that can potentially be recognized by T cells [26, 27]. Recent studies demonstrated that functionally competent CD4+ and CD8+ T cells with specificity for cancer antigens are spontaneously induced in the bone marrow of all PC patients [27, 28]. Moreover, in approximately 50% of the patients, these tumor-specific T cells are also present in the blood. Upon specific stimulation they mainly secrete the type 1 cytokine IFN-γ, which is typical of cytotoxic immune responses. The high incidence of spontaneous T-cell reactivity versus PC is in contrast to observations from numerous other cancer entities that induced cancer-reactive T cells only in 25–60% of the patients [29–31].
T-cell responses are regulated by dendritic cells (DCs), which constantly take up antigens in all tissues and upon in situ activation, stimulate naive T cells. While type I interferons, heat shock proteins, and extracellular matrix degradation products may induce DC activation in cancer tissues, immune-suppressive cytokines (IL-10/TGF-β) inhibit DC activation, and in PC the latter are produced at high concentrations by cancer-induced pancreatic stellate cells, cancer-infiltrating macrophages and mast cells [32], or Tregs [33]. Through recruitment and activation of stroma cell populations, PC generates a predominantly immune-suppressive microenvironment (Figure 2).
Figure 2.
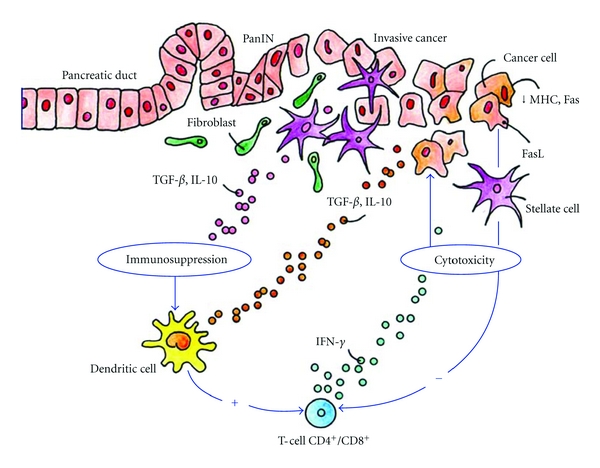
Pancreatic cancer microenvironment: interactions of immune cells with the cancer cells. Yellow: products of stellate cells; green: T-cell derived cytokines; grey: cancer cell-derived factors.
The regular induction of T-cell responses in the bone marrow of PC patients is thus intriguing.
The bone marrow is a site of T-cell priming against blood-borne antigens [34]. It can collect soluble cancer antigens released into the blood from necrotic cancer areas. Here, these are incorporated and presented by bone marrow-resident DCs in an immune-stimulatory environment. In addition, disseminated neoplastic cells detectable in many patients represent a local source of cancer antigens [28].
PC is frequently diagnosed at late stages. In this situation, large antigen amounts may reach the bone marrow. This might explain the comparably high incidence of T-cell responses in PC despite a predominantly immune-suppressive environment in the primary cancer. Once stimulated, T cells differentiate into effector T cells and enter the blood. Since cancer-reactive T cells have been found in the blood of many PC patients, these cells may infiltrate pancreatic carcinomas.
In one study, cancer-reactive CD8+ T cells specifically lysed autologous PC cells in vitro and delayed progression of xenotransplanted, autologous carcinomas [27]. Accordingly, increased numbers of cancer-infiltrating CD4+ and CD8+ T cells correlated well with improved prognosis of PC patients [35].
These findings point to a potential implication of cancer- specific T cells during cancer progression, but PC cells successfully employ various mechanisms to evade immune surveillance (Figure 2): (a) the downregulation of MHC molecules and of fas receptor, rendering neoplastic cells more resistant to recognition and cytolysis by activated effector T cells [27], (b) the recruitment and local maintenance of Tregs [36] that inhibit effector T-cell activation and function, (c) the secretion of IL-10 and TGF-β, additionally reducing local T-cell activity [27, 37], (d) the inactivation of cancer-infiltrating T cells as shown by a severe loss of CD3 zeta, [37] and (e) the expression of fas ligand on neoplastic cells, inducing apoptosis in cancer-infiltrating effector T cells [38].
Thus, PC is not characterized by a lack of specific T-cell immunity but by a potent barrier established by complex cancer-stroma interactions that inhibit T-cell activity in situ; for this purpose is most explanatory the recent results obtained by De Monte et al. [39]; they showed that thymic stromal lymphopoietin (TSLP), which favors Th2 cell polarization through myeloid DC conditioning, was secreted by cancer-associated fibroblasts (CAFs) after activation with tumor-derived TNF-α and IL-1β. Also the authors found that the ratio of GATA-3+(Th2)/T-bet+ (Th1) tumor-infiltrating T cells is an independent predictive marker of patient survival. Patients surgically treated for stage IB/III disease with a ratio inferior to the median value had a statistically significant prolonged overall survival, implying an active role for Th2 responses in disease progression.
In addition, in a mouse model in which an activating K-Ras mutation is expressed in the pancreas, preinvasive pancreatic lesions are characterized by the infiltration of immune suppressor cells rather than immune effector cells, suggesting that tumor immunity may be blocked from the inception of PC development [40].
All mice with the K-Ras mutation develop pancreatic adenocarcinoma and eventually die of disease. Finally, the finding that antagonism of negative T-cell regulators, such as cytotoxic T-lymphocyte-associated (CTLA) protein-4 and B- and T-lymphocyte attenuator (BTLA), can augment the antitumor immune response confirms that patients mount an immune-specific response to their tumor [41, 42]. Despite mounting evidence that an antitumor immune response is elicited in cancer patients, this response is ineffective and does not result in the tumor eradication, and a better understanding of the mechanisms underlying these interactions is required to develop future therapeutic strategies to employ the patient's own T-cell arsenal for efficient cancer control.
2.2. T-Cell Immunotherapy of Pancreatic Cancer
The history of vaccine trials in pancreatic cancer targeting a defined PC antigen started with the publication of a pilot study of mutant ras peptide vaccines tailored to represent the K-RAS mutations identified in biopsies from the patients with cancer [43] In this trial, immune responses specific for individual ras mutations were obtained in 2 of the 5 patients enrolled; in addition, both patients had a relatively long survival (11 and 8 months). These data shown that: (a) patients with metastatic PC were immunocompetent, (b) mutant ras vaccines were immunogenic, and (c) immune responses were correlated with survival. Furthermore, the treatment was well tolerated as no adverse effects were observed. A fine evaluation of the immune responses in these two patients [44] highlighted that peptide vaccination with a single mutant p21-ras-derived peptide induced CD4+ and CD8+ specific for nested epitopes, including the Gly/Val substitution at codon 12 and that both these T-cell subsets specifically recognize tumour cells owning to the corresponding K-ras mutation. Encouraged by these results, a second trial was performed, using intradermal vaccination of mutant ras peptides with GMCSF (Granulocyte-macrophage colony-stimulating factor) as an adjuvant [45]. 48 patients (10 surgically resected and 38 with advanced disease) were treated on an outpatient basis. Peptide-specific immunity was induced in 25 of 43 (58%) evaluable patients, indicating that the protocol used is very potent and able to elicit immune responses even in patients with end-stage disease. This study also demonstrated a strong association between immune responses and prolonged survival. Patients with advanced cancer and with immune response to the peptide vaccine showed prolonged survival from the start of treatment compared to nonresponders (median survival 148 days versus 61 days). Furthermore, the study proved long-term memory in numerous patients and entry of vaccine-specific T cells into the tumour mass.
In recent years, much work has focused on adoptive tumor immunotherapy in which the T cells of cancer patient are expanded and reinfused into the patient.
One method results in the selective expansion of T cells endogenously expressing TCRs specific for the tumor antigen of interest [46]. In a clinical study, MUC-1-specific autologous T cells, isolated from patient PBMCs (peripheral blood mononuclear cells), were expanded by incubation with a MUC-1-presenting cell line prior to administration to PC patients. The mean survival time for unresectable patients in this study was 5 months [47]. However, patients with resectable pancreatic cancer had 1-, 2- and 3-year survival rates of 83.3, 32.4, and 19.4%, respectively, and a mean survival time of 17.8 months. In a similar study, the same group isolated adherent cells from patient PBMCs to generate mature DCs that were then pulsed with MUC-1 peptide. The pulsed DCs were administered, along with autologous expanded MUC-1-specific T cells, to patients with unresectable or recurrent pancreatic cancer. Remarkably, a complete response was observed in one patient with lung metastases, and the mean survival time of the whole group was 9.8 months, suggesting that the addition of pulsed DCs may have improved the outcome [48].
A key role in future immunotherapeutic treatment of PC patients seems to be for the novel antigen PC-associated α-enolase (ENOA), a metabolic enzyme involved in the synthesis of pyruvate. In tumor cells, ENOA is upregulated and supports anaerobic proliferation (Warburg effect); also, it is expressed at the cell surface, where it promotes cancer invasion. ENOA is upregulated in different tumors, including brain, breast, cervix, colon, gastric, kidney, lung ovary, and especially pancreas [49].
In pancreatic cancer, ENOA elicits a CD4+ and CD8+ T-cell response both in vitro and in vivo [49]. Anti-MHC class I antibodies inhibited the cytotoxic activity of ENOA-stimulated CD8+ T lymphocytes against PC cells, but no MHC class I restricted peptide of ENOA has been identified so far. Moreover, in pancreatic ductal adenocarcinoma patients, production of anti-ENOA Immunoglobulin-G (IgG) is correlated with the ability of T cells to be activated in response to the protein [49], thus confirming the induction of a T- and B-cell integrated antitumor activation against ENOA. In oral squamous cell carcinoma, an HLA-DR8-restricted peptide (amino acid residues 321–336) of human ENOA recognized by CD4+ T cells and able to confer cytotoxic susceptibility has been identified [50, 51].
Most importantly, clinical correlations [52–54] propose ENOA as a novel target for cancer immunotherapy, in particular in pancreatic cancer, where pancreas-specific Ser 419 phosphorylated ENOA is upregulated and also induces the production of autoantibodies with diagnostic and prognostic value [49].
3. Gastric Cancer
3.1. Gastric Cancer-Infiltrating T Cells
Although the GC etiology has been completely obscure for many decades, several considerable advances in the knowledge of the carcinogenesis and development of gastric cancer have been made in the present era. First, it is well known that Helicobacter pylori (H. pylori) infection is associated with the GC carcinogenesis, suggesting that chronic inflammation may be implicated in the development of intestinal metaplasia and mutations in oncogenes that precede the GC development; indeed, the International Agency for Research on Cancer classified H. pylori as a class I human carcinogen in 1994 [55]. Second, the long-suspected influence of genetic susceptibility has been elucidated, and several polymorphisms of inflammatory cytokine genes have been implicated as risk factors for gastric cancer [56–60].
Although immune cells constitute an additional and prominent component of the host response to cancer, their participation in tumor pathogenesis remains unclear. In the tumor microenvironment, there is a delicate balance between antitumor immunity and tumor-originated proinflammatory activity, which weakens antitumor immunity [61–63].
It has been shown that the infiltrating grade of CD3+ tumor-infiltrating lymphocytes (TILs) was correlated with a favorable outcome in patients with several types of cancer, including gastric cancer [64]. Thus, it is imperative to understand immunoregulation in gastric cancer, in order to develop novel treatment strategies or improve the efficacy of standard therapies.
The first evidence of correlation between T-cell response and GC was the study of Ren et al. [65] that reported a shift from Th1 to Th2 pattern of cytokine secretion in gastric cancer and has suggested that this may be a critical factor in promoting growth of neoplastic cells. However, our data [66] of tumor-infiltrating and perilesional H. pylori-specific T cells failed to confirm such a Th1-Th2 shift. Rather, the major difference between the gastric T-cell clones from uncomplicated chronic gastritis and those from gastric cancer was the degree of expression of cytolytic activity. Indeed, in all patients studied, virtually all the H. pylori-specific CD4+ clones derived from gastric tumors or perilesional mucosa consistently expressed perforin-mediated cytolytic potential and Fas-Fas ligand-mediated proapoptotic activity against target cells.
Most recently, Maruyama et al. [67] investigated the distribution of Th17 (T helper 17) cells in relation to Treg as in the TILs as in peripheral blood of GC patients. They showed that in TILs from patients with early disease, the frequency of Th17 cells was significantly higher than that in the normal gastric mucosa (23.7 ± 8.9 versus 4.5 ± 3.1%). Besides, in TILs from patients with advanced disease (n = 28), the frequency of Th17 cells was also significantly higher, but lower compared to early disease, than that in the normal gastric mucosa (15.1 ± 6.2 versus 4.0 ± 2.0%). When the ratio of Th17/Treg in TILs was evaluated in individual cases, it was more markedly increased in early than in advanced disease.
In summary, the accumulation of Th17 cells as well as Tregs in the tumor microenvironment of gastric cancer occurred in early disease, and then the infiltration of Th17 cells gradually decreased according to the disease progression, in contrast to increased Tregs.
3.2. T-Cell-Based Antigastric Cancer Treatments
There are different types of T-cell-based anticancer therapy approaches, using (a) CTL, (b) TILs, or (c) Engineered T cells.
Improved CTL cell culture technology has permitted the first clinical tests for adoptive transfer of CTLs, and this technique [68] seems to result in substantial activity in patients with melanoma; CTLs were used to treat patients with metastatic melanoma, and 8 out of 20 patients had anti-tumor immune responses [68]. These results were confirmed in an independent trial in which engraftment of the CTLs, as measured by an elevated frequency of circulating T cells able to bind tetramers loaded with MART-1 peptides, was detectable up to two weeks after T-cell transfer in all patients [69].
Recently, Kim et al. [70] evaluated the antitumor activity of ex vivo expanded T cells against the human gastric cancer. For this purpose, human peripheral blood mononuclear cells were cultured with IL-2-containing medium in anti-CD3 antibody-coated flasks for 5 days, followed by incubation in IL-2-containing medium for 9 days. The resulting populations were mostly CD3+ T cells [97%] and comprised 1% CD3−CD56+, 36% CD3+CD56+, 11% CD4+, and 80% CD8+. This heterogeneous cell population was also called cytokine-induced killer (CIK) cells. CIK cells strongly produced IFN-γ, moderately TNF-α, but not IL-2 and IL-4. At an effector-target cell ratio of 30 : 1, CIK cells destroyed 58% of MKN74 human gastric cancer cells, as measured by the 51Cr-release assay. In addition, CIK cells at doses of 3 and 10 million cells per mouse inhibited 58% and 78% of MKN74 tumor growth in nude mouse xenograft assays, respectively. This study suggests that CIK cells may be used as an adoptive immunotherapy for GC patients.
The adoptive GC immunotherapy with CIK cells has been also reported in preclinical and clinical studies [71]. MHC-I restricted CTLs from GC patients recognize tumor-associated antigen and react specifically against self-tumor cells [72, 73]. One tumor-specific antigen, MG7-antigen, shows great potential for predicting early cancer as well as for inducing immune responses to GC [74, 75]. Using HLA-A-matched allogeneic gastric cancer cells to induce tumor-specific CTLs appears to be an alternative immunotherapy option for gastric cancer [76].
Also, CIK cells in combination with chemotherapy showed benefits for patients who suffer from advanced gastric cancers [77, 78]. The serum levels of the tumor markers were significantly decreased, the host immune function was increased, and the short-term curative effect, as well as the quality of life, was improved in patients treated by chemotherapy plus CIK cells compared to those in patients treated by chemotherapy alone. CIK cells killed MGC-803 GC cells by inducing apoptosis in the early stage and by inducing necrosis in the late stage through downregulation of p53, c-myc, and bcl-2 and upregulation of bax [79].
In summary, despite the introduction of immune cell-based immunotherapy, the paucity of preclinical and clinical studies has limited the broad application of immunotherapy for the treatment of GC patients with gastric cancers. Here, preclinical evidence proved that CIK cell immunotherapy can be used in patients with gastric cancer.
Adoptive transfer therapy with TILs requires the isolation of T cells from neoplastic biopsies or surgical tissue and the selection of tumor-specific T cells ex vivo (Figure 3). The adoptive transfer of TILs has been promising in preclinical models [80], but clinical experiences were almost uniformly disappointing [81, 82].
Figure 3.
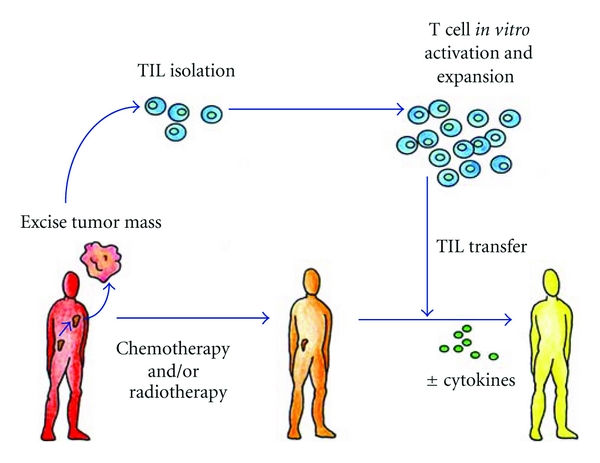
Scheme of adoptive autologous TILs transfer. T-infiltrating lymphocytes can be isolated from resected surgical samples and expanded in vitro for adoptive transfer after lymphodepleting chemotherapy. Most adoptive transfer therapy approaches using TILs have involved the use of IL-2 infusion following T-cell transfer in order to select tumor-specific T cells.
Technical difficulties in producing tumor-specific T cells currently represent a barrier to randomized clinical trials. Only 30%–40% of the biopsies yield satisfactory T-cell populations, and the whole process requires about 6 weeks before the T cells would be ready for infusion [83]. Furthermore, nearly all clinical experiences with TILs have been done in patients with melanoma, because of the easy surgical availability of the tumor tissue. However, should technical limitations of current tissue culture approaches be overcome, recent studies indicate that the presence of TILs positively correlates with patients survival in ovarian and colorectal cancer [84, 85], thus prompting the use of this protocol for other commonly encountered epithelial neoplasias. Recently we have [11] analyzed the functional properties of the T-cell response to different antigen peptides related to GC in patients with gastric adenocarcinoma. To this purpose, we have cloned and characterized TILs isolated from the neoplastic gastric tissue samples. A T-cell response specific to different peptides of gastric cancer antigens tested was documented in 17 out of 20 patients, selected for their HLA-A02 and/or -A24 alleles. Most of the cancer peptide-specific TILs expressed a Th1 profile and cytotoxic activity against target cells. The effector functions of cancer peptide-specific T-cells obtained from the peripheral blood of the same patients were also studied, and the majority of peripheral blood peptide-specific T cells also expressed the Th1 functional profile.
In conclusion, in most of patients with gastric adenocarcinoma, a specific type-1 T-cell response to GC antigens was detectable and would have the potential of hamper tumor cell growth. However, in order to get tumor cell killing in vivo, the activity and the number of cancer peptide-specific Th1 cells probably need to be enhanced by vaccination with the appropriate cancer antigenic peptides or by injection of the autologous tumor peptide-specific T cells expanded in vitro. These studies have laid the groundwork for a possible vaccination of gastric adenocarcinoma patients with specific peptides of tumor-associated antigens able to raise an effective immune response to gastric cancer.
4. Colorectal Cancer
4.1. Tumour-Infiltrating T-Cell Subsets in Colorectal Cancer
In recent years, different studies demonstrated the presence of T cell into neoplastic tissue of colorectal patients and also that the type, location, and density of tumor-infiltrating immune cells are of strong predictive impact influencing the behavior of human CRC [85, 86]. Although the exact mechanism remains uncertain, the adaptive immune system plays an important role in suppressing tumour progression [87, 88]. In the Table 1 we resumed the major studies correlating the TIL subsets and survival of CRC patients.
Table 1.
Studies correlating colorectal cancer patient survival with TIL subsets.
| N° of events | High density of T-cells (versus low density) | References | ||||||
|---|---|---|---|---|---|---|---|---|
| Sample size | OS | CS | Disease stage | T-cell subset analysed | 5-year CS, OS, or DFS, long-rank P value | CS, OS, or DFS univariate HR (95% Cl)*, P value | CS, OS, or DFS multivariate HR (95% Cl)*, P value | |
| 131 | — | — | Dukes' A–D | CD8+ | P = 0.0003 (OS) | — | 0.61 (0.41–0,89) | [139]; 58: 3491–3494 |
|
| ||||||||
| CD3+ | P = 0.004 (OS) | — |
P = 0.011 (OS) 0.40 (0.19–0.85) (OS) |
|||||
| 109 | 37 | — | II-III | CD8+ | P = 0.0008 (OS) | — | 0.33 (0.15–0.73) (OS) | [140]; 159: 297–304 |
| GZMB+ | P = 0.0001 (OS) | — | 0.23 (0.10–0.50) (OS) | |||||
|
| ||||||||
| CD8+ | 77% (versus 38%) P = 0.011 (CS) |
— | — | |||||
| 93 | 59 | 47 | Dukes' C | CD45RO+ | 66% (versus 33%) P = 0.002 (CS) |
— | — | [141]; 17: 25–29 |
| CD68+ | 60% (versus 37%) P = 0.033 (CS) |
— | — | |||||
|
| ||||||||
| 72 | — | — | I–IV | CD134+ | P = 0.02 (CS) | — | — | [142]; 183: 512–518 |
|
| ||||||||
| 41 | 25 | — | Dukes' A–D | CD4+/CD8+ ratio | 22% (versus 61%) (OS) | — | P = 0.046 (OS) | [143]; 52: 423–428 |
|
| ||||||||
| 97 | — | — | — | CD8+ | — | P = 0.01 (OS) | — | [144]; 10: 309–313. |
|
| ||||||||
| 152 | — | — | III | CD8+ | P < 0.001 (CS) | — | — | [145]; 35:808–816 |
|
| ||||||||
| 93 | — | — | II-III | CD8+ | P = 0.03 (DFS) | — | 0.56 (0.32–0.99) P = 0.04 (DFS) | [146]; 84: 493–501 |
|
| ||||||||
| 371 | — | 74 | I–IV | CD8+ | P < 0.0001 (CS) | — | 0.43 (0.23–0.83) P = 0.01 (CS) | [147]; 91: 1711–1717 |
|
| ||||||||
| 336 | 158 | — | Dukes' A–D | CD45RO+ | 65% (versus 35%) P < 0.0001 (OS) 72% (versus 50%) P < 0.001 (DFS) |
— | P = 0.02 (OS) | [148]; 353: 2654–2666 |
|
| ||||||||
| CD3+ | P < 0.05 (CS) | — | — | |||||
| 117 | — | — | Dukes' A–D | CD8+ | P < 0.25 (CS) | — | — | [149]; 4: 1351–1357 |
| CD16+ | P < 0.04 (CS) | — | — | |||||
|
| ||||||||
| CD3+ | 73% (versus 40%) P < 0.0001 (OS) 81% (versus 54%) P = 0.0012 (DFS) |
— | 0.53 (0.40–0.70) P < 0.0001 (OS) 0.42 (0.29–0.60) P < 0.0001 (DFS) |
|||||
| CD8+ | 72% (versus 50%) P < 0.0001 (OS) |
— | — | |||||
| 406 | — | — | I–III | CD45RO+ | 82% (versus 56%) P = 0.0002 (DFS) 68% (versus 33%) P < 0.0003 (OS) |
— | — | [150]; 313: 1960–1964 |
| GZMB+ | 77% (versus 37%) P = 0.002 (DFS) 72% (versus 61%) P = 0.15 (OS) |
— | — | |||||
| CD3+, CD45RO+ | 84% (versus 68%) P = 0.39 (DFS) P < 0.0001 (DFS) |
— | — | |||||
|
| ||||||||
| 587 | — | — | I-II (MSS only) | CD8+ | — | 0.47 (0.33 0.68) P < 0.001 (CS) | 0.47 (0.30–0.73) P = 0.001 (CS) | [151]; 112: 495–502 |
|
| ||||||||
| 392 | — | 226 | I-II (Rectum only) | CD8+ | — | 0.55 (0.41–0.74) P < 0.001 (CS) | 0.63 (0.45–0.88) P = 0.006 (CS) | [152]; 99: 1712–1717 |
|
| ||||||||
| 101 | — | — | II-III | CD3+/FOXP3+ ratio | 71% (versus 62%) (OS) 67% (versus 46%) (DFS) |
0.57 (0.30–1.09) P = 0.087 (OS) 0.46 (0.24–0.90) P = 0.021 (DFS) | 0.47 (0.24–0.94) P = 0.039 (DFS) | [153]; 137: 1270–1279 |
|
| ||||||||
| Node-negative P = 0.01 (CS) |
||||||||
| 286 | — | 136 | II-III | CD3+ | Node-positive P = 0.66 (CS) |
— | — | [154]; 10: 877–884 |
| Node-negative P = 0.006 (DFS) |
||||||||
| Node-positive P = 0.62 (DFS) |
||||||||
|
| ||||||||
| MSS group 162% (versus 46%) P = 0.004 (CS) |
— | 0.73 (0.60–0.90) P = 0.019 (CS) | ||||||
| 1232 | — | — | I-III | FOXP3+ | MSS group 2 (CS) 60% (versus 44%) P = 0.001 (CS) |
— | 0.70 (0.60–0.90) P = 0.007 (CS) | [155]; 126: 2635–2643 |
| MSI group (CS) 75%? (versus 63%?) P = 0.029 (CS) |
— | 0.63 (0.3–1.2) P = 0.13 (CS) | ||||||
|
| ||||||||
| CD8+ | — | 0.74 (0.67–0.82) P < 0.0001 (CS) |
NS (CS) | |||||
| 445 | — | — | II-III | CD45RO+ | — | 0.74 (0.65–0.84) P < 0.0001 (CS) |
NS (CS) | [156]; 27: 186–192 |
| FOXP3+ | — | 0.78 (0.70–0.87) P < 0.0001 (CS) |
0.54 (0.38–0.77) P = 0.001 (CS) |
|||||
|
| ||||||||
| CD8+ | P < 0.0001 (OS and DFS) | — | — | |||||
| 411 | — | — | I-II | CD45RO+ | P < 0.0001 (OS and DFS) | — | — | [157]; 27: 5944–5951 |
| CD8+ plus CD45RO+ | P < 0.0001 (OS and DFS) | — | P < 0.0001 (CS, OS and DFS) | |||||
|
| ||||||||
| CD3+ | P = 0.04 (OS) | — | 0.54 (0.18–1.59) P = 0.26 (OS) | |||||
| 209 | 100 | 100 | I–IV | CD8+ | P = 0.04 (OS) | — | 2.06 (0.67–6.39) P = 0.21 (OS) | [158]; 11: 19 |
| GZMB+ | — | — | 1.18 (0.45–3.13) P = 0.74 (OS) | |||||
|
| ||||||||
| 94 | — | — | I–IV | CD8+/FOXP3+ ratio | P = 0.01 (OS) | 0.35 (0.15–0.81) P = 0.014 (OS) | 0.40 (0.17–0.94) P = 0.035 (OS) | [159]; 59: 653–661 |
|
| ||||||||
| 57 | — | — | — | FOXP3+ | — | P = 0.0009 (DFS)P = 0.0005 (OS) | — | [160]; 33: 435–441 |
|
| ||||||||
| CD3+ |
P = 0.010 (DFS) P = 0.061 (OS) |
P = 0.003 (DFS) P = 0.039 (OS) |
0.20 (0.02–2.60) P = 0.22 (DFS) |
|||||
| 87 | — | — | II | CD25+ |
P = 0.013 (DFS) P = 0.15 (OS) |
P = 0.002 (DFS) P = 0.017 (OS) |
0.22 (0.02–2.35) P = 0.21 (DFS) |
[161]; 116:5188–5199 |
| CD45RO+ |
P = 0.049 (DFS) P = 0.16 (OS) |
P = 0.014 (DFS) P = 0.037 (OS) |
0.24 (0.02–1.10) P = 0.014 (DFS) |
|||||
| FOXP3+ |
P = 0.009 (DFS) P = 0.027 (OS) |
P = 0.005 (DFS) P = 0.040 (OS) |
0.14 (0.07–0.85) P = 0.027 (DFS) |
|||||
|
| ||||||||
| CD3+ | 79% (versus 75%) Q4 (versus Q1)† P = 0.19 (CS) |
0.73 (0.49–1.08) Q4 (versus Q1)†
P = 0.070‡ (CS) |
1.30 (0.81–2.07) Q4 (versus Q1)†
P = 0.16‡ (CS) |
|||||
| 768 | 366 | 229 | I-IV | CD8+ | 78% (versus 66%) Q4 (versus Q1)† P = 0.026 (CS) |
0.61 (0.42–0.88) Q4 (versus Q1)†
P = 0.007‡ (CS) |
0.81 (0.52–1.27) Q4 (versus Q1)†
P = 0.34‡ (CS) |
[162]; 222: 350–366 |
| CD45RO+ | 83% (versus 68%) Q4 (versus Q1)† P< 0.0001 (CS) |
0.40 (0.26–0.60) Q4 (versus Q1)†
P< 0.0001‡ (CS) |
0.51 (0.32–0.80) Q4 (versus Q1)†
P = 0.034‡ (CS) |
|||||
| FOXP3+ | 80% (versus 64%) Q4 (versus Q1)† P< 0.0001 (CS) |
0.48 (0.32–0.70) Q4 (versus Q1)†
P< 0.0001‡ (CS) |
0.89 (0.59–1.34) Q4 (versus Q1)†
P = 0.034‡ (CS) |
|||||
*HR is based on comparing high versus low score of a given T-cell subset. †Quartile of density (Q1-4, first to fourth quartile). ‡ P for trend. Cl: confidence interval; CS: colorectal cancer-specific survival; DFS: disease-free survival; HR: hazard ratio; MSI: microsatellite instability; NS: not significant; OS: overall survival.
From the above, the tumour-infiltrating T cells may be at the same time, an indicator of the host immune response versus cancer cells and an attractive target for immunotherapy [18, 89, 90].
The TILs may also reflect specific molecular alterations associated with indolent tumour behaviour. Previous studies have shown that lymphocytic infiltration is associated with microsatellite instability (MSI) in colorectal cancer [91–93]. Truncated peptides produced by frameshift mutations due to MSI may be immunogenic and contribute to the host immune response [88, 89, 94]. However, at the time, very little is known about the interrelationship between TILs, MSI, and other tumour molecular features, such as the CpG island methylator phenotype (CIMP), global DNA hypomethylation, and KRAS, BRAF, and PIK3CA mutations.
Previous studies have reported that MSI [95], CIMP [96], BRAF mutation [97], PIK3CA mutation [98], and tumour LINE-1 hypomethylation [99] are associated with prognosis and that lymphocytic infiltration is associated with many of these molecular variables [92]. As such, to define the prognostic effect of tumour-infiltrating T cells independently of those potential confounders, large studies of colorectal cancers with extensive molecular characterization are needed. Most recently, Nosho and coll. [100], using a database of 768 colorectal cancers, analyzed the subsets of TILs in relation with molecular changes in patients with CRC. They demonstrated that the densities of CD8+, CD45RO+, and FOXP3+ cells were significantly associated with patient survival in univariate analyses (P trend < 0.007). In the multivariate model, tumour-infiltrating CD45RO+-cell density, but not CD3+, CD8+, or FOXP3+-cell density, was significantly associated with survival (P = 0.0032). In multivariate linear regression analysis, MSI-high (P < 0.0001) and high-level tumour LINE-1 methylation (P = 0.0013) were independently associated with higher CD45RO+-cell density. The survival benefit associated with CD45RO+ cells was independent of MSI and LINE-1 status. In conclusion, tumour-infiltrating CD45RO+-cell density is a prognostic biomarker associated with longer survival of colorectal cancer patients, independent of clinical, pathological, and molecular features. In addition, MSI-high and tumour LINE-1 methylation level are independent predictors of CD45RO+-cell density. These results offer a possible mechanism by which MSI confers an improved clinical outcome and support efforts to augment the host immune response in the cancer microenvironment as a strategy of targeted immunotherapy.
As with all tumors analyzed so far, even for the CRC it is very important to evaluate the impact of Tregs on the specific immune responses against tumor-associated antigens (TAAs). The grade of local infiltration did not correlate with responses against well-defined TAAs as EpCAM, Her-2/neu, and CEA [101]. Depleting Tregs in PBMCs from CRC patients dramatically boosted the IFN-γ and TNF-α production in T cells, which were stimulated with a CEA peptide [102]. In spite of the unmasking of responses in opposition to other TAAs, recall antigens such as PPD were not affected suggesting a TAA-specific rather than a systemic immune suppression [103].
In an extremely ample analysis various TAA-specific Tregs were exclusively identified in CRC patients. Peptides for CEA, telomerase, Her-2/neu, and MUC-1 all led to an activation of Tregs [104]. TAA-specific Tregs were successfully identified using a p53 peptide [105]; in addition to CD4+ Tregs also CD8+CD28− Tregs could be isolated from peripheral blood, tumor tissue, and metastatic lymph nodes of CRC patients [106]. These cells suppressed T cells in an IL-10-dependent fashion and were mainly CD194+, which may have contributed to their accumulation via recruitment. A recent study identified circulating and tumor-infiltrating CD28+CD8+ Tregs with a CD25+, FOXP3+, CD152+, GITR+, CD194+, TGF-β +, and CD127− phenotype [107]. Remarkably this type of Tregs was found in 90% of the CRC specimens but was totally absent in normal colonic tissue suggesting a cancer-specific presence without contribution to the physiologic epithelial homeostasis [108]. Ligands for CD194 (e.g., CCL17 or CCL22) were in contrast to IL-6 and TGF-β not highly expressed in the tumor tissue, altogether indicating a conversion from CD8+ rather than a tumor-directed migration as the cause for the observed infiltration. In another recent study CXCL11 produced by CRC-derived CD68+ myeloid cells is suggested to be a promising chemoattractant for Tregs [109].
4.2. T-Cell-Based Immunotherapy in CRC Patients
T-cell-based immunotherapy (TCI) was first described in 1988 [110], but the decisive improvement in efficacy came in 2002 with the introduction of an immunodepleting preparative regimen given before the adoptive transfer, which could result in the clonal repopulation of patients with antitumour T cells [111]. Of patients with metastatic melanoma refractory to all other treatments, 50% will experience an objective response, some with complete responses [112]. Responses can be durable and are seen in all organ sites, including the brain. Recent studies demonstrating that normal human T cell can be genetically engineered to recognize cancer antigens and mediate cancer regression in vivo have opened opportunities for enhancing and extending the TCI approach to patients with a wide variety of cancer types [113]. These studies provide a valuable guide to the immunological principles that form the basis of effective immunotherapy for CRC patients.
Most nonhematopoietic tumors such as CRC express MHC class I molecules, but do not express MHC class II molecules, therefore it is believed that the predominant tumor-specific cell-mediated immune effector mechanism is the killing by CTL. However, the clinical history of the patient with cancer often demonstrates the failure of the immune system to eliminate the tumor [114]. It is now generally accepted that this is mostly due to poor tumor-specific MHC class II-restricted CD4+ T helper generated in tumor-bearing hosts [115–117] and that Th cells are required for priming and clonal expansion of specific CTL following reencounter with antigen [118–121].
Although at clinical level, TCI results are still preliminary [122], nevertheless the importance of including CD4+ together with CD8+ T cells to induce optimal therapeutic effects has been established [112, 123].
For this purpose and to optimize the antitumor immunological arms in terms of specificity and long-lasting memory, vaccination with tumor cells transduced with the AIR-1-encoded CIITA, the MHC class II [MHC-II] gene transactivator [124, 125], has been explored with the idea that CIITA-transfected cells may act as “surrogate APC” for optimal triggering of tumor-specific Th cells and thus facilitate the recognition of TAA presented by tumor cell MHC-II molecules. Indeed, the group of Accola showed that complete rejection and long-lasting antitumor memory could be obtained after vaccination with CIITA-expressing TS/A mammary adenocarcinoma [126–128]; Most recently, the same group [129] demonstrated that CIITA-expressing C51 colon adenocarcinoma cells are rejected in high percentage of mice or strongly reduced in growth. Induction of antitumor immunity depended on the ability of the MHC-II-positive tumor cells to trigger CD4+ T cells, which in turn induce stimulation and maturation of CTL effectors. Importantly, they showed that immune CD4+ Th cells can induce protective antitumor responses in naive mice injected with parental nontransfected tumor cells. Purified CD4+ T cells from C51-CIITA vaccinated and challenged mice were also efficacious in preventing tumor growth of C51 tumor, as 50% of the animals were protected and the remaining 50% displayed a significant growth retardation. Similar results were obtained when immune CD8+ T cells were used in adoptive transfer, even if CD4+ T cells were clearly superior to CD8+ T cells in antitumor protective function. Interestingly, the protective phenotype was associated to both a Th1 and Th2 polarization of the immune effectors.
In conclusion, these results demonstrated that tumor cell modification by CIITA may offer an alternative strategy not only for preventive vaccination but also for the generation of more efficacious TCI for CRC patients.
In recent years it has also become increasingly the cancer stem cell theory [130], the idea that cancers are composed of several types of cells, and that only a small population of cancer cells that can regenerate cancer tissues, much as normal tissue can be regenerated only by a small population of stem-like cells. Recently, cancer stem-like cells and tumor-initiating cells (CSCs/TICs) have been isolated from various types of malignancies, including colon cancer [131–135].
In colon cancer, CSCs/TICs can reinitiate tumors that resemble mother colon cancer tissues morphologically when transplanted into immunodeficient mice [132]. Furthermore, these CSCs/TICs have higher tumorigenic potential than do non-CSCs/TICs, suggesting that they are essential for tumor maintenance and distant metastasis [132].
Previous reports have shown that CSCs/TICs are resistant to a variety of treatments, including chemotherapy and radiotherapy, with varied mechanisms of resistance, including high expression of drug transporters, relative cell cycle quiescence, high levels of DNA repair machinery, and resistance to apoptosis [136].
In recent times Inoda and coll. shown that CTL specific for the tumor-associated antigen CEP55 can efficiently recognize colon CSCs/TICs both in vitro and in vivo. The authors isolated CSCs/TICs as side population (SP) cells from colon cancer cell lines SW480, HT29, and HCT15. The SP cells expressed high levels of the stem cell markers SOX2, POU5F1, LGR5, and ALDH1A1 and shown resistance to chemotherapeutic agents (irinotecan or etoposide). To evaluate the susceptibility of SP cells to CTLs, they used CTL clone 41, which is specific for the CEP55-derived antigenic peptide Cep55/c10orf3_193 [137, 138]. The SP cells expressed HLA class I and CEP55 at the same level as the main population cells. The SP cells were susceptible to CTL clone 41 at the same level as main population cells. Furthermore, adoptive transfer of CTL clone 41 inhibited tumor growth of SW480 SP cells in vivo.
These results suggest that Cep55/c10orf3_193 [137, 138] peptide-based cancer vaccine therapy or adoptive cell transfer of the CTL clone is a possible approach for targeting chemotherapy-resistant colon CSCs/TICs.
5. Conclusion
Despite advances in clinical diagnostics, surgical techniques, and development of new chemo/radiotherapy regimens the prognosis of gastrointestinal oncology remains poor, and the need for new treatment options, such as immunotherapy, is imperative.
Studies of T-cell-based immunotherapy have clearly demonstrated that the administration of highly avid anti-tumour T cells directed against a defined target can mediate the regression of large, vascularized, metastatic cancers and provide guiding principles as well as encouragement for the further development of adoptive T-cell therapy for cancer patients.
In this paper we have reported the evidence of the key role of T-cell response versus cancer of the digestive system and the results obtained in different clinical trials using T-cell immunotherapy.
We showed that for pancreatic cancer as well as for both gastric and colorectal cancer good results were obtained in some clinical settings but in order that T-cell-based immunotherapy become a real treatment for gastrointestinal oncology, several problems must be solved.
A major problem with the application of TCI is that it is a highly personalized treatment and does not easily fit into current modes of oncological practice. The treatment is expensive, labour-intensive, and requires high laboratory expertise. In essence, a new reagent needs to be created for each patient, and this patient-specific nature of the treatment makes it difficult to commercialize.
Moreover, currently the major challenge in the field is to conduct randomized clinical trials demonstrating sufficient clinical benefit to justify the logistics and costs of customized cellular therapies. In many clinical trials, patients are enrolled at an advanced cancer stage, and this aspect could determine an unfavourable outcome; thus, it would be very interesting to plan clinical trials in early-stage of cancer because it would be possible that gastrointestinal cancer immunotherapeutic approaches confer a survival advantage when applied earlier during the course of the disease, such as in the adjuvant setting.
However, the big hurdle to make immunotherapy approach successful for gastrointestinal oncology remains the immune evasion strategies set up by the tumor resulting in avoidance of both innate and adaptive immunity.
Investigations during the past few years have provided new insights into the cellular and molecular mechanisms involved in the bidirectional crosstalk between cancer cells and the immune cells. Understanding this functional dialogue and the hierarchical status of different tumor-immune escape stratagems at different stages of tumor development will guide the design of novel therapeutic strategies aiming to demolish the “tumor fortress”.
Thus, it will be of particular interest to study the kinetics of the interactions between different inhibitory molecules and endogenous factors that influence the expansion and trafficking of Tregs and tolerogenic DCs within tumor-draining lymph nodes and the tumor surroundings.
On the basis of clinical and experimental evidence, it is reasonable to conclude that successful therapy for gastrointestinal oncology must involve a combination approach, which should involve systemic chemotherapy and transplantation to reduce the burden or to eliminate immune suppressive cells, together with tailor-made immunotherapies customized to each single patient.
References
- 1.Garcia M, Jemal A, Ward EM, et al. Global Cancer Facts & Figures 2007. American Cancer Society, http://www.cancer.org.
- 2.Raimondi S, Maisonneuve P, Lowenfels AB. Epidemiology of pancreatic cancer: an overview. Nature Reviews Gastroenterology and Hepatology. 2009;6(12):699–708. doi: 10.1038/nrgastro.2009.177. [DOI] [PubMed] [Google Scholar]
- 3.American Cancer Society. Cancer Facts & Figures. American Cancer Society, 2010, http://www.cancer.org/Research/CancerFactsFigures/index.
- 4.Winter JM, Cameron JL, Campbell KA, et al. 1423 pancreaticoduodenectomies for pancreatic cancer: a single-institution experience. Journal of Gastrointestinal Surgery. 2006;10(9):1199–1210. doi: 10.1016/j.gassur.2006.08.018. [DOI] [PubMed] [Google Scholar]
- 5.Saka M, Morita S, Fukagawa T, Katai H. Present and future status of gastric cancer surgery. Japanese Journal of Clinical Oncology. 2011;41(3):307–313. doi: 10.1093/jjco/hyq240. [DOI] [PubMed] [Google Scholar]
- 6.Giuliani F, de Vita F, Colucci G, Pisconti S. Maintenance therapy in colon cancer. Cancer Treatment Reviews. 2010;36(supplement 3):S42–S45. doi: 10.1016/S0305-7372(10)70019-0. [DOI] [PubMed] [Google Scholar]
- 7.Cunningham D, Allum WH, Stenning SP, et al. MAGIC Trial Participants. Perioperative chemotherapy versus surgery alone for resectable gastroesophageal cancer. The New England Journal of Medicine. 2006;355(1):11–20. doi: 10.1056/NEJMoa055531. [DOI] [PubMed] [Google Scholar]
- 8.Catenacci DVT, Kozloff M, Kindler HL, Polite B. Personalized colon cancer care in 2010. Seminars in Oncology. 2011;38(2):284–308. doi: 10.1053/j.seminoncol.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Artinyan A, Anaya DA, McKenzie S, Ellenhorn JDI, Kim J. Neoadjuvant therapy is associated with improved survival in resectable pancreatic adenocarcinoma. Cancer. 2011;117(10):2044–2049. doi: 10.1002/cncr.25763. [DOI] [PubMed] [Google Scholar]
- 10.Touchefeu Y, Harrington KJ, Galmiche JP, Vassaux G. Review article: gene therapy, recent developments and future prospects in gastrointestinal oncology. Alimentary Pharmacology and Therapeutics. 2010;32(8):953–968. doi: 10.1111/j.1365-2036.2010.04424.x. [DOI] [PubMed] [Google Scholar]
- 11.Amedei A, Niccolai E, Bella CD, et al. Characterization of tumor antigen peptide-specific T cells isolated from the neoplastic tissue of patients with gastric adenocarcinoma. Cancer Immunology, Immunotherapy. 2009;58(11):1819–1830. doi: 10.1007/s00262-009-0693-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gaudernack G. Prospects for vaccine therapy for pancreatic cancer. Best Practice and Research: Clinical Gastroenterology. 2006;20(2):299–314. doi: 10.1016/j.bpg.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 13.Iovino F, Meraviglia S, Spina M, et al. Immunotherapy targeting colon cancer stem cells. Immunotherapy. 2011;3(1):97–106. doi: 10.2217/imt.10.87. [DOI] [PubMed] [Google Scholar]
- 14.Goedert JJ. The epidemiology of acquired immunodeficiency syndrome malignancies. Seminars in Oncology. 2000;27(4):390–401. [PubMed] [Google Scholar]
- 15.Challis GB, Stam HJ. The spontaneous regression of cancer. A review of cases from 1900 to 1987. Acta Oncologica. 1990;29(5):545–550. doi: 10.3109/02841869009090048. [DOI] [PubMed] [Google Scholar]
- 16.Zhang L, Conejo-Garcia JR, Katsaros D, et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. The New England Journal of Medicine. 2003;348(3):203–213. doi: 10.1056/NEJMoa020177. [DOI] [PubMed] [Google Scholar]
- 17.Nagorsen D, Scheibenbogen C, Marincola FM, Letsch A, Keilholz U. Natural T cell immunity against cancer. Clinical Cancer Research. 2003;9(12):4296–4303. [PubMed] [Google Scholar]
- 18.Erdman SE, Poutahidis T. Roles for inflammation and regulatory T cells in colon cancer. Toxicologic Pathology. 2010;38(1):76–87. doi: 10.1177/0192623309354110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yokokawa J, Palena C, Arlen P, et al. Identification of novel human CTL epitopes and their agonist epitopes of mesothelin. Clinical Cancer Research. 2005;11(17):6342–6351. doi: 10.1158/1078-0432.CCR-05-0596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Andersen MH, Pedersen LO, Becket JC, Straten PT. Identification of a cytotoxic T lymphocyte response to the apoptosis inhibitor protein survivin in cancer patients. Cancer Research. 2001;61(3):869–872. [PubMed] [Google Scholar]
- 21.Johnston FM, Tan MCB, Tan BR, Jr., et al. Circulating mesothelin protein and cellular antimesothelin immunity in patients with pancreatic cancer. Clinical Cancer Research. 2009;15(21):6511–6518. doi: 10.1158/1078-0432.CCR-09-0565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kotera Y, Fontenot JD, Pecher G, Metzgar RS, Finn OJ. Humoral immunity against a tandem repeat epitope of human mucin MUC-1 in sera from breast, pancreatic, and colon cancer patients. Cancer Research. 1994;54(11):2856–2860. [PubMed] [Google Scholar]
- 23.Kubuschok B, Neumann F, Breit R, et al. Naturally occurring T-cell response against mutated p21 ras oncoprotein in pancreatic cancer. Clinical Cancer Research. 2006;12(4):1365–1372. doi: 10.1158/1078-0432.CCR-05-1672. [DOI] [PubMed] [Google Scholar]
- 24.Wenandy L, Sørensen RB, Sengeløv L, Svane IM, Straten PT, Andersen MH. The immunogenicity of the hTERT540-548 peptide in cancer. Clinical Cancer Research. 2008;14(1):4–7. doi: 10.1158/1078-0432.CCR-07-4590. [DOI] [PubMed] [Google Scholar]
- 25.Yanagimoto H, Mine T, Yamamoto K, et al. Immunological evaluation of personalized peptide vaccination with gemcitabine for pancreatic cancer. Cancer Science. 2007;98(4):605–611. doi: 10.1111/j.1349-7006.2007.00429.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kubuschok B, Xie X, Jesnowski R, et al. Expression of cancer testis antigens in pancreatic carcinoma cell lines, pancreatic adenocarcinoma and chronic pancreatitis. International Journal of Cancer. 2004;109(4):568–575. doi: 10.1002/ijc.20006. [DOI] [PubMed] [Google Scholar]
- 27.Schmitz-Winnenthal FH, Volk C, Z’Graggen K, et al. High frequencies of functional tumor-reactive T cells in bone marrow and blood of pancreatic cancer patients. Cancer Research. 2005;65(21):10079–10087. doi: 10.1158/0008-5472.CAN-05-1098. [DOI] [PubMed] [Google Scholar]
- 28.Schmitz-Winnenthal FH, Escobedo LVG, Beckhove P, et al. Specific immune recognition of pancreatic carcinoma by patient-derived CD4 and CD8 T cells and its improvement by interferon-γ . International Journal of Oncology. 2006;28(6):1419–1428. doi: 10.3892/ijo.28.6.1419. [DOI] [PubMed] [Google Scholar]
- 29.Choi C, Witzens M, Bucur M, et al. Enrichment of functional CD8 memory T cells specific for MUC1 in bone marrow of patients with multiple myeloma. Blood. 2005;105(5):2132–2134. doi: 10.1182/blood-2004-01-0366. [DOI] [PubMed] [Google Scholar]
- 30.Feuerer M, Beckhove P, Bai L, et al. Therapy of human tumors in NOD/SCID mice with patient-derived reactivated memory T cells from bone marrow. Nature Medicine. 2001;7(4):452–458. doi: 10.1038/86523. [DOI] [PubMed] [Google Scholar]
- 31.Müller-Berghaus J, Ehlert K, Ugurel S, et al. Melanoma-reactive T cells in the bone marrow of melanoma patients: association with disease stage and disease duration. Cancer Research. 2006;66(12):5997–6001. doi: 10.1158/0008-5472.CAN-04-0484. [DOI] [PubMed] [Google Scholar]
- 32.Esposito I, Menicagli M, Funel N, et al. Inflammatory cells contribute to the generation of an angiogenic phenotype in pancreatic ductal adenocarcinoma. Journal of Clinical Pathology. 2004;57(6):630–636. doi: 10.1136/jcp.2003.014498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jesnowski R, Fürst D, Ringel J, et al. Immortalization of pancreatic stellate cells as an in vitro model of pancreatic fibrosis: deactivation is induced by matrigel and N-acetylcysteine. Laboratory Investigation. 2005;85(10):1276–1291. doi: 10.1038/labinvest.3700329. [DOI] [PubMed] [Google Scholar]
- 34.Feuerer M, Beckhove P, Garbi N, et al. Bone marrow as a priming site for T-cell responses to blood-borne antigen. Nature Medicine. 2003;9(9):1151–1157. doi: 10.1038/nm914. [DOI] [PubMed] [Google Scholar]
- 35.Fukunaga A, Miyamoto M, Cho Y, et al. CD8+ tumor-infiltrating lymphocytes together with CD4+ tumor-infiltrating lymphocytes and dendritic cells improve the prognosis of patients with pancreatic adenocarcinoma. Pancreas. 2004;28(1):e26–e31. doi: 10.1097/00006676-200401000-00023. [DOI] [PubMed] [Google Scholar]
- 36.Hiraoka N, Onozato K, Kosuge T, Hirohashi S. Prevalence of FOXP3+ regulatory T cells increases during the progression of pancreatic ductal adenocarcinoma and its premalignant lesions. Clinical Cancer Research. 2006;12(18):5423–5434. doi: 10.1158/1078-0432.CCR-06-0369. [DOI] [PubMed] [Google Scholar]
- 37.von Bernstorff W, Voss M, Freichel S, et al. Systemic and local immunosuppression in pancreatic cancer patients. Clinical Cancer Research. 2001;7(3):925s–932s. [PubMed] [Google Scholar]
- 38.Bernstorff WV, Spanjaard RA, Chan AK, et al. Pancreatic cancer cells can evade immune surveillance via nonfunctional Fas (APO-1/CD95) receptors and aberrant expression of functional Fas ligand. Surgery. 1999;125(1):73–84. doi: 10.1067/msy.2099.93570. [DOI] [PubMed] [Google Scholar]
- 39.De Monte L, Reni M, Tassi E, et al. Intratumor T helper type 2 cell infiltrate correlates with cancer-associated fibroblast thymic stromal lymphopoietin production and reduced survival in pancreatic cancer. Journal of Experimental Medicine. 2011;208(3):469–478. doi: 10.1084/jem.20101876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Clark CE, Beatty GL, Vonderheide RH. Immunosurveillance of pancreatic adenocarcinoma: insights from genetically engineered mouse models of cancer. Cancer Letters. 2009;279(1):1–7. doi: 10.1016/j.canlet.2008.09.037. [DOI] [PubMed] [Google Scholar]
- 41.Fong L, Small EJ. Anti-cytotoxic T-lymphocyte antigen-4 antibody: the first in an emerging class of immunomodulatory antibodies for cancer treatment. Journal of Clinical Oncology. 2008;26(32):5275–5283. doi: 10.1200/JCO.2008.17.8954. [DOI] [PubMed] [Google Scholar]
- 42.Paulos CM, June CH. Putting the brakes on BTLA in T cell-mediated cancer immunotherapy. Journal of Clinical Investigation. 2010;120(1):76–80. doi: 10.1172/JCI41811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gjertsen MK, Bakka A, Breivik J, et al. Vaccination with mutant ras peptides and induction of T-cell responsiveness in pancreatic carcinoma patients carrying the corresponding RAS mutation. The Lancet. 1995;346(8987):1399–1400. doi: 10.1016/s0140-6736(95)92408-6. [DOI] [PubMed] [Google Scholar]
- 44.Gjertsen MK, Bjørheim J, Saeterdal I, Myklebust J, Gaudernack G. Cytotoxic CD4+ and CD8+ T lymphocytes, generated by mutant p21-ras (12Val) peptide vaccination of a patient, recognize 12Val-dependent nested epitopes present within the vaccine peptide and kill autologous tumour cells carrying this mutation. International Journal of Cancer. 1997;72(5):784–790. doi: 10.1002/(sici)1097-0215(19970904)72:5<784::aid-ijc14>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 45.Gjertsen MK, Buanes T, Rosseland AR, et al. Intradermal ras peptide vaccination with granulocyte-macrophage colony-stimulating factor as adjuvant: clinical and immunological responses in patients with pancreatic adenocarcinoma. International Journal of Cancer. 2001;92(3):441–450. doi: 10.1002/ijc.1205. [DOI] [PubMed] [Google Scholar]
- 46.Hung CF, Tsai YC, He L, Wu TC. Control of mesothelin-expressing ovarian cancer using adoptive transfer of mesothelin peptide-specific CD8+ T cells. Gene Therapy. 2007;14(12):921–929. doi: 10.1038/sj.gt.3302913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kawaoka T, Oka M, Takashima M, et al. Adoptive immunotherapy for pancreatic cancer: cytotoxic T lymphocytes stimulated by the MUC1 -expressing human pancreatic cancer cell line YPK-1. Oncology Reports. 2008;20(1):155–163. [PubMed] [Google Scholar]
- 48.Kondo H, Hazama S, Kawaoka T, et al. Adoptive immunotherapy for pancreatic cancer using MUC1 peptide-pulsed dendritic cells and activated T lymphocytes. Anticancer Research. 2008;28(1):379–387. [PubMed] [Google Scholar]
- 49.Cappello P, Tomaino B, Chiarle R, et al. An integrated humoral and cellular response is elicited in pancreatic cancer by α-enolase, a novel pancreatic ductal adenocarcinoma-associated antigen. International Journal of Cancer. 2009;125(3):639–648. doi: 10.1002/ijc.24355. [DOI] [PubMed] [Google Scholar]
- 50.Sato N, Nabeta Y, Kondo H, et al. Human CD8 and CD4 T cell epitopes of epithelial cancer antigens. Cancer Chemotherapy and Pharmacology. 2000;46:S86–S90. doi: 10.1007/pl00014057. [DOI] [PubMed] [Google Scholar]
- 51.Kondo H, Sahara H, Miyazaki A, et al. Natural antigenic peptides from squamous cell carcinoma recognized by autologous HLA-DR8-restricted CD4+ T cells. Japanese Journal of Cancer Research. 2002;93(8):917–924. doi: 10.1111/j.1349-7006.2002.tb01338.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shih NY, Lai HL, Chang GC, et al. Anti-α-enolase autoantibodies are down-regulated in advanced cancer patients. Japanese Journal of Clinical Oncology. 2010;40(7):663–669. doi: 10.1093/jjco/hyq028. Article ID hyq028. [DOI] [PubMed] [Google Scholar]
- 53.Tu SH, Chang CC, Chen CS, et al. Increased expression of enolase α in human breast cancer confers tamoxifen resistance in human breast cancer cells. Breast Cancer Research and Treatment. 2010;121(3):539–553. doi: 10.1007/s10549-009-0492-0. [DOI] [PubMed] [Google Scholar]
- 54.Chang GC, Liu KJ, Hsieh CL, et al. Identification of α-enolase as an autoantigen in lung cancer: its overexpression is associated with clinical outcomes. Clinical Cancer Research. 2006;12(19):5746–5754. doi: 10.1158/1078-0432.CCR-06-0324. [DOI] [PubMed] [Google Scholar]
- 55.Schistosomes, liver flukes and Helicobacter pylori. IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Lyon, 7–14 June 1994. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans. 1994;61:1–241. [PMC free article] [PubMed] [Google Scholar]
- 56.Crusius JBA, Canzian F, Capellá G, et al. Cytokine gene polymorphisms and the risk of adenocarcinoma of the stomach in the European prospective investigation into cancer and nutrition (EPIC-EURGAST) Annals of Oncology. 2008;19(11):1894–1902. doi: 10.1093/annonc/mdn400. [DOI] [PubMed] [Google Scholar]
- 57.González CA, Sala N, Capellá G. Genetic susceptibility and gastric cancer risk. International Journal of Cancer. 2002;100(3):249–260. doi: 10.1002/ijc.10466. [DOI] [PubMed] [Google Scholar]
- 58.Hamajima N, Naito M, Kondo T, Goto Y. Genetic factors involved in the development of Helicobacter pylori-related gastric cancer. Cancer Science. 2006;97(11):1129–1138. doi: 10.1111/j.1349-7006.2006.00290.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hou L, El-Omar EM, Chen J, et al. Polymorphisms in Th1-type cell-mediated response genes and risk of gastric cancer. Carcinogenesis. 2007;28(1):118–123. doi: 10.1093/carcin/bgl130. [DOI] [PubMed] [Google Scholar]
- 60.Starzyńska T, Ferenc K, Wex T, et al. The association between the interleukin-1 polymorphisms and gastric cancer risk depends on the family history of gastric carcinoma in the study population. American Journal of Gastroenterology. 2006;101(2):248–254. doi: 10.1111/j.1572-0241.2006.00422.x. [DOI] [PubMed] [Google Scholar]
- 61.Ben-Baruch A. Inflammation-associated immune suppression in cancer: the roles played by cytokines, chemokines and additional mediators. Seminars in Cancer Biology. 2006;16(1):38–52. doi: 10.1016/j.semcancer.2005.07.006. [DOI] [PubMed] [Google Scholar]
- 62.Lin WW, Karin M. A cytokine-mediated link between innate immunity, inflammation, and cancer. Journal of Clinical Investigation. 2007;117(5):1175–1183. doi: 10.1172/JCI31537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kim R, Emi M, Tanabe K, Arihiro K. Tumor-driven evolution of immunosuppressive networks during malignant progression. Cancer Research. 2006;66(11):5527–5536. doi: 10.1158/0008-5472.CAN-05-4128. [DOI] [PubMed] [Google Scholar]
- 64.Lee HE, Chae SW, Lee YJ, et al. Prognostic implications of type and density of tumour-infiltrating lymphocytes in gastric cancer. British Journal of Cancer. 2008;99(10):1704–1711. doi: 10.1038/sj.bjc.6604738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ren Z, Pang G, Clancy R, et al. Shift of the gastric T-cell response in gastric carcinoma. Journal of Gastroenterology and Hepatology. 2001;16(2):142–148. doi: 10.1046/j.1440-1746.2001.02385.x. [DOI] [PubMed] [Google Scholar]
- 66.D’Elios MM, Amedei A, Prete GD. Helicobacter pylori antigen-specific T-cell responses at gastric level in chronic gastritis, peptic ulcer, gastric cancer and low-grade mucosa-associated lymphoid tissue (MALT) lymphoma. Microbes and Infection. 2003;5(8):723–730. doi: 10.1016/s1286-4579(03)00114-x. [DOI] [PubMed] [Google Scholar]
- 67.Maruyama T, Kono K, Mizukami Y, et al. Distribution of Th17 cells and FoxP3(+) regulatory T cells in tumor-infiltrating lymphocytes, tumor-draining lymph nodes and peripheral blood lymphocytes in patients with gastric cancer. Cancer Science. 2010;101(9):1947–1954. doi: 10.1111/j.1349-7006.2010.01624.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.June CH. Principles of adoptive T cell cancer therapy. Journal of Clinical Investigation. 2007;117(5):1204–1212. doi: 10.1172/JCI31446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yee C, Thompson JA, Byrd D, et al. Adoptive T cell therapy using antigen-specific CD8+ T cell clones for the treatment of patients with metastatic melanoma: in vivo persistence, migration, and antitumor effect of transferred T cells. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(25):16168–16173. doi: 10.1073/pnas.242600099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kim YJ, Lim J, Kang JS, et al. Adoptive immunotherapy of human gastric cancer with ex vivo expanded T cells. Archives of Pharmacal Research. 2010;33(11):1789–1795. doi: 10.1007/s12272-010-1111-7. [DOI] [PubMed] [Google Scholar]
- 71.Wu Y, Wang L, Zhang Y. Dendritic cells as vectors for immunotherapy of tumor and its application for gastric cancer therapy. Cellular & Molecular Immunology. 2004;1(5):351–356. [PubMed] [Google Scholar]
- 72.Hoshino T, Seki N, Kikuchi M, et al. HLA class-I-restricted and tumor-specific CTL in tumor-infiltrating lymphocytes of patients with gastric cancer. International Journal of Cancer. 1997;70(6):631–638. doi: 10.1002/(sici)1097-0215(19970317)70:6<631::aid-ijc1>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 73.Kono K, Rongcun Y, Charo J, et al. Identification of HER2/neu-derived peptide epitopes recognized by gastric cancer-specific cytotoxic T lymphocytes. International Journal of Cancer. 1998;78(2):202–208. doi: 10.1002/(sici)1097-0215(19981005)78:2<202::aid-ijc14>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 74.Guo DL, Dong M, Wang L, Sun LP, Yuan Y. Expression of gastric cancer-associated MG7 antigen in gastric cancer, precancerous lesions and H. pylori-associated gastric diseases. World Journal of Gastroenterology. 2002;8(6):1009–1013. doi: 10.3748/wjg.v8.i6.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wu K, Nie Y, Guo C, Chen Y, Ding J, Fan D. Molecular basis of therapeutic approaches to gastric cancer. Journal of Gastroenterology and Hepatology. 2009;24(1):37–41. doi: 10.1111/j.1440-1746.2008.05753.x. [DOI] [PubMed] [Google Scholar]
- 76.Nie Y, Wu K, Yang J, et al. Induction of T lymphocytes specific to human gastric cancer using HLA-A matched allogeneic gastric tumor cells. Journal of Immunotherapy. 2003;26(5):403–411. doi: 10.1097/00002371-200309000-00003. [DOI] [PubMed] [Google Scholar]
- 77.Jiang J, Xu N, Wu C, et al. Treatment of advanced gastric cancer by chemotherapy combined with autologous cytokine-induced killer cells. Anticancer Research. 2006;26(3):2237–2242. [PubMed] [Google Scholar]
- 78.Wu C, Jiang J, Shi L, Xu N. Prospective study of chemotherapy in combination with cytokine-induced killer cells in patients suffering from advanced non-small cell lung cancer. Anticancer Research. 2008;28(6):3997–4002. [PubMed] [Google Scholar]
- 79.Sun S, Li XM, Li XD, Yang WS. Studies on inducing apoptosis effects and mechanism of CIK cells for MGC-803 gastric cancer cell lines. Cancer Biotherapy and Radiopharmaceuticals. 2005;20(2):173–180. doi: 10.1089/cbr.2005.20.173. [DOI] [PubMed] [Google Scholar]
- 80.Alexander RB, Rosenberg SA. Long term survival of adoptively transferred tumor-infiltrating lymphocytes in mice. Journal of Immunology. 1990;145(5):1615–1620. [PubMed] [Google Scholar]
- 81.Kono K, Takahashi A, Ichihara F, et al. Prognostic significance of adoptive immunotherapy with tumor-associated lymphocytes in patients with advanced gastric cancer: a randomized trial. Clinical Cancer Research. 2002;8(6):1767–1771. [PubMed] [Google Scholar]
- 82.Rosenberg SA, Yannelli JR, Yang JC, et al. Treatment of patients with metastatic melanoma with autologous tumor- infiltrating lymphocytes and interleukin 2. Journal of the National Cancer Institute. 1994;86(15):1159–1166. doi: 10.1093/jnci/86.15.1159. [DOI] [PubMed] [Google Scholar]
- 83.Dudley ME, Wunderlich JR, Shelton TE, Even J, Rosenberg SA. Generation of tumor-infiltrating lymphocyte cultures for use in adoptive transfer therapy for melanoma patients. Journal of Immunotherapy. 2003;26(4):332–342. doi: 10.1097/00002371-200307000-00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tomšová M, Melichar B, Sedláková I, Šteiner I. Prognostic significance of CD3+ tumor-infiltrating lymphocytes in ovarian carcinoma. Gynecologic Oncology. 2008;108(2):415–420. doi: 10.1016/j.ygyno.2007.10.016. [DOI] [PubMed] [Google Scholar]
- 85.Galon J, Costes A, Sanchez-Cabo F, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;313(5795):1960–1964. doi: 10.1126/science.1129139. [DOI] [PubMed] [Google Scholar]
- 86.Pagès F, Berger A, Camus M, et al. Effector memory T cells, early metastasis, and survival in colorectal cancer. The New England Journal of Medicine. 2005;353(25):2654–2666. doi: 10.1056/NEJMoa051424. [DOI] [PubMed] [Google Scholar]
- 87.Salama P, Phillips M, Grieu F, et al. Tumor-infiltrating FOXP3+ T regulatory cells show strong prognostic significance in colorectal cancer. Journal of Clinical Oncology. 2009;27(2):186–192. doi: 10.1200/JCO.2008.18.7229. [DOI] [PubMed] [Google Scholar]
- 88.Schwitalle Y, Kloor M, Eiermann S, et al. Immune response against frameshift-induced neopeptides in HNPCC patients and healthy HNPCC mutation carriers. Gastroenterology. 2008;134(4):988–997. doi: 10.1053/j.gastro.2008.01.015. [DOI] [PubMed] [Google Scholar]
- 89.Tougeron D, Fauquembergue E, Rouquette A, et al. Tumor-infiltrating lymphocytes in colorectal cancers with microsatellite instability are correlated with the number and spectrum of frameshift mutations. Modern Pathology. 2009;22(9):1186–1195. doi: 10.1038/modpathol.2009.80. [DOI] [PubMed] [Google Scholar]
- 90.Disis ML, Bernhard H, Jaffee EM. Use of tumour-responsive T cells as cancer treatment. The Lancet. 2009;373(9664):673–683. doi: 10.1016/S0140-6736(09)60404-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Shia J, Ellis NA, Paty PB, et al. Value of histopathology in predicting microsatellite instability in hereditary nonpolyposis colorectal cancer and sporadic colorectal cancer. American Journal of Surgical Pathology. 2003;27(11):1407–1417. doi: 10.1097/00000478-200311000-00002. [DOI] [PubMed] [Google Scholar]
- 92.Jenkins MA, Hayashi S, O’Shea AM, et al. Pathology features in bethesda guidelines predict colorectal cancer microsatellite instability: a population-based study. Gastroenterology. 2007;133(1):48–56. doi: 10.1053/j.gastro.2007.04.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ogino S, Nosho K, Irahara N, et al. Lymphocytic reaction to colorectal cancer is associated with longer survival, independent of lymph node count, microsatellite instability, and CpG island methylator phenotype. Clinical Cancer Research. 2009;15(20):6412–6420. doi: 10.1158/1078-0432.CCR-09-1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Speetjens FM, Lauwen MM, Franken KL, et al. Prediction of the immunogenic potential of frameshift-mutated antigens in microsatellite instable cancer. International Journal of Cancer. 2008;123(4):838–845. doi: 10.1002/ijc.23570. [DOI] [PubMed] [Google Scholar]
- 95.Popat S, Hubner R, Houlston RS. Systematic review of microsatellite instability and colorectal cancer prognosis. Journal of Clinical Oncology. 2005;23(3):609–618. doi: 10.1200/JCO.2005.01.086. [DOI] [PubMed] [Google Scholar]
- 96.Ogino S, Nosho K, Kirkner GJ, et al. CpG island methylator phenotype, microsatellite instability, BRAF mutation and clinical outcome in colon cancer. Gut. 2009;58(1):90–96. doi: 10.1136/gut.2008.155473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Samowitz WS, Sweeney C, Herrick J, et al. Poor survival associated with the BRAF V600E mutation in microsatellite-stable colon cancers. Cancer Research. 2005;65(14):6063–6069. doi: 10.1158/0008-5472.CAN-05-0404. [DOI] [PubMed] [Google Scholar]
- 98.Ogino S, Nosho K, Kirkner GJ, et al. PIK3CA mutation is associated with poor prognosis among patients with curatively resected colon cancer. Journal of Clinical Oncology. 2009;27(9):1477–1484. doi: 10.1200/JCO.2008.18.6544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ogino S, Nosho K, Kirkner GJ, et al. A cohort study of tumoral LINE-1 hypomethylation and prognosis in colon cancer. Journal of the National Cancer Institute. 2008;100(23):1734–1738. doi: 10.1093/jnci/djn359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Nosho K, Baba Y, Tanaka N, et al. Tumour-infiltrating T-cell subsets, molecular changes in colorectal cancer, and prognosis: cohort study and literature review. Journal of Pathology. 2010;222(4):350–366. doi: 10.1002/path.2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Nagorsen D, Voigt S, Berg E, Stein H, Thiel E, Loddenkemper C. Tumor-infiltrating macrophages and dendritic cells in human colorectal cancer: relation to local regulatory T cells, systemic T-cell response against tumor-associated antigens and survival. Journal of Translational Medicine. 2007;5:p. 62. doi: 10.1186/1479-5876-5-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Yaqub S, Henjum K, Mahic M, et al. Regulatory T cells in colorectal cancer patients suppress anti-tumor immune activity in a COX-2 dependent manner. Cancer Immunology, Immunotherapy. 2008;57(6):813–821. doi: 10.1007/s00262-007-0417-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Clarke SL, Betts GJ, Plant A, et al. CD4+CD25+FOXP3+ regulatory T cells suppress anti-tumor immune responses in patients with colorectal cancer. PLoS ONE. 2006;1(1):p. e129. doi: 10.1371/journal.pone.0000129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Bonertz A, Weitz J, Pietsch DHK, et al. Antigen-specific Tregs control T cell responses against a limited repertoire of tumor antigens in patients with colorectal carcinoma. Journal of Clinical Investigation. 2009;119(11):3311–3321. doi: 10.1172/JCI39608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Bueter M, Gasser M, Schramm N, et al. T-cell response to p53 tumor-associated antigen in patients with colorectal carcinoma. International Journal of Oncology. 2006;28(2):431–438. [PubMed] [Google Scholar]
- 106.Filaci G, Fenoglio D, Fravega M, et al. CD8+CD28- T regulatory lymphocytes inhibiting T cell proliferative and cytotoxic functions infiltrate human cancers. Journal of Immunology. 2007;179(7):4323–4334. doi: 10.4049/jimmunol.179.7.4323. [DOI] [PubMed] [Google Scholar]
- 107.Chaput N, Louafi S, Bardier A, et al. Identification of CD8+CD25+Foxp3+ suppressive T cells in colorectal cancer tissue. Gut. 2009;58(4):520–529. doi: 10.1136/gut.2008.158824. [DOI] [PubMed] [Google Scholar]
- 108.Makita S, Kanai T, Oshima S, et al. CD4+CD25bright T cells in human intestinal lamina propria as regulatory cells. Journal of Immunology. 2004;173(5):3119–3130. doi: 10.4049/jimmunol.173.5.3119. [DOI] [PubMed] [Google Scholar]
- 109.Yang S, Wang B, Guan C, et al. Foxp3+IL-17+ T cells promote development of cancer-initiating cells in colorectal cancer. Journal of Leukocyte Biology. 2010;89(1):85–91. doi: 10.1189/jlb.0910506. [DOI] [PubMed] [Google Scholar]
- 110.Rosenberg SA, Packard BS, Aebersold PM, et al. Use of tumor-infiltrating lymphocyts and interleukin-2 in the immunotherapy of patients with metastatic melanoma. A preliminary report. The New England Journal of Medicine. 1988;319(25):1676–1680. doi: 10.1056/NEJM198812223192527. [DOI] [PubMed] [Google Scholar]
- 111.Dudley ME, Wunderlich JR, Robbins PF, et al. Cancer regression and autoimmunity in patients following clonal repopulation with anti-tumor lymphocytes. Science. 2002;298(5594):850–854. doi: 10.1126/science.1076514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Dudley ME, Wunderlich JR, Yang JC, et al. Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. Journal of Clinical Oncology. 2005;23(10):2346–2357. doi: 10.1200/JCO.2005.00.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Morgan RA, Dudley ME, Wunderlich JR, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314(5796):126–129. doi: 10.1126/science.1129003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nature Medicine. 2004;10(9):909–915. doi: 10.1038/nm1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ossendorp F, Mengedé E, Camps M, Filius R, Melief CJM. Specific T helper cell requirement for optimal induction of cytotoxic T lymphocytes against major histocompatibility complex class II negative tumors. Journal of Experimental Medicine. 1998;187(5):693–702. doi: 10.1084/jem.187.5.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Hung K, Hayashi R, Lafond-Walker A, Lowenstein C, Pardoll D, Levitsky H. The central role of CD4+ T cells in the antitumor immune response. Journal of Experimental Medicine. 1998;188(12):2357–2368. doi: 10.1084/jem.188.12.2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Muranski P, Restifo NP. Adoptive immunotherapy of cancer using CD4+ T cells. Current Opinion in Immunology. 2009;21(2):200–208. doi: 10.1016/j.coi.2009.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ridge JP, di Rosa F, Matzinger P. A conditioned dendritic cell can be a temporal bridge between a CD4 + T-helper and a T-killer cell. Nature. 1998;393(6684):474–478. doi: 10.1038/30989. [DOI] [PubMed] [Google Scholar]
- 119.Janssen EM, Droin NM, Lemmens EE, et al. CD4+ T-cell help controls CD8+ T-cell memory via TRAIL-mediated activation-induced cell death. Nature. 2005;434(7029):88–93. doi: 10.1038/nature03337. [DOI] [PubMed] [Google Scholar]
- 120.Bennett SRM, Carbone FR, Karamalis F, Flavell RA, Miller JFAP, Heath WR. Help for cytotoxic-T-cell responses is mediated by CD4O signalling. Nature. 1998;393(6684):478–480. doi: 10.1038/30996. [DOI] [PubMed] [Google Scholar]
- 121.Schoenberger SP, Toes REM, van der Voort EIH, Offringa R, Melief CJM. T-cell help for cytotoxic T lymphocytes is mediated by CD40-CD4OL interactions. Nature. 1998;393(6684):480–483. doi: 10.1038/31002. [DOI] [PubMed] [Google Scholar]
- 122.Rosenberg SA, Restifo NP, Yang JC, Morgan RA, Dudley ME. Adoptive cell transfer: a clinical path to effective cancer immunotherapy. Nature Reviews Cancer. 2008;8(4):299–308. doi: 10.1038/nrc2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Antony PA, Piccirillo CA, Akpinarli A, et al. CD8+ T cell immunity against a tumor/self-antigen is augmented by CD4+ T helper cells and hindered by naturally occurring T regulatory cells. Journal of Immunology. 2005;174(5):2591–2601. doi: 10.4049/jimmunol.174.5.2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Accolla RS, Jotterand-Bellomo M, Scarpellino L. aIr-1, a newly found locus on mouse chromosome 16 encoding a trans-acting activator factor for MHC class II gene expression. Journal of Experimental Medicine. 1986;164(1):369–374. doi: 10.1084/jem.164.1.369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Steimle V, Otten LA, Zufferey M, Mach B. Complementation cloning of an MHC class II transactivator mutated in hereditary MHC class II deficiency (or bare lymphocyte syndrome) Cell. 1993;75(1):135–146. [PubMed] [Google Scholar]
- 126.Meazza R, Comes A, Orengo AM, Ferrini S, Accolla RS. Tumor rejection by gene transfer of the MHC class II transactivator in murine mammary adenocarcinoma cells. European Journal of Immunology. 2003;33(5):1183–1192. doi: 10.1002/eji.200323712. [DOI] [PubMed] [Google Scholar]
- 127.Mortara L, Castellani P, Meazza R, et al. CIITA-induced MHC class II expression in mammary adenocarcinoma leads to a Th1 polarization of the tumor microenvironment, tumor rejection, and specific antitumor memory. Clinical Cancer Research. 2006;12(11):3435–3443. doi: 10.1158/1078-0432.CCR-06-0165. [DOI] [PubMed] [Google Scholar]
- 128.Mortara L, Frangione V, Castellani P, Barbaro ADL, Accolla RS. Irradiated CIITA-positive mammary adenocarcinoma cells act as a potent anti-tumor-preventive vaccine by inducing tumor-specific CD4+ T cell priming and CD8+ T cell effector functions. International Immunology. 2009;21(6):655–665. doi: 10.1093/intimm/dxp034. [DOI] [PubMed] [Google Scholar]
- 129.Frangione V, Mortara L, Castellani P, Barbaro ADL, Accolla RS. CIITA-driven MHC-II positive tumor cells: preventive vaccines and superior generators of antitumor CD4+ T lymphocytes for immunotherapy. International Journal of Cancer. 2010;127(7):1614–1624. doi: 10.1002/ijc.25183. [DOI] [PubMed] [Google Scholar]
- 130.Papailiou J, Bramis KJ, Gazouli M, Theodoropoulos G. Stem cells in colon cancer. A new era in cancer theory begins. International Journal of Colorectal Disease. 2011;26(1):1–11. doi: 10.1007/s00384-010-1022-6. [DOI] [PubMed] [Google Scholar]
- 131.Clarke MF, Dick JE, Dirks PB, et al. Cancer stem cells—perspectives on current status and future directions: AACR workshop on cancer stem cells. Cancer Research. 2006;66(19):9339–9344. doi: 10.1158/0008-5472.CAN-06-3126. [DOI] [PubMed] [Google Scholar]
- 132.Dalerba P, Dylla SJ, Park IK, et al. Phenotypic characterization of human colorectal cancer stem cells. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(24):10158–10163. doi: 10.1073/pnas.0703478104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.O’Brien CA, Pollett A, Gallinger S, Dick JE. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature. 2007;445(7123):106–110. doi: 10.1038/nature05372. [DOI] [PubMed] [Google Scholar]
- 134.Ricci-Vitiani L, Lombardi DG, Pilozzi E, et al. Identification and expansion of human colon-cancer-initiating cells. Nature. 2007;445(7123):111–115. doi: 10.1038/nature05384. [DOI] [PubMed] [Google Scholar]
- 135.Huang EH, Hynes MJ, Zhang T, et al. Aldehyde dehydrogenase 1 is a marker for normal and malignant human colonic stem cells (SC) and tracks SC overpopulation during colon tumorigenesis. Cancer Research. 2009;69(8):3382–3389. doi: 10.1158/0008-5472.CAN-08-4418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Dean M, Fojo T, Bates S. Tumour stem cells and drug resistance. Nature Reviews Cancer. 2005;5(4):275–284. doi: 10.1038/nrc1590. [DOI] [PubMed] [Google Scholar]
- 137.Inoda S, Hirohashi Y, Torigoe T, et al. Cep55/c10orf3, a tumor antigen derived from a centrosome residing protein in breast carcinoma. Journal of Immunotherapy. 2009;32(5):474–485. doi: 10.1097/CJI.0b013e3181a1d109. [DOI] [PubMed] [Google Scholar]
- 138.Inoda S, Morita R, Hirohashi Y, et al. The feasibility of Cep55/c10orf3 derived peptide vaccine therapy for colorectal carcinoma. Experimental and Molecular Pathology. 2011;90(1):55–60. doi: 10.1016/j.yexmp.2010.10.001. [DOI] [PubMed] [Google Scholar]
- 139.Naito Y, Saito K, Shiiba K, et al. CD8+ T cells infiltrated within cancer cell nests as a prognostic factor in human colorectal cancer. Cancer Research. 1998;58(16):3491–3494. [PubMed] [Google Scholar]
- 140.Guidoboni M, Gafà R, Viel A, et al. Microsatellite instability and high content of activated cytotoxic lymphocytes identify colon cancer patients with a favorable prognosis. American Journal of Pathology. 2001;159(1):297–304. doi: 10.1016/S0002-9440(10)61695-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Öberg Å, Samii S, Stenling R, Lindmark G. Different occurrence of CD8+, CD45R0+, and CD68+ immune cells in regional lymph node metastases from colorectal cancer as potential prognostic predictors. International Journal of Colorectal Disease. 2002;17(1):25–29. doi: 10.1007/s003840100337. [DOI] [PubMed] [Google Scholar]
- 142.Petty JK, He K, Corless CL, Vetto JT, Weinberg AD. Survival in human colorectal cancer correlates with expression of the T-cell costimulatory molecule OX-40 (CD134) American Journal of Surgery. 2002;183(5):512–518. doi: 10.1016/s0002-9610(02)00831-0. [DOI] [PubMed] [Google Scholar]
- 143.Diederichsen ACP, Hjelmborg JVB, Christensen PB, Zeuthen J, Fenge C. Prognostic value of the CD4+/CD8+ ratio of tumour infiltrating lymphocytes in colorectal cancer and HLA-DR expression on tumour cells. Cancer Immunology, Immunotherapy. 2003;52(7):423–428. doi: 10.1007/s00262-003-0388-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Funada Y, Noguchi T, Kikuchi R, Takeno S, Uchida Y, Gabbert HE. Prognostic significance of CD8+ T cell and macrophage peritumoral infiltration in colorectal cancer. Oncology Reports. 2003;10(2):309–313. [PubMed] [Google Scholar]
- 145.Prall F, Dührkop T, Weirich V, et al. Prognostic role of CD8+ tumor-infiltrating lymphocytes in stage III colorectal cancer with and without microsatellite instability. Human Pathology. 2004;35(7):808–816. doi: 10.1016/j.humpath.2004.01.022. [DOI] [PubMed] [Google Scholar]
- 146.Menon AG, Janssen-Van Rhijn CM, Morreau H, et al. Immune system and prognosis in colorectal cancer: a detailed immunohistochemical analysis. Laboratory Investigation. 2004;84(4):493–501. doi: 10.1038/labinvest.3700055. [DOI] [PubMed] [Google Scholar]
- 147.Chiba T, Ohtani H, Mizoi T, et al. Intraepithelial CD8+ T-cell-count becomes a prognostic factor after a longer follow-up period in human colorectal carcinoma: possible association with suppression of micrometastasis. British Journal of Cancer. 2004;91(9):1711–1717. doi: 10.1038/sj.bjc.6602201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Pagès F, Berger A, Camus M, et al. Effector memory T cells, early metastasis, and survival in colorectal cancer. The New England Journal of Medicine. 2005;353(25):2654–2666. doi: 10.1056/NEJMoa051424. [DOI] [PubMed] [Google Scholar]
- 149.Baeten CIM, Castermans K, Hillen HFP, Griffioen AW. Proliferating endothelial cells and leukocyte infiltration as prognostic markers in colorectal cancer. Clinical Gastroenterology and Hepatology. 2006;4(11):1351–1357. doi: 10.1016/j.cgh.2006.08.005. [DOI] [PubMed] [Google Scholar]
- 150.Galon J, Costes A, Sanchez-Cabo F, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;313(5795):1960–1964. doi: 10.1126/science.1129139. [DOI] [PubMed] [Google Scholar]
- 151.Zlobec I, Minoo P, Baumhoer D, et al. Multimarker phenotype predicts adverse survival in patients with lymph node-negative colorectal cancer. Cancer. 2008;112(3):495–502. doi: 10.1002/cncr.23208. [DOI] [PubMed] [Google Scholar]
- 152.Zlobec I, Baker K, Terracciano L, et al. Two-marker protein profile predicts poor prognosis in patients with early rectal cancer. British Journal of Cancer. 2008;99(10):1712–1717. doi: 10.1038/sj.bjc.6604729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Sinicrope FA, Rego RL, Ansell SM, Knutson KL, Foster NR, Sargent DJ. Intraepithelial effector (CD3+)/regulatory (FoxP3+) T-cell ratio predicts a clinical outcome of human colon carcinoma. Gastroenterology. 2009;137(4):1270–1279. doi: 10.1053/j.gastro.2009.06.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Laghi L, Bianchi P, Miranda E, et al. CD3+ cells at the invasive margin of deeply invading (pT3-T4) colorectal cancer and risk of post-surgical metastasis: a longitudinal study. The Lancet Oncology. 2009;10(9):877–884. doi: 10.1016/S1470-2045(09)70186-X. [DOI] [PubMed] [Google Scholar]
- 155.Frey DM, Droeser RA, Viehl CT, et al. High frequency of tumor-infiltrating FOXP3+ regulatory T cells predicts improved survival in mismatch repair-proficient colorectal cancer patients. International Journal of Cancer. 2010;126(11):2635–2643. doi: 10.1002/ijc.24989. [DOI] [PubMed] [Google Scholar]
- 156.Salama P, Phillips M, Grieu F, et al. Tumor-infiltrating FOXP3+ T regulatory cells show strong prognostic significance in colorectal cancer. Journal of Clinical Oncology. 2009;27(2):186–192. doi: 10.1200/JCO.2008.18.7229. [DOI] [PubMed] [Google Scholar]
- 157.Pagès F, Kirilovsky A, Mlecnik B, et al. In situ cytotoxic and memory T cells predict outcome in patients with early-stage colorectal cancer. Journal of Clinical Oncology. 2009;27(35):5944–5951. doi: 10.1200/JCO.2008.19.6147. [DOI] [PubMed] [Google Scholar]
- 158.Deschoolmeester V, Baay M, van Marck E, et al. Tumor infiltrating lymphocytes: an intriguing player in the survival of colorectal cancer patients. BMC Immunology. 2010;11:p. 19. doi: 10.1186/1471-2172-11-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Suzuki H, Chikazawa N, Tasaka T, et al. Intratumoral CD8+ T/FOXP3+ cell ratio is a predictive marker for survival in patients with colorectal cancer. Cancer Immunology, Immunotherapy. 2010;59(5):653–661. doi: 10.1007/s00262-009-0781-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Correale P, Rotundo MS, del Vecchio MT, et al. Regulatory (FoxP3+) T-cell tumor infiltration is a favorable prognostic factor in advanced colon cancer patients undergoing chemo or chemoimmunotherapy. Journal of Immunotherapy. 2010;33(4):435–441. doi: 10.1097/CJI.0b013e3181d32f01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Lee WS, Park S, Lee WY, Yun SH, Chun HK. Clinical impact of tumor-infiltrating lymphocytes for survival in stage II colon cancer. Cancer. 2010;116(22):5188–5199. doi: 10.1002/cncr.25293. [DOI] [PubMed] [Google Scholar]
- 162.Nosho K, Baba Y, Tanaka N, et al. Tumour-infiltrating T-cell subsets, molecular changes in colorectal cancer, and prognosis: cohort study and literature review. Journal of Pathology. 2010;222(4):350–366. doi: 10.1002/path.2774. [DOI] [PMC free article] [PubMed] [Google Scholar]