

Abstract
Mycoplasma, which was used to create the first “synthetic life”, has been an important species in the emerging field, synthetic biology. However, essential genes, an important concept of synthetic biology, for both M. mycoides and M. capricolum, as well as 14 other Mycoplasma with available genomes, are still unknown. We have developed a gene essentiality prediction algorithm that incorporates information of biased gene strand distribution, homologous search and codon adaptation index. The algorithm, which achieved an accuracy of 80.8% and 78.9% in self-consistence and cross-validation tests, respectively, predicted 5880 essential genes in the 16 Mycoplasma genomes. The intersection set of essential genes in available Mycoplasma genomes consists of 153 core essential genes. The predicted essential genes (available from pDEG, tubic.tju.edu.cn/pdeg) and the proposed algorithm can be helpful for studying minimal Mycoplasma genomes as well as essential genes in other genomes.
The year 2010 saw the creation of the first artificial self-replicating bacterial cells1. In this famous work, Venter's group designed, synthesized and assembled JCVI-syn1.0, a 1.08 Mb Mycoplasma mycoides genome, which was then transplanted into a M. capricolum recipient cell. These efforts resulted in the creation of new M. mycoides cells, whose genetic materials only contain the synthetic chromosomes1. This is a technical milestone in the emerging field, synthetic biology, because conceptually, it means a synthetic life can be designed and made2.
An important concept of synthetic biology is the minimal genome, which contains all essential genes of an organism3,4. The minimal genome can serve as a chassis in which interchangeable elements are inserted to create organisms with desirable traits5,6,7. Mycoplasma has been an important species for synthetic biology, mainly because of their small genome sizes. The first genome-scale gene essentiality screen was performed in a Mycoplasma genome8. However, the essential genes for both M. mycoides and M. capricolum, as well as those for 14 other Mycoplasma with available genomes are not known. The goal of the current study was to develop a novel and reliable algorithm to predict essential genes in the 16 Mycoplasma genomes.
Identification of essential genes in silico is important and necessary, not only because their experimental determination is highly labor-intensive and time-consuming, but also because the speed for genome sequencing far outpaces that of the genome-wide gene essentiality studies. Although experimental techniques in identifying essential genes have been dramatically improved, genome-wide gene essentiality data are only available in 15 bacterial genomes9. In contrast, the number of available genomes has reached 1000, and the projects of sequencing 4000 more bacterial genomes are underway. With the increasing ability for genome sequencing, the in silico prediction of essential genes will be more and more important.
Various algorithms have been proposed to predict essential genes. Most algorithms are based on various genomic features, which include connectivity in protein-protein interaction network, fluctuation in mRNA expression, evolutionary rate, phylogenetic conservation, GC content, codon adaptation index (CAI), predicted sub-cellular localization and codon usages10,11,12,13,14,15,16. Because bacterial essential gene products comprise attractive drug targets for developing antibiotics, some studies are aimed at identifying essential genes that could serve as drug targets. These studies mainly rely on homologous search against available essential genes, for instance, through homologous searches against DEG (database of essential genes)9,17, based on the notion that those homologous to known essential genes are likely to be essential also. These bacterial pathogens include: Pseudomonas aeruginosa18, Burkholderia pseudomallei19, H. pylori20, Aeromonas hydrophila21, Neisseria gonorrhoeae22, Aeromonas hydrophila23 and Wolbachia24. Very recently, Duffield and coworkers, by using a modified down-selectoin computational tool, predicted 52 essential genes that are conserved in 7 or more genomes in DEG, and 7 of the 8 genes that were experimentally validated in Yersinia pseudotuberculosis were found to be esesntial25.
Essential genes have been known to be biasedly distributed in leading and lagging strands in E. coli and B. subtilis26. We then confirmed this phenomenon in 10 genomes in which gene essentiality screens had been performed27. However, the information of the biased essential gene distribution has not been effectively integrated into the gene essentiality prediction programs. With the availability of DoriC28, the database that contains replication origins for almost all bacterial genomes, such information (gene distribution in leading and lagging strands), if can be effectively used, will be helpful for the essential gene prediction for most bacterial genomes.
We developed an algorithm that integrates the information of biased distribution of essential genes in leading and lagging strands, in addition to homologous search and CAI values. The algorithm, which is simple and reliable, achieved an accuracy of 80.8% in predicting essential genes in M. pulmonis genome (self-consistence test), and achieved an accuracy of 78.9% and 78.1% in predicting those in S. aureus and Bacillus subtilis genomes, respectively (cross validation tests). Second, we then predicted 5880 essential genes in 16 Mycoplasma genomes. The detailed information of the genes is organized into a Database of predicted Essential Genes (pDEG) (http://tubic.tju.edu.cn/pdeg). The intersection set of essential genes in 18 Mycoplasma genomes (5880 predicted in the 16 Mycoplasma genomes, 379 and 310 experimentally determined in M. genitalium and M. pulmonis, respectively), consists of 153 core essential genes. The proposed algorithm and the prediction results will be helpful for studying essential genes in Mycoplasma as well as in other genomes. In particular, it is helpful for designing various Mycoplasma chassis used in synthetic biology.
Results
Training procedure and the self-consistence test
The training set included 379 and 310 essential genes for M. genitalium G37 (M. gen) and M. pulmonis UAB CTIP (M. pul), respectively. The training procedure could be performed in one of the two manners: essential genes of M. pul are predicted based on those of M. gen; or conversely, essential genes of M. gen are predicted based on those of M. pul. Since the average size of the 16 Mycoplasma genomes is about 1 Mb (see Table 1), the M. gen genome did not seem to be a suitable representative, because it has the smallest genome size (0.58 Mb). Therefore, we chose to train the parameters based on the first manner, i.e., essential genes of M. pul (genome size about 1 Mb), were predicted based on the experimentally determined ones of M. gen. The highest prediction accuracy achieved in the training procedure represents the self-consistence test accuracy that the present algorithm can reach. The parameters obtained following the training procedure can then be used to predict essential genes in the 16 Mycoplasma genomes.
Table 1. Detailed prediction and related information for the 16 Mycoplasma genomesa.
| Predicted essential genes | Total genes | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Organism | Abbr. | Size (Mb) | GC (%) | Leading | Lagging | Both | Leading | Lagging | Both | RefSeq |
| Mycoplasma agalactiae | Mag | 1.01 | 29.0 | 259 | 118 | 377 | 513 | 300 | 813 | NC_013948 |
| Mycoplasma agalactiae PG2 | MagPG2 | 0.88 | 29.7 | 253 | 115 | 368 | 452 | 290 | 742 | NC_009497 |
| Mycoplasma arthritidis 158L3-1 | Mar | 0.82 | 30.7 | 215 | 103 | 318 | 386 | 245 | 631 | NC_011025 |
| Mycoplasma capricolum subsp. capricolum ATCC 27343 | Mca | 1.01 | 23.8 | 282 | 78 | 360 | 591 | 221 | 812 | NC_007633 |
| Mycoplasma conjunctivae HRC/581 | Mco | 0.85 | 28.6 | 218 | 108 | 326 | 469 | 222 | 691 | NC_012806 |
| Mycoplasma crocodyli MP145 | Mcr | 0.93 | 27.0 | 232 | 127 | 359 | 404 | 285 | 689 | NC_014014 |
| Mycoplasma gallisepticum str. R(low) | Mga | 1.01 | 31.5 | 341 | 72 | 413 | 604 | 159 | 763 | NC_004829 |
| Mycoplasma genitalium G37 | Mge | 0.58 | 31.7 | 317 | 62 | 379 | 385 | 92 | 477 | NC_000908 |
| Mycoplasma hominis | Mho | 0.67 | 27.0 | 219 | 91 | 310 | 343 | 180 | 523 | NC_013511 |
| Mycoplasma hyopneumoniae 232 | Mhy232 | 0.89 | 28.6 | 187 | 156 | 343 | 366 | 325 | 691 | NC_006360 |
| Mycoplasma hyopneumoniae 7448 | Mhy7448 | 0.92 | 28.5 | 183 | 163 | 346 | 346 | 311 | 657 | NC_007332 |
| Mycoplasma hyopneumoniae J | MhyJ | 0.90 | 28.5 | 185 | 161 | 346 | 343 | 314 | 657 | NC_007295 |
| Mycoplasma mobile 163K | Mmo | 0.78 | 25.0 | 245 | 118 | 363 | 401 | 232 | 633 | NC_006908 |
| Mycoplasma mycoides subsp. mycoides SC str. PG1 | Mmy | 1.21 | 24.0 | 286 | 115 | 401 | 647 | 369 | 1016 | NC_005364 |
| Mycoplasma penetrans HF-2 | Mpe | 1.36 | 25.7 | 344 | 56 | 400 | 849 | 188 | 1037 | NC_004432 |
| Mycoplasma pneumoniae M129 | Mpn | 0.82 | 40.0 | 404 | 90 | 494 | 546 | 143 | 689 | NC_000912 |
| Mycoplasma pulmonis UAB CTIP | Mpu | 0.96 | 26.6 | 208 | 102 | 310 | 484 | 298 | 782 | NC_002771 |
| Mycoplasma synoviae 53 | Msy | 0.80 | 28.5 | 202 | 154 | 356 | 334 | 325 | 659 | NC_007294 |
aBold figures denote essential genes that are experimentally identified. Note the biased distribution of essential genes between leading and lagging strands.
Comparing the prediction with essential genes identified experimentally in the M. pul genome, parameters were determined such that the prediction accuracy reached the best value. The detailed training procedure is described in Fig. 1. We intended to keep the sensitivity Sn being roughly equal to the specificity Sp (Fig. 2a). The corresponding ROC curve is shown in Fig. 2b, where the AUC (Area Under the Curve) value was 0.812. The detailed prediction accuracy in terms of leading and lagging strands is listed in Table 2. Overall, the accuracy was 80.8% (Sn = 0.78 and Sp = 0.83), which may be considered as the highest self-consistence test accuracy that the present algorithm can reach.
Figure 1. The flow chart of the proposed algorithm in training and prediction phases.

Figure 2. Accuracy indices and the ROC curve for the current algorithm.

(A) Sensitivity, specificity and positive prediction rate in relation to the parameter s defined in eq. (8). The value of s (s ≥ s0) was chosen such that the sensitivity Sn is roughly equal to the specificity Sp. (B) The ROC curve (blue) and AUC (Area Under Curve). The red line denotes an extrapolation of the ROC curve to the point where 1 − Sp = 1. The AUC value is found to be 0.812.
Table 2. The self-consistence test accuracya.
| Organism | Strand | Sn | Sp | S+ | A |
|---|---|---|---|---|---|
| Mycoplasma pulmonis UAB CTIP (M. pul) | Leading | 80.3% | 82.6% | 77.7% | 81.4% |
| Lagging | 74.5% | 84.2% | 71.0% | 79.3% | |
| Both | 78.4% | 83.3% | 75.5% | 80.8% |
aThe bold figure denotes the overall prediction accuracy.
Cross-validation test
In addition to the self-consistence tests, the algorithm should also be evaluated by an independent data set. That is, once the parameters are determined, they should be tested by using a genome whose essential genes are experimentally determined, but M. gen and M. pul genomes should be excluded. However, so far M. gen and M. pul have been the only 2 genomes in the Mycoplasma family that have genome wide gene essentiality studies performed. Therefore, instead of using the information of essential genes of a third Mycoplasma genome, which is unavailable, we chose to use two bacterial genomes closely related to the two Mycoplasma genomes, Bacillus subtilis str. 168 and Staphylococcus aureus N315, whose essential genes were identified experimentally29,30,31.
Using the parameters in the training procedure of the algorithm, we predicted the essential genes for B. subtilis str. 168 and S. aureus N315. We find that instead of merely using the information of the 379 essential genes in the M. gen genome, the prediction accuracy can be improved using the combined set of the 379 and 310 essential genes in genomes of M. gen and M. pul, respectively. The prediction results are listed in Table 3. The average AUC value equals to (0.813 + 0.778)/2 = 0.796. The average prediction accuracy (78.1% + 78.9%)/2 = 78.5% may be deemed as the cross-validation test accuracy of the present algorithm. Because these two genomes do not belong to the Mycoplasma family, it is likely that overall accuracy of the present algorithm in predicting Mycoplasma essential genes is in the interval: 78.5% < accuracy ≤ 80.8%. For some of the 16 Mycoplasma genomes under study, it is possible that the prediction accuracy exceeds 80.8%, because they are much more closely related to M. gen and M. pul than B. subtilis and S. aureus.
Table 3. The cross-validation test accuracy a.
| Organism | Strand | Sn | Sp | A |
|---|---|---|---|---|
| Bacillus subtilis 168 | Leading | 69.8% | 86.6% | 78.2% |
| Lagging | 36.8% | 94.1% | 65.5% | |
| Both | 67.5% | 88.7% | 78.1% | |
| Staphylococcus aureus N315 | Leading | 73.3% | 85.5% | 79.4% |
| Lagging | 41.4% | 93.3% | 67.3% | |
| Both | 70.2% | 87.6% | 78.9% |
aBold figures denote the overall prediction accuracy.
Prediction of essential genes in the 16 Mycoplasma genomes
Based on the parameters obtained in the training procedure and the aggregate set of the 379 and 310 essential genes for M. genitalium G37 and M. pulmonis UAB CTIP, respectively, essential genes for the 16 Mycoplasma genomes were predicted. A total of 5880 essential genes were predicted, with on average 368 essential genes in each genome. The overall prediction results are listed in Table 1. The detailed information for each of the predicted essential gene is described in a database of predicted essential genes (pDEG), which is accessible from the website: http://tubic.tju.edu.cn/pdeg/. The database pDEG is organized with the same form as DEG. In pDEG, the detailed information of all the predicted essential genes can be obtained, including their names, functions, DNA and protein sequences and COG codes. If a predicted essential gene codes for an enzyme, the EC number and the KEGG linkage32 describing the involved metabolic pathway are also provided. Users can search for a predicted essential gene by their functions and names, and can also browse and download all the records in pDEG.
Core essential genes for the Mycoplasma family
The phylogenetic tree of the 18 Mycoplasma genomes was drawn based on the 16S rRNA (Fig. 3), where the abbreviations of 18 bacteria are shown in Table 1. We then obtained the intersection set of genes and essential genes based on reciprocal homolog searches between genomes. For example, the number of intersection genes between the genomes of M. mycoides and M. capricolum was 679. The number of overall intersection genes among the 18 Mycoplasma genomes was 191. Similarly, the numbers of intersection essential genes between two genomes or two genome clusters are shown in Fig. 3b. Note that the essential genes of the M. genitalium and M. pulmonis genome are identified experimentally, whereas the essential genes of remaining 16 bacterial genomes are predicted in the present study.
Figure 3. The phylogenetic tree of the 18 Mycoplasma genomes based on the 16S rRNA.

The intersection set of (A) genes and (B) essential genes in the 18 Mycoplasma genomes. The numbers on the left indicate gene numbers in intersection sets between genomes, whereas those on the right denote total gene number in a genome. The intersection set of the 5880 predicted essential genes and those experimentally identified in M. genitalium and M. pulmonis genomes consists of 153 core essential genes for the Mycoplasma family.
The intersection set of the essential genes in the 18 Mycoplasma genomes (5880 predicted in the 16 Mycoplasma genomes, 379 and 310 experimentally determined in M. genitalium and M. pulmonis, respectively) consists of 153 genes, which are called core essential genes for the Mycoplasma family. The core essential genes likely encode functions that are absolutely required for the survival of Mycoplasma, and their homologues in other bacteria likely have critical functions as well. Detailed information of the 153 core essential genes is available from pDEG.
Discussion
Essential genes are those indispensable for the survival of an organism under certain conditions, and the essential-gene concept is especially important for the burgeoning field, synthetic biology. A goal in synthetic-biology field is to develop the cellular chassis, which, composed of essential genes, contains all necessary components for cell survival. Based on the chassis, other gene circuits can be inserted to create experimental organisms with desirable traits that serve human needs. We here put forward two concepts: pan essential genes and core essential genes. For Mycoplasma species, pan essential genes are the combined essential gene set, while core essential genes are the intersection set of essential genes among Mycoplasma species. Based on the current dataset, the number of Mycoplasma pan essential genes is 6569 (5880 predicted, 379 and 310 experimentally determined in M. genitalium and M. pulmonis, respectively). However, we hypothesize that although the number of pan essential genes will continue to increase with more Mycoplasma genomes, the number of core essential genes (153) will largely remain the same. The core essential genes are likely needed for all Mycoplasma genomes and are likely all needed for the Mycoplasma chassis.
Indeed, the core essential genes are generally functionally important, and are involved in critical cellular processes. Based on COG functional classification33, core essential genes, compared to non-core essential and non-essential genes, had a higher proportion of genes involved in information storage and processing (Fig. 4a), and most of the core ones (55%) are involved in translation, ribosomal structure, transcription and replication (Fig. 4b). For example, they include most genes coding for 30S, 50S ribosomal proteins and aminoacyl-tRNA synthetases. They include those involved in replication, such as replication initiation protein (dnaA), replication DNA helicase (dnaB), DNA gyrase subunit A (gyrA) and subunit B (gyrB), DNA ligase (ligA), DNA polymerase III subunit-related proteins (dnaX, polC) and DNA primase (dnaG). They include genes of 4 protein synthesis elongation factors G, P, Ts and Tu (fusA, efp, tsf and tuf) and 2 translation initiation factors IF-2 (infB) and IF-3 (infC), and transcription related genes, such as DNA-directed RNA polymerase subunit alpha (rpoA) and beta (rpoB) and RNA polymerase sigma factor RpoD (rpoD). They also include almost all subunits of F0F1 ATP synthase (atpA, atpB, atpD, atpE and atpG) and many enzymes involved in energy production and metabolism. For details, refer to http://tubic.tju.edu.cn/pdeg/core/.
Figure 4. Functional classification of genes in the M. genitalium genome based on COG.

(A) COG classification of core-essential, non-core-essential and non-essential genes in M. genitalium. (B) Distribution of COG classification of the 153 core-essential genes.
It is noteworthy that some core essential genes do not have clearly defined functions. For instance, MG_423 encodes a hypothetical protein (accession number NP_073094) in the M. genitalium genome. Blast searches suggested that this gene likely encodes ribonucleas J, which plays a key in mRNA degradation34. Being a core essential gene prioritizes this gene to be further functionally characterized.
In summary, we here have predicted essential genes of the16 Mycoplasma genomes currently available in GenBank, based on experimentally identified essential genes of the M. genitalium and M. pulmonis genomes. The algorithm is simple and effective. The cross-validation test shows that the sensitivity Sn and the specificity Sp of the algorithm are all roughly equal to 80%. This accuracy means that about 80% of the essential genes in the Mycoplasma genomes under study are correctly predicted as essential; likewise, about 80% of the non-essential genes in these genomes are correctly predicted as non-essential. The high accuracy achieved is mainly due to the homologous mapping among evolutionally closely related bacteria, together with other information including biased distribution of essential genes in leading and lagging strands and CAI values. Mycoplasma has been an important species in the field of synthetic biology. The prediction results and the proposed algorithm can be useful in studying the minimal genomes of Mycoplasma, and in gene essentiality studies for other genomes. In particular, it is helpful for designing various Mycoplasma chassis used in synthetic biology.
Methods
The genomic RefSeq protein sequences for all the 18 Mycoplasma genomes were downloaded from the NCBI website (ftp://ftp.ncbi.nih.gov/genomes/Bacteria). The alignment program BLAST+ was downloaded from the same website (version Blast-2.2.23+, ftp://ftp.ncbi.nih.gov/blast)35. There are 379 and 310 experimentally determined essential genes for M. genitalium G3736 and M. pulmonis UAB CTIP37, respectively. It is noteworthy that definition of essential genes depends on certain experimental conditions, such as in rich growth medium38. In addition, synthetic lethal (lethality due to inactivation of more than 1 gene) is not considered in single gene knockout experiments. The detailed information for each of the 18 Mycoplasma genomes is listed in Table 1.
Following parameters were used in the present study to assess the performance of the algorithm.
 |
where TP, FN, FP and TN denote true positives, false negatives, false positives and true negatives, respectively. The sensitivity Sn represents the proportion of essential genes that have been correctly predicted as essential. The specificity Sp represents the proportion of non-essential genes that have been correctly predicted as non-essential. The positive prediction rate S+ represents the percentage of essential genes over the predicted ones. The accuracy A is the average of the sensitivity and specificity.
The prediction is partially based on the alignment of protein primary sequences to be predicted against those from closely related organisms in DEG, using the program Blastp. For each query protein sequence, we define
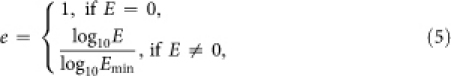 |
where E is the expectation value of the best scoring alignment in Blastp (with default parameters), and Emin is the smallest E value other than 0 of all genes from the 18 Mycoplasma genomes.
The prediction is also based on the strand-bias of essential genes26. We define
where b is a real number and β1, β2 ∈ [−1, 1]. The replication origin and terminus are determined based on the DoriC database28.
Finally, the prediction is also partially based on the CAI value of a gene to be predicted13,14,16. The CAI values were calculated using the CodonW software (http://codonw.sourceforge.net). We define
where c is a real number and γ ∈ [0,1]. Accordingly, we define the prediction parameter s by
Using an iterative procedure (Fig. 1), the parameters β and γ were determined based on the training set. For each gene to be predicted we calculate the set of parameters (e, b, c), and finally the prediction parameter s. We further look for a threshold s0 such that if s ≥ s0, the gene is predicted to be essential, otherwise, if s < s0, the gene is predicted to be non-essential. Detailed prediction results are available from the website http://tubic.tju.edu.cn/pdeg/, and programs are available upon request.
Author Contributions
Conceived and designed the experiments: RRZ. Performed the experiments: YL. Analyzed the data: RRZ and YL. Wrote the paper: RRZ.
Acknowledgments
We would like to thank Prof. CT Zhang for invaluable assistance and inspiring discussion. The present work was supported in part by a fund (176412) from Wayne State University to R.R.Z., and by the National Natural Science Foundation of China (Grant No. 90408028).
References
- Gibson D. G. et al.. Creation of a bacterial cell controlled by a chemically synthesized genome. Science 329, 52–6 (2010). [DOI] [PubMed] [Google Scholar]
- Pennisi E. Synthetic genome brings new life to bacterium. Science 328, 958–9 (2010). [DOI] [PubMed] [Google Scholar]
- Itaya M. An estimation of minimal genome size required for life. FEBS Lett 362, 257–60 (1995). [DOI] [PubMed] [Google Scholar]
- Koonin E. V. How many genes can make a cell: the minimal-gene-set concept. Annu Rev Genomics Hum Genet 1, 99–116 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Editorial. . Unbottling the genes. Nat Biotechnol 27, 1059 (2009). [DOI] [PubMed] [Google Scholar]
- Henkel J. & Maurer S. M. Parts, property and sharing. Nat Biotechnol 27, 1095–8 (2009). [DOI] [PubMed] [Google Scholar]
- Koonin E. V. Comparative genomics, minimal gene-sets and the last universal common ancestor. Nat Rev Microbiol 1, 127–36 (2003). [DOI] [PubMed] [Google Scholar]
- Hutchison C. A. et al.. Global transposon mutagenesis and a minimal Mycoplasma genome. Science 286, 2165–9 (1999). [DOI] [PubMed] [Google Scholar]
- Zhang R. & Lin Y. DEG 5.0, a database of essential genes in both prokaryotes and eukaryotes. Nucleic Acids Res 37, D455–8 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong H. & Barabasi A. L. Prediction of proteni essentiality based on genomic data. ComPlexUs 1, 19–28 (2003). [Google Scholar]
- Chen Y. & Xu D. Understanding protein dispensability through machine-learning analysis of high-throughput data. Bioinformatics 21, 575–81 (2005). [DOI] [PubMed] [Google Scholar]
- Saha S. & Heber S. In silico prediction of yeast deletion phenotypes. Genet Mol Res 5, 224–32 (2006). [PubMed] [Google Scholar]
- Seringhaus M., Paccanaro A., Borneman A., Snyder M. & Gerstein M. Predicting essential genes in fungal genomes. Genome Res 16, 1126–35 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafson A. M., Snitkin E. S., Parker S. C., DeLisi C. & Kasif S. Towards the identification of essential genes using targeted genome sequencing and comparative analysis. BMC Genomics 7, 265 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plaimas K., Eils R. & Konig R. Identifying essential genes in bacterial metabolic networks with machine learning methods. BMC Syst Biol 4, 56 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng J. et al.. Investigating the predictability of essential genes across distantly related organisms using an integrative approach. Nucleic Acids Res 39, 795–807 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R., Ou H. Y. & Zhang C. T. DEG: a database of essential genes. Nucleic Acids Res 32, D271–2 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakharkar K. R., Sakharkar M. K. & Chow V. T. A novel genomics approach for the identification of drug targets in pathogens, with special reference to Pseudomonas aeruginosa. In Silico Biol 4, 355–60 (2004). [PubMed] [Google Scholar]
- Chong C. E., Lim B. S., Nathan S. & Mohamed R. In silico analysis of Burkholderia pseudomallei genome sequence for potential drug targets. In Silico Biol 6, 341–6 (2006). [PubMed] [Google Scholar]
- Dutta A. et al.. In silico identification of potential therapeutic targets in the human pathogen Helicobacter pylori. In Silico Biol 6, 43–7 (2006). [PubMed] [Google Scholar]
- Sharma V., Gupta P. & Dixit A. In silico identification of putative drug targets from different metabolic pathways of Aeromonas hydrophila. In Silico Biol 8, 331–8 (2008). [PubMed] [Google Scholar]
- Barh D. & Kumar A. In silico identification of candidate drug and vaccine targets from various pathways in Neisseria gonorrhoeae. In Silico Biol 9, 225–31 (2009). [PubMed] [Google Scholar]
- Barh D., Kumar A. & Misra A. N. Genomic Target Database (GTD): a database of potential targets in human pathogenic bacteria. Bioinformation 4, 50–1 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holman A. G., Davis P. J., Foster J. M., Carlow C. K. & Kumar S. Computational prediction of essential genes in an unculturable endosymbiotic bacterium, Wolbachia of Brugia malayi. BMC Microbiol 9, 243 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffield M. et al.. Predicting conserved essential genes in bacteria: in silico identification of putative drug targets. Mol Biosyst 6, 2482–9 (2010). [DOI] [PubMed] [Google Scholar]
- Rocha E. P. & Danchin A. Essentiality, not expressiveness, drives gene-strand bias in bacteria. Nat Genet 34, 377–8 (2003). [DOI] [PubMed] [Google Scholar]
- Lin Y., Gao F. & Zhang C. T. Functionality of essential genes drives gene strand-bias in bacterial genomes. Biochem Biophys Res Commun 396, 472–6 (2010). [DOI] [PubMed] [Google Scholar]
- Gao F. & Zhang C. T. DoriC: a database of oriC regions in bacterial genomes. Bioinformatics 23, 1866–7 (2007). [DOI] [PubMed] [Google Scholar]
- Kobayashi K. et al.. Essential Bacillus subtilis genes. Proc Natl Acad Sci U S A 100, 4678–83 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsyth R. A. et al.. A genome-wide strategy for the identification of essential genes in Staphylococcus aureus. Mol Microbiol 43, 1387–400 (2002). [DOI] [PubMed] [Google Scholar]
- Ji Y. et al.. Identification of critical staphylococcal genes using conditional phenotypes generated by antisense RNA. Science 293, 2266–9 (2001). [DOI] [PubMed] [Google Scholar]
- Ogata H. et al.. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res 27, 29–34 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatusov R. L. et al.. The COG database: an updated version includes eukaryotes. BMC Bioinformatics 4, 41 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Even S. et al.. Ribonucleases J1 and J2: two novel endoribonucleases in B.subtilis with functional homology to E.coli RNase E. Nucleic Acids Res 33, 2141–52 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camacho C. et al.. BLAST+: architecture and applications. BMC Bioinformatics 10, 421 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass J. I. et al.. Essential genes of a minimal bacterium. Proc Natl Acad Sci U S A 103, 425–30 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- French C. T. et al.. Large-scale transposon mutagenesis of Mycoplasma pulmonis. Mol Microbiol 69, 67–76 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdes S. et al.. Essential genes on metabolic maps. Curr Opin Biotechnol 17, 448–56 (2006). [DOI] [PubMed] [Google Scholar]