

p31comet opposes the activities of the Mad2 spindle assembly checkpoint protein, localizes to unattached kinetochores, and like many checkpoint proteins, turns over rapidly at that site. Depletion of p31comet prevents timely passage into anaphase, showing that mitotic progression requires an active mechanism for silencing the spindle checkpoint.
Abstract
The spindle assembly checkpoint links the onset of anaphase to completion of chromosome-microtubule attachment and is mediated by the binding of Mad and Bub proteins to kinetochores of unattached or maloriented chromosomes. Mad2 and BubR1 traffic between kinetochores and the cytosol, thereby transmitting a “wait anaphase” signal to the anaphase-promoting complex. It is generally assumed that this signal dissipates automatically upon kinetochore-microtubule binding, but it has been shown that under conditions of nocodazole-induced arrest p31comet, a Mad2-binding protein, is required for mitotic progression. In this article we investigate the localization and function of p31comet during normal, unperturbed mitosis in human and marsupial cells. We find that, like Mad2, p31comet traffics on and off kinetochores and is also present in the cytosol. Cells depleted of p31comet arrest in metaphase with mature bipolar kinetochore-microtubule attachments, a satisfied checkpoint, and high cyclin B levels. Thus p31comet is required for timely mitotic exit. We propose that p31comet is an essential component of the machinery that silences the checkpoint during each cell cycle.
INTRODUCTION
The spindle assembly checkpoint (SAC) prevents errors in chromosome segregation by linking the dissolution of sister chromatid cohesion to the formation of bipolar kinetochore-microtubule (MT) attachments (for review, see Musacchio and Salmon, 2007). The Mad, Bub, and Mps1 components of the checkpoint were first isolated in Saccharomyces cerevisiae and are highly conserved among eukaryotes (Hoyt et al., 1991; Li and Murray, 1991; Hardwick et al., 1996). During prometaphase and metaphase, binding of spindle checkpoint proteins to unattached or maloriented kinetochores initiates a signaling cascade that inhibits Cdc20, an essential activator of the anaphase-promoting complex/cyclosome (APC/C). The APC/C is an E3 ubiquitin ligase the targets of which include cyclin B, securin, and other proteins the degradation of which is necessary for cells to transition from metaphase to anaphase. Mad2 and BubR1 associate with each other in a large mitotic checkpoint complex (MCC) that also contains Cdc20 and Bub3 (Hardwick et al., 2000; Sudakin et al., 2001). Rapid shuttling of Mad2, BubR1, and Cdc20 between kinetochores and the cytosolic MCC (Kallio et al., 2002; Howell et al., 2004; Shah et al., 2004) is thought to transmit the “wait anaphase” signal that links the state of kinetochore attachment to APC/C activity (Ciliberto and Shah, 2009; Kulukian et al., 2009). The precise nature of this signal remains unclear, but structural and biochemical data point to a switch between open and closed conformations of Mad2 (Luo et al., 2004; De Antoni et al., 2005): Mad2 is closed (C-Mad2) when bound to Mad1 at kinetochores or to Cdc20. Transient dimerization between C-Mad2 and an open, cytosolic Mad2 conformation (O-Mad2) is thought to promote binding to and inhibition of Cdc20 (Mapelli et al., 2007; Yang et al., 2008; Simonetta et al., 2009). Mad2 and BubR1 also appear to have a kinetochore-independent role in regulating APC/C early in mitosis before the establishment of an active spindle checkpoint in early prometaphase (Meraldi et al., 2004).
The SAC is traditionally regarded as a negative regulator of mitotic exit with Mad2 playing a particularly direct role in inhibiting proteins that control anaphase onset. Much less is known about processes that silence the checkpoint, activate APC/C, and promote mitotic progression when all chromosomes are correctly spindle-bound (Ciliberto and Shah, 2009). Historically, it has been assumed that spontaneous decay of an inhibitory checkpoint signal is involved. Recent work, however, has posited a role for the E2 ubiquitin ligase UbcH10 in disassembling the MCC-APC/C via ubiquitination of Cdc20 (Reddy et al., 2007) and consequent formation of active APC/C. In vitro biochemical studies have also implicated p31comet in the dissociation of anaphase-inhibitory complexes (Reddy et al., 2007; Teichner et al., 2011). Consistent with this view, RNA interference (RNAi)-mediated depletion of p31comet, a protein that binds C-Mad2 (Habu et al., 2002; Xia et al., 2004; Mapelli et al., 2006; Yang et al., 2007), reduces the rate at which cells recover from nocodazole-induced mitotic arrest (Xia et al., 2004). As such, a role for p31comet in exit from an unperturbed mitosis has not yet been studied. The distinction between nocodazole-induced mitotic arrest and normal mitotic progression is important: recent work has demonstrated that Ube2S, an E2 involved in ubiquitin chain extension, behaves differently in nocodazole-arrested cells than in normally growing cells (Garnett et al., 2009). It is an open question as to whether p31comet behaves in this way. Here we examine these questions by combining RNAi-mediated protein depletion of p31comet and other checkpoint proteins, live-cell imaging, and quantitative modeling. We find that p31comet binds to unattached kinetochores in mitosis in a Mad2-dependent manner, that this binding is highly dynamic, and that p31comet is essential in every cell cycle for rapid inactivation of the SAC and for timely passage from metaphase into anaphase. Thus mitotic progression requires an active mechanism for silencing the spindle checkpoint.
RESULTS
p31comet is required for mitotic exit in each cell cycle
The time of anaphase onset relative to nuclear breakdown (NBD; defined as T = 0) was determined by live-cell imaging of HeLa cells stably expressing monomeric red fluorescent protein (mRFP) or enhanced green fluorescent protein (EGFP) fusions to Histone2B. In unperturbed HeLa cells anaphase onset times have a characteristic skew-normal distribution with a modal (peak) value of (T =) 26 ± 1.5 min (Meraldi et al., 2004) and few cells (∼5%) enter anaphase before 18 min. In contrast, when cells were depleted of Mad2 (Figure 1C), ∼90% entered anaphase prematurely (before 18 min; see Figure 1A). Premature anaphase onset was phenocopied by overexpression of GFP-tagged p31comet: >80% of cells transiently expressing GFP-p31comet from a cytomegalovirus promoter entered anaphase prematurely (Figure 1A; Yang et al., 2007).
FIGURE 1:

p31comet and Mad2 act in opposition to promote anaphase onset. (A) p31comet overexpression and Mad2 depletion cause premature anaphase. HeLa Histone 2B-mRFP cells were transfected with GFP or GFP-p31comet, and HeLa Histone 2B-GFP cells were transfected with Mad2 siRNA or control siRNA and followed by time-lapse microscopy. The times represent minutes after NBD; scale bar, 10 μm. The graph shows the fraction of cells undergoing premature anaphase, defined as chromosome segregation at T < 18 min. (B) p31comet depletion and Mad2 overexpression arrest cells in mitosis. As in (A), HeLa Histone 2B-GFP cells were transfected with p31comet or control siRNA, and HeLa Histone 2B-mRFP cells were transfected with GFP-Mad2. The graph depicts a fraction of cells arrested in mitosis, defined as still in metaphase at T = 45 min. (C) Immunoblots of whole-cell lysates from HeLa cells treated with siRNA against lamin A (control), p31comet, or Mad2. Blots were probed with antibodies as indicated.
Few unperturbed HeLa cells were still in metaphase at T = 45 min (∼5%). When cells were depleted of p31comet by RNAi (Figure 1C and Supplemental Figure 1), however, we observed that ∼95% were in metaphase at 45 min as compared with 10% of control RNAi-treated cells (Figure 1B; the difference between 5 and 10% is presumed to represent the nonspecific effects of small interfering RNA [siRNA] transfection). This phenotype resembled that of transient overexpression of GFP-Mad2 (∼60% mitotic; Figure 1B) or nocodazole exposure (>95% mitotic; Meraldi et al., 2004), both of which are known to arrest cells in metaphase (Sotillo et al., 2007). Some overexpression or depletion phenotypes for p31comet have been described previously, but direct comparison with phenotypes associated with changes in Mad2 levels makes the pattern of opposing effects clear: overexpression of p31comet or depletion of Mad2 causes premature mitotic exit, whereas depletion of p31comet or overexpression of Mad2 causes mitotic arrest. Mad2 is well known as an anaphase inhibitory factor, and p31comet therefore acts as an anaphase promoting factor.
Why did cells depleted of p31comet arrest in mitosis? Immunofluorescence revealed that p31comet-depleted cells had congressed chromosomes, high levels of cyclin B, and low levels of cyclin A (Figure 2A). To determine whether chromosomes were bound correctly to MTs, cells were assayed for interkinetochore stretching and for the levels of Mad2 and Bub proteins. Following chromosome congression, bipolar attachment of spindle MTs to paired sisters creates pulling forces that can be assayed by measuring distances between puncta of core kinetochore proteins (Shelby et al., 1996). Using a recently developed kinetochore-tracking assay and HeLa cells that express the centromeric histone CENP-A (Jaqaman et al., 2010), we found that sister centromeres in metaphase cells depleted of p31comet were highly stretched and that they had therefore achieved bipolar attachment. If anything, interkinetochore distances were higher in p31comet-depleted than in control cells (1.13 ± 0.23 μm vs. 1.01 ± 0.25 μm, representing the mean and SD of >240 kinetochore pairs for each condition; Figure 2B). We conclude that kinetochore–microtubule attachment is normal in the absence of p31comet and that spindles can generate normal pulling forces. As a further assay for correct spindle assembly we asked whether the SAC was activated as assayed by measuring the levels of kinetochore-bound Mad and Bub proteins. In normal mitosis, checkpoint proteins are enriched on prometaphase kinetochores but then dissociate as spindle microtubules bind to kinetochores during metaphase (Hoffman et al., 2001). When Mad1, Mad2, Bub1, and BubR1 staining was compared in p31comet-depleted and control cells using immunofluorescence microscopy, levels of kinetochore-bound checkpoint proteins were indistinguishable (Figure 2C). Mad1 and Mad2 levels, when compared relative to staining of CREST (syndrome of calcinosis, Raynaud's, esophageal dysmotility, sclerodactyly, and telangiectasia) antiserum, were undetectable in metaphase cells treated with control or p31comet RNAi (Supplemental Figure 2). Similar results were observed for checkpoint proteins Mps1 and Bub3 and for CENP-E and CENP-F (Supplemental Figure 3). Depleting p31comet does not interfere with chromosome-MT attachment nor does it prevent normal dissociation of checkpoint proteins from kinetochores as mitosis proceeds. Thus p31comet loss arrests cells subsequent to kinetochore-MT attachment at a point in the cell cycle that is distinct from a checkpoint-mediated arrest.
FIGURE 2:

Cell cycle arrest following p31comet depletion is not due to defective kinetochore-MT attachments or sustained kinetochore signaling. (A) Cells depleted of p31comet arrest in mitosis. HeLa cells were treated with control or p31comet siRNA and stained with DAPI (blue) and anti-cyclin B or anti-cyclin A (green). (B) Kinetochores in p31comet-depleted cells are under tension. HeLa EGFP-CENP-A cells were transfected with siRNAs against p31comet or a control, and interkinetochore distances were measured in metaphase cells using a kinetochore-tracking assay. Interkinetochore distances are represented as whisker plots based on 240 (p31comet) and 319 (control) sister-kinetochore pairs in two independent experiments. (C) Checkpoint proteins localize normally in p31comet-depleted cells. HeLa cells depleted for p31comet or Lamin A control were stained with DAPI (blue) and antibodies against the indicated checkpoint proteins (red and green). Scale bars, 10 μm.
Binding of p31comet to kinetochores
To determine the subcellular localization of p31comet, cells were costained with affinity-purified anti-p31comet antibodies and CREST antiserum to identify centromeres (Figure 3A and Supplemental Figure 1). p31comet was found at high levels on unattached kinetochores in prometaphase, at significantly lower levels as kinetochores became MT-bound during metaphase and in the cytosol at all cell-cycle stages. Both kinetochore and cytosolic staining by anti-p31comet antibodies were specific insofar as they were reduced significantly by RNAi directed against p31comet (Figure 3, B and C, and Supplemental Figure 1), and the pattern was identical to those of Mad1 and Mad2 (Chen et al., 1998; Howell et al., 2004; Shah et al., 2004); these data confirm and extend previous observations from Habu et al. (2002). RNAi-mediated depletion of Mad2 reduced levels of p31comet staining on prometaphase kinetochores to the same extent as depletion of p31comet itself (>10-fold; Figure 3, B and C). p31comet is therefore among the few proteins known whose binding to kinetochores is completely dependent on the presence of Mad2 (Vigneron et al., 2004).
FIGURE 3:

p31comet localizes to unattached kinetochores in a Mad2-dependent manner. (A) p31comet localizes to unattached kinetochores. HeLa cells were stained with DAPI, anti-p31comet antibodies, and CREST serum. Scale bar, 5 μm. (B) p31comet binds to kinetochores in prometaphase and requires Mad2. HeLa cells treated with the indicated siRNA were stained with DAPI, CREST, and anti-p31comet antiserum. Scale bar, 2.5 μm. Indicated sister kinetochore pairs are magnified at right; scale bar, 0.5 μm. (C) Quantitation of p31comet loss from kinetochores. p31comet/CREST signal ratios from individual kinetochores are plotted as dots; bars represent mean ratio values. (D) Images captured from a time-lapse sequence showing p31comet–EYFP localization during mitosis. Kinetochore localization (arrows) is most apparent in early prometaphase when chromosomes are unattached. Upon attachment a portion of the fluorescence accumulates at spindle poles (arrowheads) and in the spindle region (triangles). After all chromosomes no longer have kinetochore staining, anaphase onset begins (dashed lines mark chromosome masses). Time points are in minutes. Scale bar, 10 μm.
To observe the precise dynamics of p31comet localization, we generated PtK2 cells that stably expressed an enhanced yellow fluorescent protein (EYFP) fusion to the human protein. Live-cell analysis of these cells confirmed localization data obtained from immunostained HeLa cells: p31comet-EYFP was present on unattached kinetochores in early prophase/prometaphase and then was lost as kinetochores became MT-bound (Figure 3D, arrows, and Supplemental Movie 1). Cells entered anaphase only after levels of kinetochore-associated p31comet-EYFP had fallen below the level of detection (Figure 3D), a behavior similar to what has previously been observed for both Mad1 and Mad2. As PtK2 cells progressed into late metaphase and kinetochores became MT-bound, p31comet-EYFP was observed to accumulate at spindle poles and to remain there until late anaphase, at which point it dissociated and fluorescence was lost (Figure 3D, short arrowheads). In HeLa cells, anti-p31comet antibodies did not appreciably stain spindle poles. It has previously been established, however, that whereas Mad2 localizes to spindle poles late in mitosis in PtK2, it does not in HeLa cells (Howell et al., 2000; Shah et al., 2004). Thus, whatever the origins of differential spindle staining between PtK2 and Hela cells, similar results have been obtained with Mad2 and p31comet: levels of Mad2 and p31comet on individual kinetochores were highly correlated in both cell types.
Kinetochore-dependent and -independent p31comet functions
The fact that RNAi and overexpression phenotypes of Mad2 and p31comet are mirror images of each other and that localization of p31comet to kinetochores requires Mad2 raises the question of whether the arrest of p31comet-depleted cells is Mad2-dependent. In cotransfection experiments we observed that, whereas 70 ± 20% of p31comet-depleted cells (cotransfected with second control siRNA) were in mitosis 45 min after NBD, only 12 ± 4% of cells depleted of both Mad2 and p31comet were similarly arrested (Figure 4A). Codepletion phenotypes were slightly weaker than single-depletion phenotypes (e.g., with Mad2 RNAi alone, no cells were in mitosis at 45 min) presumably due to competition between cotransfected siRNA oligos. Even so, we conclude that the arrest of cells following depletion of p31comet requires Mad2.
FIGURE 4:
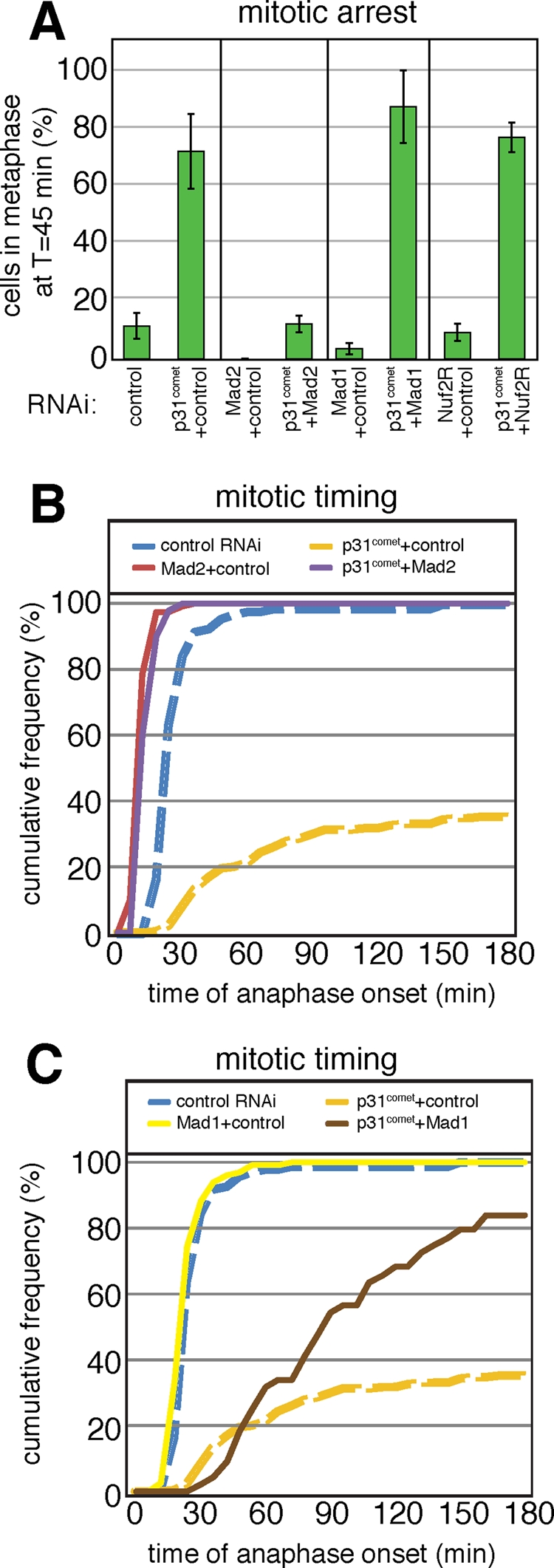
p31comet acts downstream of the timer functions of Mad2 but upstream of kinetochore functions. (A) Mitotic arrest of HeLa Histone 2B-GFP cells was measured as in Figure 1B. Bars indicate mean and range of at least two independent experiments. (B and C) Cumulative frequency graphs of anaphase times in cells treated with siRNA as indicated. Dashed lines for control RNAi and p31comet/control RNAi are identical between B and C.
To determine which other components of the checkpoint are involved in the mitotic arrest caused by loss of p31comet, we codepleted p31comet with Mad1 and Nuf2R. Mad1 is a checkpoint protein that binds to and recruits Mad2 to kinetochores (Chen et al., 1998), and Nuf2R is a structural component of kinetochores required for kinetochore binding by Mad1, Mad2, and other checkpoint proteins (DeLuca et al., 2003). Single depletion of Mad1 (4 ± 3% mitotic cells at t = 45 min) or Nuf2R (10 ± 4% mitotic cells) inactivated the checkpoint, as described previously (Meraldi et al., 2004; McAinsh et al., 2006). The mitotic arrest caused by p31comet depletion, however, was not overcome by codepletion with either Mad1 (87 ± 18% mitotic cells) or Nuf2R (77 ± 7% mitotic cells). Thus, even though depletion of Mad1, Mad2, or Nuf2 abrogates the checkpoint, mitotic arrest caused by loss of p31comet is overcome only by Mad2 depletion (Figure 4A). Because depletion of either Mad1 or Nuf2 causes Mad2 to dissociate from kinetochores, our data suggest that p31comet is also acting via cytosolic Mad2, a fraction of which is associated with the MCC. The results also serve as further evidence that p31comet depletion does not simply arrest cells in a checkpoint-dependent manner.
Cumulative frequency graphs that capture the timing of mitotic exit in single cells provide a kinetic picture of cell-cycle progression and a more nuanced view of function. We observed that half of all cells transfected with control RNAi progressed from NBD to anaphase at 21 min (the modal time), in line with previous data (Figure 4, B and C). The modal time of anaphase entry was not affected by Mad1 depletion, but depletion of Mad2 cut the time in half, to ∼12 min, consistent with previous data showing that Mad2 but not Mad1 is involved in the timing of mitotic exit independent of checkpoint control (Meraldi et al., 2004). Kinetic analysis also revealed that cells depleted of p31comet do not arrest permanently in metaphase but instead proceeded slowly into anaphase such that roughly 40% had exited mitosis by t = 180 min (Figure 4, B and C, orange line). This finding contrasts with the phenotype of a classical checkpoint arrest provoked by depletion of essential spindle components in which cells enter a near-complete arrest of six or more hours in duration (Meraldi et al., 2004). As mentioned earlier in the text, Mad2 depletion was epistatic to p31comet depletion, and codepletion phenocopied the kinetics of Mad2 depletion alone (half recovery time [t1/2] ∼11 min; Figure 4B; purple line). In contrast, codepleting cells of Mad1 and p31comet resulted in a phenotype midway between control RNAi and p31comet-depletion alone, causing cells to exit mitosis with a t1/2 ∼ 90 min (Figure 4C; brown line). Although the intermediate phenotype could be due to incomplete Mad1 knockdown, we would expect that at least a subset of cells would show a faster mitotic transit if in fact p31comet is downstream of Mad1. Instead, we interpret this intermediate phenotype as reflecting a role for p31comet in antagonizing both the kinetochore-dependent Mad2 functions (this manifests itself as the difference between the depletion phenotypes for p31comet alone vs. p31comet in combination with Mad1 or Nuf2) and the kinetochore-independent functions of Mad2 (this manifests itself as the difference between the double depletion phenotypes for p31comet and Mad1 or Nuf2 vs. p31comet and Mad2).
Two pools of Mad2 are found on kinetochores: a rapidly cycling pool of Mad2 that transiently associates with a stable pool of Mad2 that is tightly bound to Mad1 (Shah et al., 2004). To assay the rate of turnover of p31comet on kinetochores we performed fluorescence recovery after photobleaching experiments (FRAP) on PtK2 cells stably expressing p31comet-EYFP. Figure 5A illustrates a representative FRAP sequence in which one of a pair of unattached kinetochores is rapidly bleached and its recovery observed. In unperturbed mitoses the mean t1/2 of p31comet-EYFP at unattached kinetochores was 10 ± 0.9 s (SEM, n = 26 kinetochores from 15 cells) with a total percentage recovery of 79 ± 3% (SEM). In cells arrested with nocodazole (300 nM), the mean t1/2 was 12 ± 1.3 s (SEM, n = 11 kinetochores from 10 cells) with a total percentage recovery of 81 ± 5% (SEM), values that are indistinguishable from normal prometaphase kinetochores. These recovery kinetics are similar to those for the rapidly exchanging pool of Mad2 (t1/2 = 10.8 ± 1.2 s) (Shah et al., 2004). To determine the rate of dissociation of p31comet from a pool of Mad2 that is not dynamic, we examined the interphase nuclear envelope, which carries stable Mad1–Mad2 complexes (Shah et al., 2004). The fraction of p31comet bound to the nuclear envelope was observed to dissociate at a relatively slow rate (t1/2 = 56 ± 12 s SEM, percent recovery = 67.1 ± 2.46%, n = 13 cells; Figure 5, D and E), consistent with tight binding of C-Mad2 to Mad1. In contrast, the nucleoplasmic fraction of p31comet exhibited a mean t1/2 of 0.53 ± 0.36 s (SEM) with a percent recovery of 32.9% ± 2.46% (SEM), consistent with a rapid diffusible nucleoplasmic pool of p31comet. Thus p31comet traffics on and off kinetochores at nearly the same rate as the dynamic pool of Mad2. Moreover, rapid association/dissociation of p31comet from a Mad1–Mad2 scaffold is a special property of kinetochores, because Mad1–Mad2 complexes on the nuclear envelope bind relatively stably to p31comet.
FIGURE 5:

Trafficking of p31comet on and off kinetochores. (A) An image sequence showing FRAP of p31comet–EYFP at an unattached kinetochore. The inset shows the bleached kinetochore recovering in detail. Time points are in seconds. Scale bar, 10 μm. (B) Recovery curve for p31comet–EYFP at unattached kinetochores in untreated PtK2 cells. (C) Recovery curve for p31comet–EYFP at unattached kinetochores in nocodazole-arrested PtK2 cells. (D) An image sequence showing FRAP of p31comet–EYFP in the nucleoplasm and nuclear envelope. Time points are in seconds. Scale bar, 5 μm. (E) Recovery curve for p31comet–EYFP at the nuclear envelope in PtK2 cells shown as a percentage of the original fluorescence intensity over time. A biphasic curve had the best fit to the data. The fast phase is the diffusible nucleoplasmic fraction of p31comet–EYFP, whereas the slow fraction is p31comet at the nuclear envelope.
DISCUSSION
Activation of the SAC by unattached kinetochores in normally dividing cells and by maloriented kinetochores (particularly those generated by spindle poisons) has been widely studied in higher and lower eukaryotes, but the mechanisms that extinguish checkpoint signaling at the end of metaphase are less well understood (Ciliberto and Shah, 2009). Historically, it has been assumed that the SAC comprises only negative regulators of mitotic progression. Data showing delayed recovery of p31comet-depleted cells from nocodazole-mediated arrest (Xia et al., 2004) and in vitro studies of p31comet binding proteins (Reddy et al., 2007; Teichner et al., 2011), however, have highlighted a role for p31comet in APC/C activation and mitotic progression. The ubiquitin-conjugating enzymes UbcH10 (Reddy et al., 2007) and Ube2S (Garnett et al., 2009; Williamson et al., 2009; Wu et al., 2010) also have been implicated in silencing the spindle assembly checkpoint. In this article we examine the role of p31comet in cells that have not been exposed to spindle poisons. We find that RNAi-mediated depletion of p31comet dramatically slows the entry of cells into anaphase, showing that p31comet is required in each cell cycle for timely mitotic exit. Although arrest is not absolute, the phenotype is strong with ∼80% of p31comet-depleted cells still in metaphase 3 h after nuclear envelope breakdown as compared with progression of 100% of cells within 30 min under normal circumstances.
A remarkable feature of forced changes in Mad2 and p31comet levels is that the resulting phenotypes are mirror images of each other: Overexpression of Mad2 or depletion of p31comet blocks or dramatically slows mitosis, whereas depletion of Mad2 or overexpression of p31comet causes rapid progression into anaphase. In p31comet-depleted cells sister chromatids have achieved normal bipolar attachment, and SAC proteins including Mad2 have dissociated from kinetochores, showing that checkpoint conditions have been satisfied at kinetochores. Our findings suggest that p31comet is a cytoplasmic checkpoint activator required for mitotic progression after the loss of checkpoint proteins from kinetochores.
Early in mitosis, before the completion of chromosome-MT attachment, we find that p31comet is similar to Mad2 and other checkpoint proteins in localizing to kinetochores. Previous work by Habu and colleagues (2002) demonstrated p31comet localization to a chromatin domain roughly coincident with centromeres; using new p31comet antibodies and a GFP fusion protein, we show that puncta of p31comet colocalize with core kinetochore proteins. Kinetochore localization by p31comet is Mad2-dependent and is lost when chromosomes bind spindle MTs. The recruitment of Mad2 to kinetochores is a complex process thought to play an integral role in generation of the “wait-anaphase” signal. Mad2 in the closed conformation is bound directly to Mad1 and serves as a template for conversion of open O-Mad2 into its active and closed C-Mad2 form after which it dissociates and binds to Cdc20 in the cytosol, thereby blocking APC/C activity (Xia et al., 2004; Mapelli et al., 2006; Simonetta et al., 2009). The conversion of O-Mad2 into C-Mad2 at unattached kinetochores results in association and dissociation of a pool of Mad2 from the stable Mad2–Mad1 oligomer with a half-life ∼11 s (as measured by FRAP) (Shah et al., 2004). It has been estimated that this turnover permits ∼60 molecules of C-Mad2 to be generated per detached kinetochore per second (Ciliberto and Shah, 2009). Remarkably, we find that p31comet traffics through the kinetochore with kinetics indistinguishable from that of Mad2 (FRAP recovery t1/2 ∼11 s), implying similarly rapid association and dissociation kinetics. This observation argues against the simplest model for p31comet function, namely that it extinguishes the wait anaphase signal by acting as a stable cap on Mad1/C-Mad2 complexes that functions by blocking binding to and activation of O-Mad2 (Mapelli et al., 2006). The capping model found initial support from the observation that p31comet has ∼40-fold greater affinity for C-Mad2 than for O-Mad2 (Vink et al., 2006). Indeed, we find that p31comet binds Mad1/C-Mad2 complexes present on the interphase nuclear envelope and that turnover is slow (FRAP recovery t1/2 ∼60 s) like the turnover of stable Mad1/C-Mad2 complexes themselves (Shah et al., 2004). The function of the interphase Mad1/C-Mad2 complex is not known, but its ability to stably bind p31comet argues that avid p31comet–C-Mad2 association can occur in vivo. The binding of p31comet to unattached kinetochores therefore appears to be considerably weaker than binding to the nuclear envelope, implying either that p31comet or Mad2 is modified in some way or that it is stable Mad2-p31comet complexes that traffic on and off kinetochores.
We have previously demonstrated that the consequences of dissociating Mad2 from kinetochores, for example, by depleting cells of Mad1 or the kinetochore structural component Nuf2, is distinct from the phenotype of depleting Mad2. In all three cases SAC function is inactivated, but the cytosolic Mad2 present in Mad1- or Nuf2-depleted cells has a role in ensuring the correct timing of mitosis that is functionally (if not necessarily biochemically) distinct from its role in sensing the presence of maloriented chromosomes. It is presumed that this function involves the MCC. In this article we show that codepletion of p31comet and Mad2 phenocopies Mad2 depletion alone: Cells enter anaphase so rapidly (t1/2 for NBD to anaphase ∼ 12 min) as to indicate that both the SAC and timing functions of Mad2 are abrogated. The situation is different following codepletion of p31comet and either Mad1 or Nuf2, however. In these cases we observe an intermediate phenotype with cells slowly exiting mitosis over the course of ∼3 h. Together the combined RNAi phenotypes imply a function for p31comet as a regulator of the cytosolic Mad2 present in the MCC. Kinetochores are unable to generate active Mad2 in the absence of Mad1 or Nuf2, and we presume that this is also the case in doubly depleted p31comet/Mad1 or p31comet/Nuf2 cells. A relatively small pool of active Mad2, however, is present at the start of mitosis and forms the MCC timer (Sudakin et al., 2001; Meraldi et al., 2004). We speculate that in the absence of p31comet the timer portion of C-Mad2 is only slowly extinguished and the transition into anaphase is therefore delayed. When the checkpoint is able to function normally, then much more C-Mad2 can be generated and the consequences of p31comet depletion are correspondingly more severe. In the absence of Mad2 itself, no inhibitor to mitotic progression is generated and the combined p31comet/Mad2 depletion phenocopies Mad2 depletion.
To begin to develop a quantitative understanding of p31comet activities we created a simple (toy) computational model of checkpoint signaling. This model includes several competing processes the rates of which have been estimated experimentally (see Materials and Methods) including: 1) kinetochore generation of C-Mad2 from a kinetochore scaffold (K-C-Mad2); 2) interaction of C-Mad2 with APC/C (or MCC-APC/C); 3) generation of C-Mad2 from a cytoplasmic pool of C-Mad2, a potential amplification process (Simonetta et al., 2009); and 4) binding of p31comet to C-Mad2 so as to block subsequent binding and conversion of O-Mad2 and to potentiate the dissociation of C-Mad2-APC/C complexes (Reddy et al., 2007; Teichner et al., 2011).
When p31comet levels were varied in simulation from 0 to 120 nM (endogenous levels are estimated to be ∼100 nM based on calibrated Western blots) and steady-state levels of APC/C plotted, we observed two regimes: when [p31comet] < 60 nM, active APC/C levels were low and the checkpoint chronically active, as expected, whereas with [p31comet] > 90 nM, APC/C was chronically active and the checkpoint was bypassed, a state that mimics p31comet overexpression in cells (Figure 6A). Similar behavior was observed when kinetochore-dependent reactions were turned off, mimicking the consequences of kinetochore–MT attachment and checkpoint silencing (Figure 6B). Simulation of [p31comet] < 60 nM in the absence of checkpoint-active kinetochores mimics the strong block to mitotic progression observed following RNAi of p31comet, whereas checkpoint resolution was observed at [p31comet] > 80 nM in the absence of active kinetochores. The strong dependence of APC/C activity on p31comet levels shows that robust regulation can be achieved if we postulate a modest twofold increase in p31comet between early and late mitosis (Figure 6C). There is no evidence, however, that total p31comet levels change through mitosis, and we therefore speculate that a change in protein activity is involved (this is modeled by changing active p31comet from 50 nM [dashed lines] to 100 nM [solid lines]). With a twofold increase in the concentration or activity of active p31comet we observe a twofold increase in the concentration of capped, kinetochore-localized C-Mad2 (p31-K-C-Mad2 in Figure 6C). In addition, a dynamic equilibrium between two inhibited APC/C species: C-Mad2:APC/C and p31:C-Mad2:APC/C at [active p31comet] = 50 nM is replaced by the rapid decay of inhibited APC/C at [active p31comet] = 100 nM and formation of functional APC/C. Our admittedly simple simulations therefore integrate a number of biochemical processes to suggest that both of the postulated functions of p31comet are important to its biological function: blocking the conversion of O-Mad2 to C-Mad2 (Vink et al., 2006) and promoting the dissociation of the inhibited MCC-APC/C (Reddy et al., 2007; Teichner et al., 2011).
FIGURE 6:

Computer-driven simulations demonstrating that p31comet levels can control APC/C activity. APC/C activation state as a function of active p31comet concentration in the presence (A) or absence (B) of kinetochore signaling reveals a dependence of p31comet on APC/C activation. (C) Checkpoint protein complex dynamics are shown for two concentrations of p31comet. The dashed line illustrates the addition of a low (50 nM) concentration of p31comet, and the solid line the addition of p31comet to a high level (100 nM). The high levels of p31comet result in a rapid 80% inhibition of the kinetochore pool (1 nM K-C-Mad2 present) but much lower (40%) in the lower p31comet simulation. Through the combined action of C-Mad2 binding and C-Mad2–APC dissociation, the APC/C is partitioned between two interconverting inactive pools of p31:C-Mad2:APC/C and C-Mad2:APC/C. The result is that the APC/C is rapidly activated at the high p31comet level, whereas it is inactive at the low level.
On the basis of available experimental data and the results of quantitative simulations, we propose a model whereby p31comet affinity for Mad2 is low, and therefore inhibitor dissociation is reduced, during active checkpoint signaling, but that after kinetochores attach, p31comet avidly binds Mad2 resulting in the increased dissociation of C-Mad2 from APC/C. At a threshold level of activity, which need only be less than twofold greater than at the start of mitosis, p31comet is able to overwhelm all active C-Mad2–generating mechanisms and dissociate all inhibited APC/C. Because we have shown that p31comet actively traffics through kinetochores early in mitosis, it is appealing to propose that trafficking is associated with p31comet regulation. p31comet and Mad2 are quite similar in structure (Yang et al., 2007) making conformational regulation of p31comet a possibility, with posttranslational modification (perhaps by a checkpoint kinase) an obvious alternative.
MATERIALS AND METHODS
Generation of plasmids and cell lines
Mad2 (EST GenBank ID R10991) was subcloned by PCR into pGEX-6P-2 (Amersham Biosciences. Little Chalfont, UK) and pEGFPC1 (Clontech, Mountain View, CA). p31comet (IMAGE clone 321778; ATCC, Manassas, VA) was subcloned into pEGFPC1 and pFBnHis10HA. The cDNA for mRFP, a gift of R. Tsien (University of California, San Diego, San Diego, CA), was fused to histone H2B in pCDNA3.1 (Invitrogen, Carlsbad, CA). All plasmids were confirmed by sequencing. Human p31comet cDNA was amplified from a HeLa cDNA library with EcoRI and BamHI restriction sites by standard methods and the following primers: (5′-atgcatgcatGAATTCATGGCGGCGCCGGAGGCGGAG-3′ and 5′-atgcatgcatGGATCCCTCGCGGAAGCCTTTAAATGT-3′). This EcoRI–BamHI PCR product was ligated into the C2 and N3 frames of the Clontech EYFP plasmid to produce both N-terminal and C-terminal fusion cDNAs, respectively. PtK2 cells stably expressing EYFP fusions to human p31comet were generated via retroviral transduction as previously described (Shah et al., 2004). PtK2 cell lines were maintained in CO2-dependent culture medium and switched to a Leibovitz l-15–based HEPES-buffered medium for live imaging (Shah et al., 2004).
Cell culture, antibodies, and immunoblotting
HeLa, Histone 2B-GFP HeLa, HeLa Histone 2B-mRFP, and HeLa EGFP-CENP-A cells were generated and cultured as described (Meraldi et al., 2004; Jaqaman et al., 2010). All siRNA oligos were purchased from Dharmacon Research (Lafayette, CO). The sequences for the p31comet siRNA duplexes are p31comet-1 (GGAGUUCUAUGAACUGGAC) and p31comet-3 (CUGUAAUCAUCGCUGAACA). Duplexes and RNAi for Lamin A (Elbashir et al., 2001), Mad1 and Mad2 (Martin-Lluesma et al., 2002), and hNuf2R (Meraldi et al., 2004) have been described. HeLa cells were transfected with siRNA as described (Elbashir et al., 2001) and analyzed 48 h after transfection. GFP-p31comet and GFP-Mad2 were transiently expressed in HeLa Histone 2B-mRFP cells using Fugene 6 (Roche Diagnostics, Pleasanton, CA), and cells were analyzed 16 h after transfection.
In synchrony-release experiments that maximize protein depletion two of six tested anti-p31comet siRNA oligos resulted in ∼90–95% protein depletion as judged by immunoblotting and immunofluorescence using polyclonal anti-p31 antibodies (see Supplemental Figure 1). In contrast, transfection of cells with siRNA directed against Lamin A, a well-studied control for mitotic timing experiments, caused no changes in p31comet levels.
Polyclonal antibodies were raised in NZW rabbits against human p31comet residues 1–138 expressed in Escherichia coli (Covance, Princeton, NJ). A column of immobilized p31comet (1–138) was made with the SulfoLink kit (Pierce, Rockford, IL) for affinity purification of immune sera. Whole-cell extracts were prepared by boiling cells in 2× SDS sample buffer with 15% β-mercaptoethanol before SDS–PAGE and immunoblotting.
Microscopy
Cells were fixed, permeabilized, and blocked as described (Kapoor et al., 2000). Cross-adsorbed secondary antibodies were used (Molecular Probes, Eugene, OR). HeLa images were acquired as described (Martinez-Exposito et al., 1999), and kinetochore fluorescence intensities were calculated as described (Meraldi et al., 2004). Live cell imaging was performed in ΔT 0.15-mm dishes (Bioptechs, Butler, PA) in CO2-independent medium (GibcoBRL, Life Technologies, Carlsbad, CA) at 37°C. Exposures (0.2 s) were acquired every 3 min for 6 h using a 20× NA 0.75 objective on a Nikon Applied Precision Deltavision microscope equipped with a Mercury 100W lamp, GFP long pass filter set (for Histone 2B-GFP-Hela cells) or a Sedat filter set (to follow GFP-proteins in Histone 2B-Red-Hela cells; Chroma, Bellows Falls, VT) and a CoolSNAP HQ camera (Roper Scientific, Tucson, AZ). Point visitation was used to follow cells in multiple fields. For measurements of interkinetochore distances EGFP–CENP-A cells were recorded live with a 100× 1.35 N.A. objective on an Olympus DeltaVision microscope (Applied Precision, Issaquah, WA) equipped with a CoolSNAP HQ2 camera (Roper Scientific) and an EGFP filter set (Chroma) at a temporal resolution of 7.5 s, and the time-lapse images were subjected to kinetochore tracking assay analysis (Jaqaman et al., 2010).
Mitotic frequency plots were generated as described (Meraldi et al., 2004). Anaphase entry half-lives were calculated by fitting cumulative frequency data from live-cell imaging by nonlinear least squares regression in the nlintool of Matlab 7.0 (The Mathworks, Natick, MA). Observed half-lives were considered to be the sum of the observed lag and the calculated t1/2. The ratio of Mad1 or Mad2 levels versus CREST signal at kinetochores was determined as previously described (McAinsh et al., 2006).
For FRAP and live PtK2 studies, imaging was carried out on a Nikon TE-2000-E2 microscope equipped with an incubation chamber (in Vivo Scientific, St. Louis, MO) for environmental control, CoolSNAP HQ camera and IPLab software (BD Biosciences, San Jose, CA) for microscope-, camera-, and shutter-based acquisition. Fluorescence recovery (FRAP) experiments were carried out on the same microscopy system with the addition of a 3W argon–krypton laser tuned to 514 nm and attenuated to 10 mW at the back port of the microscope. The laser beam was expanded to slightly overfill the back aperture of a 60× high numerical aperture (NA = 1.4) objective. Laser exposure was restricted by a shutter (LUDL Electronic Products, Hawthorne, MY) controlled by IPLab software, and images were acquired after bleaching at 500-ms to 1-s intervals. Fluorescence recovery intensity measurements and curve fitting were carried out according to previously described methods (Shah et al., 2004).
Mathematical model
A mathematical model was constructed using published biochemical reactions, rate constants, and affinities for Mad2-p31comet and APC/C–MCC interactions (Tables 1 and 2). All other rate constants were chosen based on qualitative affinities or rates that have been reported in the literature (Mapelli et al., 2006; Vink et al., 2006). Whereas previous work has used a spatial model for simulations of checkpoint activity (Ciliberto and Shah, 2009), here we use a set of ordinary differential equations to represent the biochemical interactions produced at the kinetochore and that occur in the cytoplasm. The scheme of the model has a set of reactions that have C-Mad2 in the context of Mad1 (Km2c) representing the kinetochore reactions. The biochemical reactions modeled are:
TABLE 1:
Species concentrations used in simulations.
| Species | Concentration | Initial condition |
|---|---|---|
| Total Mad2 (m2c, m2o) | 120 nM | All Mad2-O |
| Kinetochore (Km2c) | 1 nM | Constitutive |
| p31comet (p) | Adjusted | 0 nM |
| APC/C (a) | 90 nM | Active APC/C |
TABLE 2:
Rate constants used in simulations.
| Rate constant | Value | Unit | Reference |
|---|---|---|---|
| kmm | 1 | nM−1s−1 | Vink et al., 2006 |
 |
0.25 | s−1 | Estimated from Howell et al., 2000; Shah et al., 2004; Simonetta et al., 2009 |
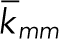 |
1000 | s−1 | Vink et al., 2006 |
| kpm | 1 | nM−1s−1 | Vink et al., 2006 |
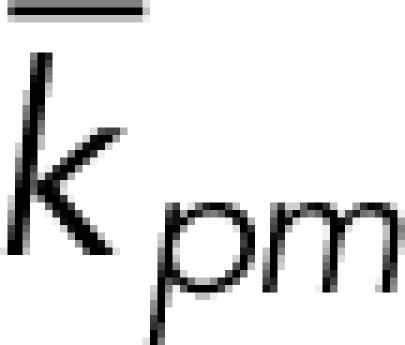 |
25 | s−1 | Vink et al., 2006 |
| cam | 1 | nM−1s−1 | M.S.M. and J.V.S., unpublished data |
 |
0.0001 | s−1 | M.S.M. and J.V.S., unpublished data |
| cmm | 1 | nM−1s−1 | Vink et al., 2006 |
 |
0.25 | s−1 | Simonetta et al., 2009 |
 |
1000 | s−1 | Vink et al., 2006 |
| cpm | 1 | nM−1s−1 | Vink et al., 2006 |
 |
0.01 | s−1 | Estimated from Reddy et al., 2007 |
 |
25 | s−1 | Vink et al., 2006 |
 |
 |
 |
 |
 |
 |
with m2o representing open Mad2; m2c closed Mad2 (or MCC); Km2c “kinetochore” Mad1-Mad2c; p p31comet; a APC/C; and : complex formation.
Note added in proof.
Work from Jia et al. (2011) has also replicated the delay due to loss of p31comet during an unperturbed mitosis.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health grants CA084179 and GM51464 (to P.K.S.) and GM77238 (to J.V.S.).
Abbreviations used:
- APC/C
anaphase-promoting complex/cyclosome
- CREST
calcinosis, Raynaud's, esophageal dysmotility, sclerodactyly, and telangiectasia
- EGFP
enhanced green fluorescent protein
- EYFP
enhanced yellow fluorescent protein
- FRAP
fluorescence recovery after photobleaching
- MCC
mitotic checkpoint complex
- mRFP
monomeric red fluorescent protein
- NBD
nuclear breakdown
- RNAi
RNA interference
- SAC
spindle assembly checkpoint
- siRNA
small interfering RNA
- t1/2
half recovery time
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E11-03-0216) on September 30, 2011.
REFERENCES
- Chen RH, Shevchenko A, Mann M, Murray AW. Spindle checkpoint protein Xmad1 recruits Xmad2 to unattached kinetochores. J Cell Biol. 1998;143:283–295. doi: 10.1083/jcb.143.2.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciliberto A, Shah JV. A quantitative systems view of the spindle assembly checkpoint. EMBO J. 2009;28:2162–2173. doi: 10.1038/emboj.2009.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Antoni A, et al. The Mad1/Mad2 complex as a template for Mad2 activation in the spindle assembly checkpoint. Curr Biol. 2005;15:214–225. doi: 10.1016/j.cub.2005.01.038. [DOI] [PubMed] [Google Scholar]
- DeLuca JG, Howell BJ, Canman JC, Hickey JM, Fang G, Salmon ED. Nuf2 and Hec1 are required for retention of the checkpoint proteins Mad1 and Mad2 to kinetochores. Curr Biol. 2003;13:2103–2109. doi: 10.1016/j.cub.2003.10.056. [DOI] [PubMed] [Google Scholar]
- Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001;411:494–498. doi: 10.1038/35078107. [DOI] [PubMed] [Google Scholar]
- Garnett MJ, Mansfeld J, Godwin C, Matsusaka T, Wu J, Russell P, Pines J, Venkitaraman AR. UBE2S elongates ubiquitin chains on APC/C substrates to promote mitotic exit. Nat Cell Biol. 2009;11:1363. doi: 10.1038/ncb1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habu T, Kim SH, Weinstein J, Matsumoto T. Identification of a MAD2-binding protein, CMT2, and its role in mitosis. EMBO J. 2002;21:6419–6428. doi: 10.1093/emboj/cdf659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardwick KG, Johnston RC, Smith DL, Murray AW. MAD3 encodes a novel component of the spindle checkpoint which interacts with Bub3p, Cdc20p, and Mad2p. J Cell Biol. 2000;148:871–882. doi: 10.1083/jcb.148.5.871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardwick KG, Weiss E, Luca FC, Winey M, Murray AW. Activation of the budding yeast spindle assembly checkpoint without mitotic spindle disruption. Science. 1996;273:953–956. doi: 10.1126/science.273.5277.953. [DOI] [PubMed] [Google Scholar]
- Hoffman DB, Pearson CG, Yen TJ, Howell BJ, Salmon ED. Microtubule-dependent changes in assembly of microtubule motor proteins and mitotic spindle checkpoint proteins at PtK1 kinetochores. Mol Biol Cell. 2001;12:1995–2009. doi: 10.1091/mbc.12.7.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell BJ, Hoffman DB, Fang G, Murray AW, Salmon ED. Visualization of Mad2 dynamics at kinetochores, along spindle fibers, and at spindle poles in living cells. J Cell Biol. 2000;150:1233–1250. doi: 10.1083/jcb.150.6.1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell BJ, Moree B, Farrar EM, Stewart S, Fang G, Salmon ED. Spindle checkpoint protein dynamics at kinetochores in living cells. Curr Biol. 2004;14:953–964. doi: 10.1016/j.cub.2004.05.053. [DOI] [PubMed] [Google Scholar]
- Hoyt MA, Totis L, Roberts BT. S. cerevisiae genes required for cell cycle arrest in response to loss of microtubule function. Cell. 1991;66:507–517. doi: 10.1016/0092-8674(81)90014-3. [DOI] [PubMed] [Google Scholar]
- Jaqaman K, et al. Kinetochore alignment within the metaphase plate is regulated by centromere stiffness and microtubule depolymerases. J Cell Biol. 2010;188:665–679. doi: 10.1083/jcb.200909005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia L, Li B, Warrington RT, Hao X, Wang S, Yu H. Defining pathways of spindle checkpoint silencing: functional redundancy between Cdc20 ubiquitination and p31comet. Mol Biol Cell. 2011;22:4227–4235. doi: 10.1091/mbc.E11-05-0389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kallio MJ, Beardmore VA, Weinstein J, Gorbsky GJ. Rapid microtubule-independent dynamics of Cdc20 at kinetochores and centrosomes in mammalian cells. J Cell Biol. 2002;158:841–847. doi: 10.1083/jcb.200201135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor TM, Mayer TU, Coughlin ML, Mitchison TJ. Probing spindle assembly mechanisms with monastrol, a small molecule inhibitor of the mitotic kinesin, Eg5. J Cell Biol. 2000;150:975–988. doi: 10.1083/jcb.150.5.975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulukian A, Han J, Cleveland D. Unattached kinetochores catalyze production of an anaphase inhibitor that requires a Mad2 template to prime Cdc20 for BubR1 binding. Dev Cell. 2009;16:105–117. doi: 10.1016/j.devcel.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Murray AW. Feedback control of mitosis in budding yeast. Cell. 1991;66:519–531. doi: 10.1016/0092-8674(81)90015-5. [DOI] [PubMed] [Google Scholar]
- Luo X, Tang Z, Xia G, Wassmann K, Matsumoto T, Rizo J, Yu H. The Mad2 spindle checkpoint protein has two distinct natively folded states. Nat Struct Mol Biol. 2004;11:338–345. doi: 10.1038/nsmb748. [DOI] [PubMed] [Google Scholar]
- Mapelli M, et al. Determinants of conformational dimerization of Mad2 and its inhibition by p31comet. EMBO J. 2006;25:1273–1284. doi: 10.1038/sj.emboj.7601033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mapelli M, Massimiliano L, Santaguida S, Musacchio A. The mad2 conformational dimer: structure and implications for the spindle assembly checkpoint. Cell. 2007;131:730–743. doi: 10.1016/j.cell.2007.08.049. [DOI] [PubMed] [Google Scholar]
- Martinez-Exposito MJ, Kaplan KB, Copeland J, Sorger PK. Retention of the BUB3 checkpoint protein on lagging chromosomes. Proc Natl Acad Sci USA. 1999;96:8493–8498. doi: 10.1073/pnas.96.15.8493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Lluesma S, Stucke VM, Nigg EA. Role of Hec1 in spindle checkpoint signaling and kinetochore recruitment of Mad1/Mad2. Science. 2002;297:2267–2270. doi: 10.1126/science.1075596. [DOI] [PubMed] [Google Scholar]
- McAinsh AD, Meraldi P, Draviam VM, Toso A, Sorger PK. The human kinetochore proteins Nnf1R and Mcm21R are required for accurate chromosome segregation. EMBO J. 2006;25:4033–4049. doi: 10.1038/sj.emboj.7601293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meraldi P, Draviam VM, Sorger PK. Timing and checkpoints in the regulation of mitotic progression. Dev Cell. 2004;7:45–60. doi: 10.1016/j.devcel.2004.06.006. [DOI] [PubMed] [Google Scholar]
- Musacchio A, Salmon ED. The spindle-assembly checkpoint in space and time. Nat Rev Mol Cell Biol. 2007;8:379–393. doi: 10.1038/nrm2163. [DOI] [PubMed] [Google Scholar]
- Reddy SK, Rape M, Margansky WA, Kirschner MW. Ubiquitination by the anaphase-promoting complex drives spindle checkpoint inactivation. Nature. 2007;446:921–925. doi: 10.1038/nature05734. [DOI] [PubMed] [Google Scholar]
- Shah JV, Botvinick E, Bonday Z, Furnari F, Berns M, Cleveland DW. Dynamics of centromere and kinetochore proteins; implications for checkpoint signaling and silencing. Curr Biol. 2004;14:942–952. doi: 10.1016/j.cub.2004.05.046. [DOI] [PubMed] [Google Scholar]
- Shelby RD, Hahn KM, Sullivan KF. Dynamic elastic behavior of alpha-satellite DNA domains visualized in situ in living human cells. J Cell Biol. 1996;135:545–557. doi: 10.1083/jcb.135.3.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonetta M, Manzoni R, Mosca R, Mapelli M, Massimiliano L, Vink M, Novak B, Musacchio A, Ciliberto A. The influence of catalysis on mad2 activation dynamics. PLoS Biol. 2009;7:e10. doi: 10.1371/journal.pbio.1000010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotillo R, Hernando E, Diaz-Rodriguez E, Teruya-Feldstein J, Cordon-Cardo C, Lowe SW, Benezra R. Mad2 overexpression promotes aneuploidy and tumorigenesis in mice. Cancer Cell. 2007;11:9–23. doi: 10.1016/j.ccr.2006.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudakin V, Chan GK, Yen TJ. Checkpoint inhibition of the APC/C in HeLa cells is mediated by a complex of BUBR1, BUB3, CDC20, and MAD2. J Cell Biol. 2001;154:925–936. doi: 10.1083/jcb.200102093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teichner A, Eytan E, Sitry-Shevah D, Miniowitz-Shemtov S, Dumin E, Gromis J, Hershko A. p31comet promotes disassembly of the mitotic checkpoint complex in an ATP-dependent process. Proc Natl Acad Sci USA. 2011;108:1–6. doi: 10.1073/pnas.1100023108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigneron S, Prieto S, Bernis C, Labbe JC, Castro A, Lorca T. Kinetochore localization of spindle checkpoint proteins: who controls whom? Mol Biol Cell. 2004;15:4584–4596. doi: 10.1091/mbc.E04-01-0051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vink M, et al. In vitro FRAP identifies the minimal requirements for Mad2 kinetochore dynamics. Curr Biol. 2006;16:755–766. doi: 10.1016/j.cub.2006.03.057. [DOI] [PubMed] [Google Scholar]
- Williamson A, Wickliffe KE, Mellone BG, Song L, Karpen GH, Rape M. Identification of a physiological E2 module for the human anaphase-promoting complex. Proc Natl Acad Sci USA. 2009;106:18213–18218. doi: 10.1073/pnas.0907887106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu T, Merbl Y, Huo Y, Gallop JL, Tzur A, Kirschner MW. UBE2S drives elongation of K11-linked ubiquitin chains by the anaphase-promoting complex. Proc Natl Acad Sci USA. 2010;107:1355–1360. doi: 10.1073/pnas.0912802107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia G, Luo X, Habu T, Rizo J, Matsumoto T, Yu H. Conformation-specific binding of p31(comet) antagonizes the function of Mad2 in the spindle checkpoint. EMBO J. 2004;23:3133–3143. doi: 10.1038/sj.emboj.7600322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M, Li B, Liu C-J, Tomchick DR, Machius M, Rizo J, Yu H, Luo X. Insights into Mad2 regulation in the spindle checkpoint revealed by the crystal structure of the symmetric Mad2 dimer. Plos Biol. 2008;6:e50. doi: 10.1371/journal.pbio.0060050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M, Li B, Tomchick DR, Machius M, Rizo J, Yu H, Luo X. p31(comet) blocks Mad2 activation through structural mimicry. Cell. 2007;131:744–755. doi: 10.1016/j.cell.2007.08.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.