

Cadherin–catenin interactions play an important role in cadherin adhesion. In the cadherin complex, α-catenin contributes to the binding strength of another catenin, p120, to the same complex. The data suggest that α-catenin–p120 contact within the cadherin–catenin complex can regulate cadherin trafficking.
Abstract
Cadherin–catenin interactions play an important role in cadherin-mediated adhesion. Here we present strong evidence that in the cadherin–catenin complex α-catenin contributes to the binding strength of another catenin, p120, to the same complex. Specifically, we found that a β-catenin–uncoupled cadherin mutant interacts much more weakly with p120 than its full-size counterpart and that it is rapidly endocytosed from the surface of A-431 cells. We also showed that p120 overexpression stabilizes this mutant on the cell surface. Examination of the α-catenin–deficient MDA-MB-468 cells and their derivates in which α-catenin was reintroduced showed that α-catenin reinforces E-cadherin–p120 association. Finally, a cross-linking analysis of the cadherin–catenin complex indicated that a large loop located in the middle of the p120 arm-repeat domain is in close spatial vicinity to the amino-terminal VH1 domain of α-catenin. The six amino acid–long extension of this loop, caused by an alternative splicing, weakens p120 binding to cadherin. The data suggest that α-catenin–p120 contact within the cadherin–catenin complex can regulate cadherin trafficking.
INTRODUCTION
Cadherin is a transmembrane adhesive receptor whose adhesive and signaling activities are under the control of cytosolic proteins collectively called catenins. These proteins associate with two distinct intracellular cadherin domains. The transmembrane-proximal (juxtamembrane) domain interacts with the p120 members of arm-repeat-domain proteins, whereas the carboxy-terminal domain associates with β-catenin or plakoglobin, two other members of the same protein family. The latter proteins link cadherin to α-catenin (Gumbiner, 1996; Provost and Rimm, 1999; Nelson, 2008). Catenins control cadherin adhesion through a variety of mechanisms, one of which is cadherin endocytosis (Bryant and Stow, 2004; Delva and Kowalczyk, 2009; Yap et al., 2007). Deregulation of this mechanism has been found in malignant tumors, in which cadherin is lost or relocalized to the cytosol (Carpenter et al., 2002; Reynolds and Carnahan, 2004). Although recent studies identified catenin structures and detailed their interactions with the cadherin intracellular region (Pokutta and Weis, 2007; Ishiyama et al., 2010), relatively limited information is available about the general organization of the cadherin–catenin complex and, in particular, about the interactions between p120 and other catenins within the same complex. Such interactions, however, may have an important regulatory function.
The expression of cadherin on the cell surface is controlled by a balance between exocytic and endocytic cadherin transport (Bryant and Stow, 2004; Delva and Kowalczyk, 2009). P120 is a key regulator of cadherin endocytosis. The binding of p120 to cadherin has been suggested to suppress endocytic elements—K738 and a dileucine motif—that are present at the juxtamembrane cadherin domain (Miyashita and Ozawa, 2007a; Hong et al., 2010). The absence of p120 exposes these motifs to clathrin adaptors, resulting in cadherin endocytosis and degradation (Xiao et al., 2003, 2005; Reynolds and Carnahan, 2004; Ishiyama et al., 2010). The blockage of clathrin-dependent endocytosis or the point inactivation of these endocytic motifs was shown to stabilize p120-uncoupled cadherin mutants on the cell surface and to rescue their adhesive function (Troyanovsky et al., 2007; Chiasson et al., 2009; Hong et al., 2010).
β-Catenin binding to cadherin has also been shown to be critical for cadherin stability and cell surface localization. A lack of β-catenin and plakoglobin (Fukunaga et al., 2005) or selective inactivation of the β-catenin–binding domain (Chen et al., 1999; Miyashita and Ozawa, 2007b) resulted in cadherin targeting to lysosomes and degradation. Remarkably, this cadherin destabilization is based on the same dileucine motif present in the p120-binding juxtamembrane domain. Point inactivation of this motif significantly increased the cell surface level of β-catenin–uncoupled cadherin mutants (Miyashita and Ozawa, 2007b). Although it is not clear how β-catenin regulates the dileucine motif, which is located at the juxtamembrane domain, this observation suggests some level of cooperation between two distinct catenin-binding cadherin domains. Some other observations also point to a cross-talk between p120 and β- or α- catenins. For example, p120 deficiency increased the severity of morphogenetic defects caused by a hypomorphic mutation in the α-catenin gene of Caenorhabditis elegans (Pettitt et al., 2003). It was also reported that overexpression of p120 rescues the adhesive properties of the β-catenin–uncoupled cadherin mutant in mouse L cells (Ohkubo and Ozawa, 1999) and that β-catenin–uncoupled mutants exhibit low binding to p120 in coimmunoprecipitation experiments (Shibamoto et al., 1995). Finally, our recent work suggests that p120 is positioned close to α-catenin in the E-cadherin–catenin complex (Kiss et al., 2008).
To better understand the cross-talk between p120 and other catenins, we studied the mechanisms of instability and abnormal localization of the β-catenin–uncoupled cadherin mutants. Our work suggests that weak interaction between p120 and α-catenin is important for preventing cadherin endocytosis. This interaction can be an important regulator of cadherin trafficking, RhoA activity, and various dynamic properties of adherens junctions.
RESULTS
Deletion of β-catenin–binding site destabilized cadherin on the cell surface
To understand the role of catenins in adherens junction dynamics, we studied A-431 cells stably expressing the E-cadherin nonfunctional mutant Ec1M-Δ844. This mutant harbored a 38 amino acid–long C-terminal deletion that encompassed a critical portion of the β-catenin–binding site (Supplemental Figure S1). This deletion did not affect the p120-binding site of E-cadherin mapped to the E-cadherin region Tyr-755–Leu-772 (Thoreson et al., 2000; Ishiyama et al., 2010). Double immunofluorescence microscopy, using anti-myc and anti–β-catenin antibodies, showed that this mutant was not recruited into the adherens junctions (Figure 1, A and A′). Instead, consistent with previous observations (Chen et al., 1999; Miyashita and Ozawa, 2007b), a large pool of this mutant was localized to the intracellular vesicles. To determine whether an increased rate of cadherin endocytosis contributed to this abnormal distribution, we examined the turnover of the Ec1M-Δ844 mutant using cell-surface biotinylation assays. Figure 2A shows that the Ec1M-Δ844 mutant degraded much more rapidly than did the endogenous cadherin present in the same cells. Furthermore, the surface-biotinylated Ec1M-Δ844 mutant was almost completely internalized during the 15-min period (Figure 2B). Only ∼5% of the endogenous cadherin was endocytosed during the same time. To exclude the role of the myc tag in such rapid internalization of the Ec1M-Δ844 mutant, we examined the endocytosis of the myc-tagged form of the full-size cadherin, Ec1M. These experiments showed that both myc-tagged and untagged cadherins present in Ec1M-expressing A-431 cells exhibited approximately the same internalization rates (Figure 2C). Therefore the ablation of the β-catenin–binding site destabilized E-cadherin on the cell surface.
FIGURE 1:

p120 overexpression recruits the Ec1M-Δ844 mutants into the junctions of A-431 cells. (A, A′) A-431 cells stably expressing β-catenin–uncoupled mutant Ec1M-Δ844 were double stained for the mutant (Δ844) using anti-myc antibody and for adherens junctions using anti–β-catenin (βCat) antibody. Note that the mutant mostly resides in the cytosol. (B, B′) The Ec1M-Δ844–expressing A-431 cells were stably transfected by the construct encoding p120-F under Mf-inducible promoter. After overnight induction, the cell were stained as in A and A′. Note that the p120-F induction targets the mutant into the adherens junctions. (C, C′) The same cells as in B and B′ but stained for the Ec1M-Δ844 mutant (Δ844) and for p120-F (p120F) using anti-FLAG antibody. Note a codistribution of the mutant and p120-F. (D, D′) A-431 cells coexpressing the Ec1M-Δ844 mutant and p120-ARM-F stained for the mutant (Δ844) by anti-myc and for p120-ARM-F (arm-F) by anti-FLAG. Note that expression of p120-ARM-F is sufficient to target the Ec1M-Δ844 mutant into the cell–cell contacts.
FIGURE 2:
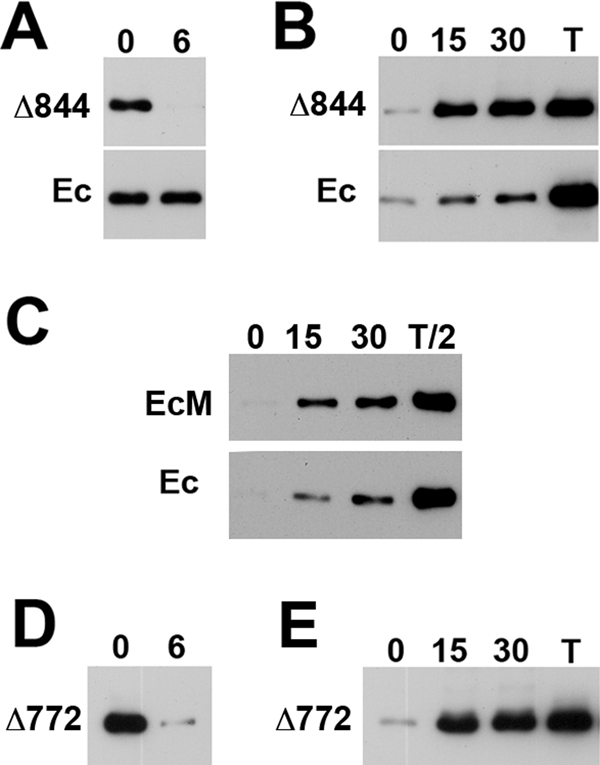
Degradation and internalization rates of the β-catenin–uncoupled cadherin mutants in A-431 cells. (A) Degradation rates of the Ec1M-Δ844 mutant (Δ844) and endogenous cadherin (Ec) in Ec1M-Δ844–expressing A-431 cells as measured by a biotin degradation assay. Lane 0 shows the amount of the biotinylated proteins at time 0; lane 6 shows their amounts after 6 h. Note a complete degradation of the mutant but not the endogenous cadherin during the chase. (B) The internalization rates of the Ec1M-Δ844 mutant (Δ844) and endogenous cadherin (Ec) in Ec1M-Δ844–expressing cells as measured by a biotin internalization assay. Numbers above the lanes show chase periods (min). T, total amount of the biotinylated proteins. Note that nearly an entire pool of the biotinylated Ec1M-Δ844 mutant but only minor fraction of the endogenous cadherin pool was internalized during 15 min. (C) The biotin internalization assay with Ec1M-expressing A-431 cells. Experiment was performed as in B, but lanes containing total biotinylated proteins (lanes T/2) were loaded with half the volume of the other lanes. Note that the internalization rates of the endogenous and myc-tagged forms of cadherin are approximately the same. (D) Degradation and (E) internalization rates of the EcM-Δ772 cadherin mutant. The experiments were performed as in A and B, respectively. Note that degradation and internalization rates of this mutant are very similar to those of the Ec1M-Δ844 mutant.
The binding of p120 to cadherin had been proposed to suppress endocytic signals present in the juxtamembrane domain (Miyashita and Ozawa, 2007a; Delva and Kowalczyk, 2009; Hong et al., 2010). The rapid endocytosis and degradation of the Ec1M-Δ844 mutant, which contains an intact juxtamembrane domain, has two possible causes. First, the remaining portion of the β-catenin–binding site in the Ec1M-Δ844 mutant may expose additional endocytic signals, which become available for endocytic machinery once the β-catenin–binding site is obliterated. Indeed, this portion contains several motifs sharing some similarities to the Tyr-based endocytic motifs of other proteins (Chen et al., 1999). Second, despite the presence of the intact p120-binding site, the binding of the Ec1M-Δ844 mutant to p120 could be compromised.
To test the first possibility—whether the remaining portion of the β-catenin–binding site exhibits additional endocytic signals—we examined the internalization of another cadherin mutant, EcM-Δ772. This mutant has the intact juxtamembrane domain but lacks all of the portions of the β-catenin–binding site that had been mapped between E-cadherin residues 782 and 881 (Supplemental Figure S1; Huber and Weis, 2001). We found that this mutant had essentially the same localization, internalization rate, and turnover rate (Figure 2, D and E) as did the Ec1M-Δ844 mutant. Therefore the remaining portion of the β-catenin–binding site is not essential for the mutant endocytosis. The fast turnover of the Ec1M-Δ844 mutant apparently is mediated by the endocytic signals present in the juxtamembrane domain. This finding is intriguing because it indicates that the juxtamembrane domain can sense the abnormalities in the β- or α-catenin binding to cadherin.
Overexpression of p120 protected the Ec1M-Δ844 mutant from degradation
The simplest explanation for the data described is that the binding between p120 and β-catenin–uncoupled cadherin mutants is weak and is not sufficient to block the endocytic signals located in the mutant's juxtamembrane domain. To test this hypothesis, we examined whether p120 overexpression rescued junctional localization of the Ec1M-Δ844 mutant. We overexpressed FLAG-tagged p120-3AB isoform (p120-F) in the Ec1M-Δ844–expressing cells using a mifepristone (Mf) inducible system. In A-431 cells, as we showed previously (Klingelhöfer et al., 2003), the recombinant proteins began to accumulate 2 h after Mf administration and reached a plateau at ∼6 h. The overnight p120 induction notably increased the total level of the Ec1M-Δ844 mutant (Figure 3A). Biotinylation experiments revealed that such induction also stabilized the mutant on the cell surface (Figure 3B) and significantly decreased its internalization rate (Figure 3C). Of interest, such dramatic changes were specific to Ec1M-Δ844 and were not noted for endogenous cadherin (Figure 3, A–C, blots Ec). Finally, in the p120-overexpressing cells, the mutant became coclustered with the endogenous cadherin–catenin complex in the cell–cell junctions (Figure 1, B and C). Thus a high level of p120 prolonged the lifetime of the EcM-Δ844 mutant and facilitated its recruitment into cell–cell junctions. Of importance, this effect required direct binding of p120 to the mutant because it was completely abolished by the inactivation of the mutant's p120-binding site (Supplemental Figures S1 and S2).
FIGURE 3:
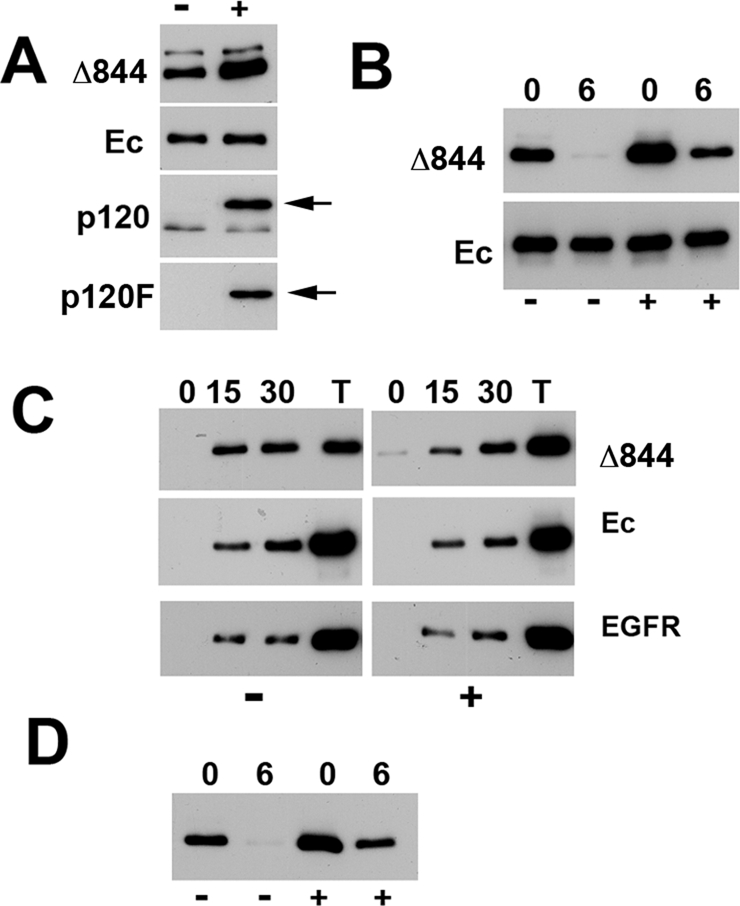
p120 overexpression stabilizes the Ec1M-Δ844 mutant. (A) Cell lysates of Ec1M-Δ844–expressing cells with (+) or without (−) overnight induction of p120-F were analyzed for the total levels of the mutant (Δ844), the endogenous E-cadherin (Ec), p120 (p120), and p120-F (p120F) using antibodies against myc, E-cadherin, p120, and FLAG, correspondingly. Arrows, p120-F. Note that the expression of p120-F increased the level of the Ec1M-Δ844 mutant but not that of the endogenous E-cadherin. (B) Degradation rates of the Ec1M-Δ844 mutant (Δ844) and endogenous cadherin (Ec) in response to p120-F induction. See Figure 2A for other abbreviations. (C) Internalization rates of Ec1M-Δ844 (Δ844), endogenous E-cadherin (Ec), and EGF receptor (EGFR) with (+) or without (–) the 120-F induction. Abbreviations as in Figure 2B. Note that in uninduced cells (left), the amounts of the internalized mutant are comparable with the total amount of the surface-biotinylated cadherin (lane T). P120-F induction (right) significantly decreases Ec1M-Δ844 mutant endocytosis. Note also that the rates of endocytosis of other tested proteins, endogenous E-cadherin, and EGF receptor were unaffected. (D) Degradation rate of the Ec1M-Δ844 mutant in response to the expression of p120-ARM-F. Abbreviations as in B.
It is possible that the amino-terminal or carboxy-terminal domains of p120-3AB caused this dramatic effect. These domains are known for altering cadherin dynamics in cell–cell junctions (Xia et al., 2006; Fukumoto et al., 2008). Another possibility is that the elevated level of p120 increased p120 binding to the cadherin mutant, thereby shielding the mutant from endocytosis. To test these possibilities, we repeated our overexpression experiments using a construct that encodes the arm-repeat domain of p120 (p120-ARM-F). Because this domain is sufficient to bind cadherins (Ireton et al., 2002), it would also protect the Ec1M-Δ844 mutant from degradation if its degradation is based on accessibility of the cadherin endocytic signals located in the juxtamembrane domain. Indeed, the subcellular localization of the Ec1M-Δ844 mutant (Figure 1D) and its stability on the cell surface (Figure 3 D) in cells overexpressing p120-ARM-F and p120-F were indistinguishable. These data suggested that abnormally weak interactions between this mutant and p120 caused the high instability of the Ec1M-Δ844 mutant and its poor recruitment into the junctions.
The EcM-Δ844 mutant has low affinity to p120 due to the absence of α-catenin in the complex
To verify the idea that the deletion of the β-catenin–binding site weakens p120 binding to cadherin, we probed p120-cadherin interactions by anti-myc coimmunoprecipitation assay. We recently showed that the digitonin cell lysis buffer, in contrast to Triton X-100–based buffers, preserves well the cadherin–p120 interaction (Kiss et al., 2008). Figure 4A shows that the Ec1M-Δ844 mutant did coimmunoprecipitate a slightly smaller amount of p120 than Ec1M. This difference became much more dramatic upon the exposure of the immunoprecipitates to high salt or to Triton-X100: p120 almost completely dissociated from Ec1M-Δ844 but not from Ec1M by washing buffers containing 0.45 M NaCl or 0.2% Triton X-100 (Figure 4A).
FIGURE 4:
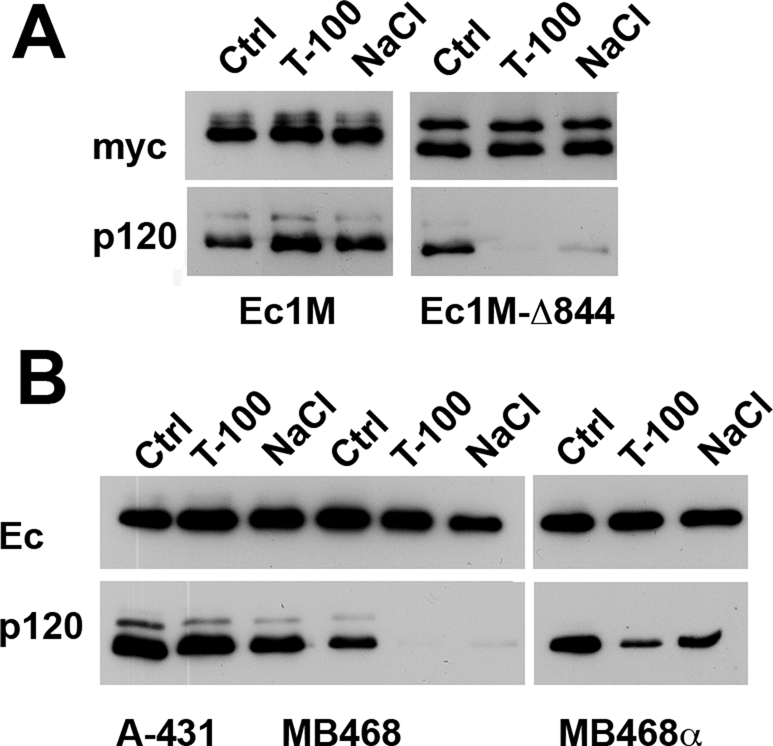
Presence of α-catenin in the cadherin–catenin complex strengthens cadherin–p120 interactions. (A) Digitonin cell lysates of the Ec1M- and EcM-Δ844–expressing cells (Ec1M and Ec1M-Δ844, respectively) were immunoprecipitated by the anti-myc antibody, and the resulting immunoprecipitates were washed with the digitonin buffer (Ctrl) or the same buffer containing either 0.2% Triton X-100 (T-100) or 0.45 M NaCl (NaCl). The immunoprecipitates were probed using anti-myc (myc) or anti-p120 (p120) antibodies. Note that the addition of Triton X-100 or NaCl into the washing buffer nearly completely removed p120 from the Ec1M-Δ844 but not Ec1M immunoprecipitates. (B) The endogenous E-cadherin was immunoprecipitated as in A using anti-E-cadherin SHE78-7 antibody from α-catenin–positive A-431 cells (A431), α-catenin–deficient MDA-MB-468 cells (MB468), and MDA-MB-468α cells (MBD468α), in which α-catenin expression was reconstituted. Blots were stained for the immunoprecipitated E-cadherin (Ec) or coimmunoprecipitated p120 (p120).
Our previous cross-linking studies showed that p120 and α-catenin are in close spatial contact within the E-cadherin–catenin complex (Kiss et al., 2008). On the basis of this observation, we proposed that these two proteins physically interact and that this interaction contributes to the strength of cadherin–p120 binding. The lack of α-catenin association with the Ec1M-Δ844 mutant could, therefore, weaken the binding of this mutant to p120. To reveal the role of α-catenin in cadherin–p120 interactions, we studied the E-cadherin–catenin complex in MDA-MB-468 cells. These cells are α-catenin deficient but express normal amounts of E-cadherin, β-catenin, plakoglobin, and p120 (Supplemental Figure S3A; see also Hazan et al., 1997; Hiraguri et al., 1998). Despite the absence of α-catenin, these cells exhibited adherens junction–like structures but were unable to form normal epithelial colonies (Supplemental Figure S3B). Reexpression of α-catenin restored the epithelial phenotype of these cells, as well as resulted in a more organized distribution of adherens junctions, desmosomes, and tight junctions (Supplemental Figure S3B). Figure 4B shows that an anti–E-cadherin antibody immunoprecipitated approximately the same amounts of E-cadherin–p120 complex from the A-431 and MDA-MB-468 cell lysates. However, Triton X-100 (0.2%) or NaCl (0.45 M) completely removed p120 from the E-cadherin–catenin complex obtained from MDA-MB-468 cells but not from A-431 cells. Finally, we showed that p120–cadherin interactions in MDA-MB-468 cells strengthened upon α-catenin reconstitution (Figure 4B).
The loop between arm repeats 5 and 6 of p120 is in contact with VH1 domain of α-catenin
p120-3AB contains 10 cysteine residues (numbered from 1 to 10 in Figure 5A), all of which are present in its arm-repeat domain. Cysteine-specific cross-linking of p120 to α-catenin suggested that at least one of these cysteines is in close proximity to α-catenin (Kiss et al., 2008). To identify such residue(s), we constructed and expressed in A-431 cells 10 p120-F point mutants, each of which harbored a point Ala substitution for one of these cysteines (Figure 5A). The A-431 subclones were selected to express approximately the same levels of these p120 mutants upon overnight Mf induction. An anti-FLAG coimmunoprecipitation assay showed that all of the mutants, except mutant p120Cys4-F, formed complexes with E-cadherin (Figure 5B). In parallel experiments, the mutant-expressing cells were lysed and cross-linked with cysteine-specific cross-linker BM[PEO]3. Western blot analyses of these lysates using anti-FLAG antibody showed that most of the mutants, as predicted, produced a high–molecular weight band corresponding to the adduct consisting of a p120 mutant and α-catenin (Figure 5C). As expected, the mutant p120Cys4-F did not form such an adduct because of its cadherin-binding defect. The only other mutant that was unable to produce this adduct was p120Cys9-F. Because this mutant had no defects in binding to E-cadherin, one may suggest that Cys9 (Cys618 according to NP_001078927.1), which resides within a large loop located between p120 arm repeats 5 and 6 (5/6 loop), actually mediates cross-linking between p120 and α-catenin.
FIGURE 5:

Identification of protein domains involved in cross-linking of p120 and α-catenin by BM[PEO]3. (A) A cysteine map of p120-F. The protein consists of three major domains—the central arm-repeat domain (individual arm repeats are depicted by open boxes, numbered) flanked by the amino-terminal (N) and carboxy-terminal (C) domains. Note that all 10 cysteines of p120 (indicated by numbered arrows) are located at the arm-repeat domain, one of which, Cys-9, is present in a large loop separating arm repeats 5 and 6. This loop may contain a small insert encoding by the alternative exon C (filled circle). FLAG epitope (F) is indicated by an open circle. (B) Recruitment of the p120-F cysteine mutants into the cadherin–catenin complex. The p120-F mutants were expressed in A-431 cells and immunoprecipitated with anti-FLAG agarose. The immunoprecipitates were probed for the p120 mutants (p120 mutants; the number of the mutant is indicated above the lanes) and for α-catenin (α-cat) using anti-FLAG or anti–α-catenin antibodies. Note that except for Cys-4, all Cys mutants interact with α-catenin. (C) Digitonin cell lysates of A-431 cells expressing p120-F (left) and its different cysteine mutants (right; the mutant's numbers are indicated above the lanes) were cross-linked by BM[PEO]3 and analyzed by Western blotting for p120-F or its mutants (p120F, p120F-mutants) and for α-catenin (α-cat). Note that the addition of the cross-link (+) resulted in appearance of p120–α-catenin adducts (arrow) that were absent in the control lysates (−). Arrowhead, p120 monomers. Note also that only two Cys mutants, Cys-4 and Cys-9, did not produce such adducts. (D) A cysteine map of the α-catenin-myc protein. The protein consists of three major domains—VH1, VH2, and VH3—separated by linker regions. Myc is indicated by an open circle (M). The protein contains 12 cysteine residues (arrows) located at the four regions. Four Cys mutants were constructed (numbered), each of which lacked all cysteines in a specific region. (E) The anti-myc coimmunoprecipitation assay with A-431 cells expressing Cys mutants of α-catenin-myc. Note that all Cys mutants (indicated by numbers) are recruited into the E-cadherin–catenin complex. Mutants and E-cadherin–catenin complex were stained by anti-myc (myc) and anti–β-catenin (β-cat), respectively. (F) Cell lysates of A-431 cells expressing different α-catenin Cys mutants were cross-linked and analyzed as in C. Blot was stained by anti-myc (myc). Note that only the mutant 1, lacking cysteines in the VH1 domain, does not show a high–molecular weight band (arrow).
In reverse experiments, we identified a domain of α-catenin that is in proximity to p120. The intact α-catenin contains 12 Cys residues, which are located in four different regions: in its three vinculin-homology domains (VH1-VH3) and in the VH1-VH2-linker region (Figure 5D). To simplify our experiments, we constructed four α-catenin myc-tagged mutants, each of which lacked all cysteines within one of these regions (see Figure 5D for detail). A-431 clones expressing these α-catenin mutants were first used in anti-myc coimmunoprecipitation experiments that verified that all four mutants were equally recruited into the cadherin–catenin complex (Figure 5E). Digitonin lysates of these cells were then cross-linked and analyzed by Western blotting as described earlier. This experiment (Figure 5F) indicated that at least one of the cysteines present in the VH1 domain of α-catenin is responsible for cross-linking to p120.
The C exon weakens binding of p120 to the intact E-cadherin but not to its β-catenin–uncoupled mutant
Of interest, alternative splicing of exon C produces p120C isoforms that contain a small, six amino acid–long insertion (Asp-625–Arg-630) approximately in the middle of the 5/6 loop, just a few amino acids away from Cys-618. To address the question of whether the exon C changes the structural organization of the cadherin–catenin complex, we expressed the p120-3ABC isoform of p120 (p120C-F) in A-431 cells and studied its subcellular localization and its interaction with cadherin. Remarkably, this small insertion dramatically changed both of these parameters. In contrast to p120-F, which predominantly localized at the adherens junctions (Figure 6A), the major pool of p120C-F was cytosolic (Figure 6B). Consistent with such abnormal distribution, only a small fraction of p120C-F coimmunoprecipitated with E-cadherin (Figure 7, A and B). Furthermore, the cadherin–p120C-F complex, which could be extracted from the cells, appeared to be hypersensitive to Triton X-100 or NaCl (Figure 7B). Taken together, these data suggest that the insertion encoded by exon C weakens p120 binding to cadherin by preventing intracomplex p120–α-catenin interactions.
FIGURE 6:

Role of the 5/6 loop in p120 subcellular distribution. A-431 cells expressing either (A) p120-F (p120F) or (B) p120C-F (p120CF) isoforms were stained with anti-FLAG antibody. Note that the p120C-F mutant is predominantly cytosolic. (C, C′, D, D′) The p120C-F isoform of p120 was expressed in Ec1M-Δ844–expressing A-431 cells under Mf-inducible promoter. Control (C, C′) or Mf-induced (D, D′) cells were double stained for (C, D) Ec1M-Δ844 (Δ844) and for (C′, D′) adherens junctions (β-cat) using anti-myc and anti–β-catenin antibodies. Note that the Mf induction redistributed the cadherin mutant toward adherens junctions.
FIGURE 7:
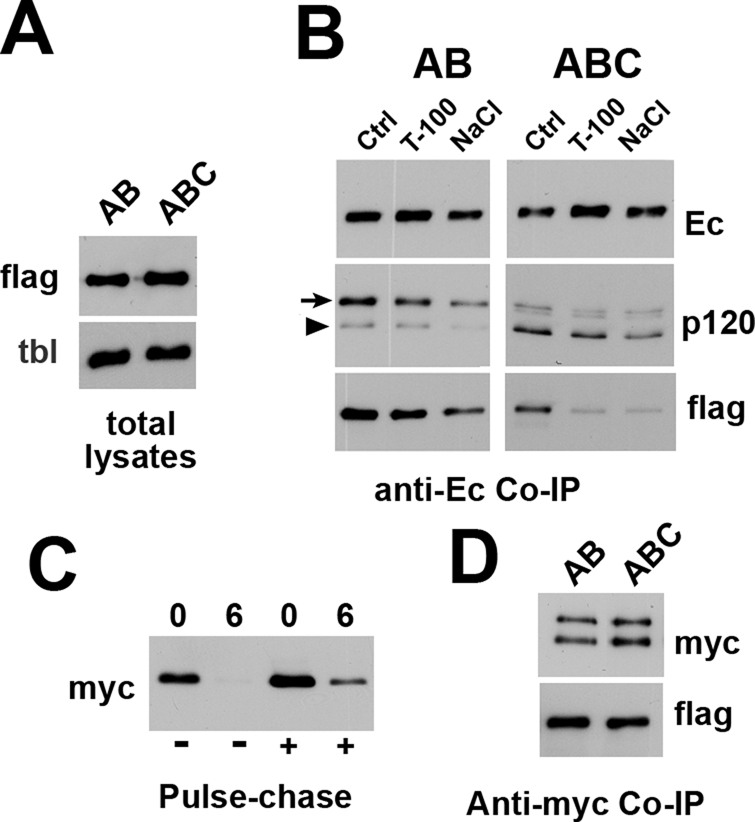
Carboxy-terminal truncation of E-cadherin abrogates differences in binding of cadherin to p120-F and p120C-F. (A) Total lysates of the Mf-induced A-431 cells expressing p120-F (lane AB) and p120C-F (lane ABC) proteins (FLAG). Anti-tubulin (tbl) staining is a loading control. Note that the expression levels of p120F and p120C-F are similar. (B) p120-F–expressing (AB column) and p120C-F–expressing (ABC column) A-431 cells were immunoprecipitated using anti–E-cadherin (SHE78-7) antibody and the resulting immunoprecipitates were washed with the digitonin IP buffer alone (Ctrl) or containing 0.2% Triton X-100 (T-100) or 0.45 M NaCl (NaCl). The blots were stained for E-cadherin (Ec), p120 (p120), and FLAG (flag) using corresponding antibodies. The FLAG-tagged and endogenous forms of p120 are indicated by an arrow and an arrowhead, respectively. Note that p120-F is the major form of p120 in the cadherin–catenin complex in the p120-F–expressing cells. By contrast, the amounts of p120C-F form are negligible. The C exon–encoded insert also destabilizes p120–cadherin interactions in 0.2% Triton or in 0.45 M NaCl. (C) Degradation rate of the Ec1M-Δ844 mutant in response to p120C-F induction. See Figure 3 for abbreviations. (D) A-431 cells coexpressing the Ec1M-Δ844 mutant and either p120-F (AB) or p120C-F (ABC) were immunoprecipitated by anti-myc and probed with anti-myc (myc) for the presence of the cadherin mutant and anti-FLAG (flag) for the presence of the FLAG-tagged p120 isoforms.
To substantiate this hypothesis, we tested whether the p120-C isoform is able to cluster β-catenin–uncoupled cadherin mutant Ec1M-Δ844. Indeed, if the exon C insert reduced p120 binding to cadherin through a defect in p120 binding to α-catenin, this exon would not change p120 binding to the β-catenin–uncoupled cadherin mutants. In agreement with this hypothesis, overexpression of the p120C-F in Ec1M-Δ844–expressing cells produced exactly the same effect as expression of p120-F: it stabilized the cadherin mutant on the cell surface (Figure 7C) and triggered clustering of the mutant at cell–cell junctions (Figure 6C, D). Furthermore, coimmunoprecipitation experiments revealed no differences in interactions between these forms of p120 and the Ec1M-Δ844 mutant (Figure 7D). These results showed that the inactivation of the β-catenin–binding site of cadherin completely eliminates the differences in cadherin binding to p120-3AB and p120-3ABC isoforms of p120.
DISCUSSION
Cadherin trafficking has emerged as one of the central regulatory processes of cadherin adhesion (Bryant and Stow, 2004; Troyanovsky et al., 2006; Delva and Kowalczyk, 2009). The binding of p120 to the juxtamembrane cadherin domain is the key regulatory step in this regulatory mechanism. In the p120-free state, the juxtamembrane domain initiates cadherin internalization (Xiao et al., 2003, 2005; Davis et al., 2003). At least two endocytic motifs that are involved in cadherin endocytosis were recently identified in this domain (Miyashita and Ozawa, 2007a; Hong et al., 2010). Point mutation of one of them, the dileucine motif, was shown to prevent cadherin internalization in the absence of p120, suggesting that p120 binding obstructs this motif from being recognized by the endocytic machinery. Much less is known about the role of β-catenin (or plakoglobin) in cadherin trafficking. In the absence of these catenins or upon deletion of the cadherin carboxy-terminal β-catenin–binding domain, cadherin was shown to be retained in the intracellular compartments and targeted to lysosomes (Chen et al., 1999; Fukunaga et al., 2005; Miyashita and Ozawa, 2007b). Surprisingly, the same dileucine motif appears to orchestrate lysosomal targeting of such cadherin mutants (Miyashita and Ozawa, 2007b). This observation suggests a potentially very important regulatory process: a cross-talk between two functionally distinct cadherin domains—the juxtamembrane, p120-binding domain and the carboxy-terminal, β-catenin–binding domain. The present work addresses the mechanism of this cross-talk and illuminates its role in the regulation of cadherin turnover.
In agreement with previous observations (Chen et al., 1999; Miyashita and Ozawa, 2007b), we found that the β-catenin–uncoupled cadherin mutant Ec1M-Δ844 is mostly intracellular and unstable. However, in contrast to previous experiments performed on MDCK cells, our experiments with A-431 cells showed that these abnormalities are based on the rapid endocytosis of this mutant. This is evident from the fact that nearly the entire surface-biotinylated pool of the mutant became intracellular in 15 min, whereas only a small fraction of the endogenous cadherin was internalized during the same period. It is possible that MDCK cells internalize β-catenin–uncoupled cadherin mutants almost instantly, thereby preventing the mutant detection on the cell surface in previous experiments. Of importance, we found that overexpression of p120 in A-431 cells stabilizes the Ec1M-Δ844 mutant on the cell surface. This stabilization requires a direct binding of p120 to the mutant because the stabilization effect is completely abolished by inactivation of the mutant p120-binding site. In addition, the arm-repeat domain of p120 that was shown to be sufficient for cadherin binding (Ireton et al., 2002) is as effective as full-size p120 in stabilizing the Ec1M-Δ844 mutant.
These data clearly show that the endogenous level of p120 is not sufficient to maintain the cell surface stability of the β-catenin–uncoupled cadherin mutants. The simplest explanation for this phenomenon is that such mutants have a reduced affinity to p120. In this case, the juxtamembrane domain of the mutants exhibits elevated endocytic activity because the mutants cannot compete with the endogenous cadherin for the limited pool of p120. We first tested this hypothesis by comparing p120 binding to Ec1M and Ec1M-Δ844. Indeed, we found that washing buffers containing 0.45 M NaCl or 0.2% Triton-X100 strongly reduced the amounts of p120 in the complex with Ec1M-Δ844 but not with Ec1M. This finding shows that, in addition to the minimal p120-binding domain, which is the same in Ec1M-Δ844 and Ec1M, cadherin exhibits another activity controlling p120 binding. Such activity might be associated either directly with the cadherin region deleted in the Ec1M-Δ844 mutant or with α- or β-catenins. Our experiments with α-catenin–deficient MD-MB-468 cells clearly demonstrated that α-catenin is a key factor stabilizing cadherin–p120 interactions. Taken together with the fact that p120 and α-catenin can be cross-linked within the same cadherin–catenin complex (Kiss et al., 2008), our data suggest that a direct interaction between these two proteins reinforces cadherin–p120 association. The α-catenin–p120 interaction can be weak and thus be significant only in the contents of the same complex. The potential weakness of this interaction can explain why it failed to be detected in previous direct in vitro binding experiments (Jou et al., 1995).
To gain further structural insights into the α-catenin–p120 interaction, we mapped the cysteine residues that are responsible for the cross-linking between p120 and α-catenin. Cysteine-specific mutagenesis of these two proteins showed that a large loop located between arm repeats 5 and 6 of the p120 arm-repeat domain is in close contact with the VH1 domain of α-catenin. This result was unexpected because published experiments with various p120 deletion mutants in SW48 cells showed that the 5/6 loop is not essential for p120 binding to E-cadherin (Ireton et al., 2002). It is possible, however, that the p120-deficient SW48 cell system, in which p120 mutants do not compete with the endogenous p120, did not allow minor abnormalities in the cadherin–p120 binding to be detected. Therefore we reinvestigated the role of the 5/6 loop in the cadherin–p120 interactions in A-431 cells expressing the endogenous p120. We used two splice forms of p120: p120-ABC and p120-AB. The p120-ABC splice form contains an additional six amino acid–long insertion in the middle of the 5/6 loop. Our experiments clearly showed that the C exon–encoded insertion reduces p120–cadherin binding: in contrast to the p120-AB, the p120-ABC form of p120 cannot compete with endogenous p120 for binding to cadherin. As a result, it is cytosolic, whereas p120-AB is recruited into cell–cell junctions. Of importance, both p120 splice forms equally bind to the β-catenin–uncoupled Ec1M-Δ844 cadherin mutant and equally prevent its endocytosis. This observation is fully consistent with structural works showing that the 5/6 loop is flexible and does not directly contribute to the E-cadherin–binding interface of p120 (Choi and Weis, 2005; Ishiyama et al., 2010). Taken together, our experiments suggest a possibility that the 5/6 loop serves as a molecular “bridge” between p120 and α-catenin reinforcing p120–cadherin association.
This finding is very interesting in light of the role of p120 as an inhibitor of RhoA. Several studies showed that the 5/6 loop is critical for binding to RhoA. These studies also show that p120 bindings to cadherin and to RhoA are mutually exclusive (Anastasiadis et al., 2000; Charrasse et al., 2002; Yanagisawa et al., 2008). Identification of the 5/6 loop as a partner of the α-catenin in the cadherin–catenin complex suggests a simple model for these mutually exclusive interactions: the association of p120 with cadherin results in the engagement of the 5/6 loop with α-catenin, thereby preventing p120 from interacting with RhoA. However, taking into consideration the data suggesting that RhoA may also directly interact with α-catenin (Magie et al., 2002), we see that the interplay between RhoA, p120, and α-catenin can be much more complex.
It is also notable that in the cadherin–catenin complex, the 5/6 loop of p120 is in close contact with the VH1 domain of α-catenin. This α-catenin domain is known to participate in two other mutually exclusive events: in α-catenin homodimerization and in α-catenin binding to β-catenin (Pokutta and Weis, 2007; Nelson, 2008). It has been proposed that the interplay between these two events mediates dynamic interactions between cadherin and the cytoskeleton. Therefore the 5/6 loop connection to the VH1 domain can potentially contribute to the switch between homodimerization and β-catenin–binding modes of α-catenin. New structural and biochemical studies are essential to uncovering the intricate network of interactions among various components of the cadherin–catenin complex.
In conclusion, our examination of the β-catenin–uncoupled cadherin mutants shows that α-catenin contributes to the strength of p120 association with the cadherin–catenin complex. We also showed that the 5/6 loop of p120, which was previously identified as the binding site for RhoA, actually appears to reinforce p120 binding to cadherin through direct contact with α-catenin. Such a network of weak catenin–catenin interactions in the cadherin–catenin complex may play a critical role in many dynamic processes regulating cadherin endocytosis and cadherin anchorage to the actin cytoskeleton.
MATERIALS AND METHODS
Plasmid construction, cell culture, DNA transfections, and antibodies
A-431 (human epidermoid carcinoma) and MDA-MB-468 (human breast carcinoma) cells were cultured in DMEM containing 10% fetal calf serum. MDA-MB-468α cells expressing α-catenin have been described (Klingelhöfer et al., 2003). Stable clones of A-431 expressing Ec1M, EcM-Δ748, and Ec1M-Δ844 cadherin mutants were used in our previous work (Chitaev and Troyanovsky, 1998). Cadherin sequences are numbered according to Z13009. Ec1M and Ec1M-D844 proteins contained a small, 18-residue-long internal deletion between p120 and catenin-binding sites (Chitaev and Troyanovsky, 1998). This deletion does not change cadherin adhesive and binding properties. The human cDNAs of p120-3AB and p120-3ABC (kindly provided by A. Reynolds, Vanderbilt University, Nashville, TN) were tagged C-terminally by FLAG epitope–encoding sequence and expressed using the mifepristone-inducible GeneSwitch System (Invitrogen, Life Technologies, Carlsbad, CA). A plasmid expressing p120-ARM-F was constructed using PCR-directed mutagenesis. The PCR-derived portions of the resulting constructs were verified by the sequencing. The expression of p120-Fl was induced by adding mifepristone at a final concentration of 10 nM.
Stable transfection using plasmid DNA and immunofluorescence microscopy of the cells were performed as described (Troyanovsky et al., 2006). The following mouse antibodies were used: anti–E-cadherin (C20820), anti-p120, anti–β-catenin, and anti–epidermal growth factor (EGF) receptor (all from BD Bioscience, San Jose, CA); anti–α-catenin and anti–E-cadherin SHE78-7 (Zymed Laboratories, South San Francisco, CA); anti–myc 9E10 (Covance, Princeton, NJ); and anti-FLAG (Sigma-Aldrich, St. Louis, MO). Anti-myc rabbit antibody (Santa Cruz Biotechnology, Santa Cruz, CA) was used for double staining.
Biotinylation of cell surface proteins
The cells of one 6-cm dish reaching near-confluent growth were washed with ice-cold phosphate-buffered saline (PBS) containing 0.2 mM CaCl2 (PBS-Ca). The plate was incubated at 4°C with 2 ml of 0.5 mg/ml sulfo-NHC-LC-biotin (Pierce Chemical, Rockford, IL) in PBS-Ca for 10 min. The reaction was stopped by washing the cells in ice-cold glycine solution (200 mM glycine/200 mM Tris, pH 7.5). To determine the rate of the biotinylated protein degradation (biotin degradation assay), cells were then chased in the same media for the indicated periods of time. At the end of the chase, cells were lysed in IP-lysis buffer containing 0.2% SDS and 1% Triton X-100. Biotinylated proteins were then precipitated by streptavidin–agarose and analyzed by immunoblotting. The overall rate of the mutant endocytosis was determined essentially as described earlier (biotin internalization assay; Troyanovsky et al., 2006). In brief, cell surface proteins were biotinylated by sulfo-NHC-LC-SS-biotin (Pierce Chemical) as indicated earlier. The cells were then incubated in standard medium (37°C) for various durations to resume endocytosis. Noninternalized biotin was then stripped from the surface by two 20-min-long washes with 50 mM glutathione at 4°C. Remaining biotinylated proteins were then analyzed as indicated earlier.
Immunoprecipitation
To analyze cadherin–p120 interactions, we used a digitonin-based coimmunoprecipitation assay, which preserves these interactions (Kiss et al., 2008). In brief, cells were extracted with 1 ml of 1% digitonin (Calbiochem, San Diego, CA)–containing immunoprecipitation (IP-D) lysis buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 2 mM EDTA, and 0.5 mM 4-(2-aminoethyl)-benzenesulfonyl fluoride) and then subjected to subsequent incubations, first with either anti-myc (9E10) or anti–E-cadherin (SHE78-7) antibodies and then with protein A–Sepharose. The immunoprecipitation of p120-F was performed using anti-FLAG agarose (Sigma-Aldrich). To analyze the strength of the cadherin–p120 interaction, the resulting immunoprecipitates were incubated for 5 min with 500 μl of the IP-D buffer supplemented with additional amounts of NaCl (final concentration 0.45 M) or with 0.2% Triton X-100.
Supplementary Material
Acknowledgments
We are grateful to A. Reynolds for providing p120 cDNA. This work was supported by Grants AR044016 and AR057992 from the National Institutes of Health.
Abbreviations used:
- BM[PEO]3
1,8-bis-maleimidotriethyleneglycol
- Mf
mifepristone
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E11-03-0250) on September 21, 2011.
REFERENCES
- Anastasiadis PZ, Moon SY, Thoreson MA, Mariner DJ, Crawford HC, Zheng Y, Reynolds AB. Inhibition of RhoA by p120 catenin. Nat Cell Biol. 2000;2:637–644. doi: 10.1038/35023588. [DOI] [PubMed] [Google Scholar]
- Bryant DM, Stow JL. The ins and outs of E-cadherin trafficking. Trends Cell Biol. 2004;14:427–434. doi: 10.1016/j.tcb.2004.07.007. [DOI] [PubMed] [Google Scholar]
- Carpenter PM, Al-Kuran RA, Theuer CP. Paranuclear E-cadherin in gastric adenocarcinoma. Am J Clin Pathol. 2002;118:887–894. doi: 10.1309/EKFB-0HJT-AB1D-5LJB. [DOI] [PubMed] [Google Scholar]
- Charrasse S, Meriane M, Comunale F, Blangy A, Gauthier-Rouvière C. N-cadherin-dependent cell-cell contact regulates Rho GTPases and beta-catenin localization in mouse C2C12 myoblasts. J Cell Biol. 2002;158:953–965. doi: 10.1083/jcb.200202034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YT, Stewart DB, Nelson WJ. Coupling assembly of the E-cadherin/beta-catenin complex to efficient endoplasmic reticulum exit and basal-lateral membrane targeting of E-cadherin in polarized MDCK cells. J Cell Biol. 1999;144:687–699. doi: 10.1083/jcb.144.4.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiasson CM, Wittich KB, Vincent PA, Faundez V, Kowalczyk AP. p120-catenin inhibits VE-cadherin internalization through a Rho-independent mechanism. Mol Biol Cell. 2009;20:1970–1980. doi: 10.1091/mbc.E08-07-0735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chitaev NA, Troyanovsky SM. Adhesive but not lateral E-cadherin complexes require calcium and catenins for their formation. J Cell Biol. 1998;142:837–846. doi: 10.1083/jcb.142.3.837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi HJ, Weis WI. Structure of the armadillo repeat domain of plakophilin 1. J Mol Biol. 2005;346:367–376. doi: 10.1016/j.jmb.2004.11.048. [DOI] [PubMed] [Google Scholar]
- Davis MA, Ireton RC, Reynolds AB. A core function for p120-catenin in cadherin turnover. J Cell Biol. 2003;163:525–534. doi: 10.1083/jcb.200307111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delva E, Kowalczyk AP. Regulation of cadherin trafficking. Traffic. 2009;10:259–267. doi: 10.1111/j.1600-0854.2008.00862.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukumoto Y, Shintani Y, Reynolds AB, Johnson KR, Wheelock MJ. The regulatory or phosphorylation domain of p120 catenin controls E-cadherin dynamics at the plasma membrane. Exp Cell Res. 2008;314:52–67. doi: 10.1016/j.yexcr.2007.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukunaga Y, Liu H, Shimizu M, Komiya S, Kawasuji M, Nagafuchi A. Defining the roles of beta-catenin and plakoglobin in cell-cell adhesion: isolation of beta-catenin/plakoglobin-deficient F9 cells. Cell Struct Funct. 2005;30:25–34. doi: 10.1247/csf.30.25. [DOI] [PubMed] [Google Scholar]
- Gumbiner BM. Cell adhesion: the molecular basis of tissue architecture and morphogenesis. Cell. 1996;84:345–357. doi: 10.1016/s0092-8674(00)81279-9. [DOI] [PubMed] [Google Scholar]
- Hazan RB, Kang L, Roe S, Borgen PI, Rimm DL. Vinculin is associated with the E-cadherin adhesion complex. J Biol Chem. 1997;272:32448–32453. doi: 10.1074/jbc.272.51.32448. [DOI] [PubMed] [Google Scholar]
- Hiraguri S, et al. Mechanisms of inactivation of E-cadherin in breast cancer cell lines. Cancer Res. 1998;58:1972–1977. [PubMed] [Google Scholar]
- Hong S, Troyanovsky RB, Troyanovsky SM. Spontaneous assembly and active disassembly balance adherens junction homeostasis. Proc Natl Acad Sci USA. 2010;107:3528–3533. doi: 10.1073/pnas.0911027107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber AH, Weis WI. The structure of the beta-catenin/E-cadherin complex and the molecular basis of diverse ligand recognition by beta-catenin. Cell. 2001;105:391–402. doi: 10.1016/s0092-8674(01)00330-0. [DOI] [PubMed] [Google Scholar]
- Ireton RC, et al. A novel role for p120 catenin in E-cadherin function. J Cell Biol. 2002;159:465–476. doi: 10.1083/jcb.200205115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishiyama N, Lee SH, Liu S, Li GY, Smith MJ, Reichardt LF, Ikura M. Dynamic and static interactions between p120 catenin and E-cadherin regulate the stability of cell-cell adhesion. Cell. 2010;141:117–128. doi: 10.1016/j.cell.2010.01.017. [DOI] [PubMed] [Google Scholar]
- Jou TS, Stewart DB, Stappert J, Nelson WJ, Marrs JA. Genetic and biochemical dissection of protein linkages in the cadherin-catenin complex. Proc Natl Acad Sci USA. 1995;92:5067–5071. doi: 10.1073/pnas.92.11.5067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiss A, Troyanovsky RB, Troyanovsky SM. p120-catenin is a key component of the cadherin-gamma-secretase supercomplex. Mol Biol Cell. 2008;19:4042–4050. doi: 10.1091/mbc.E08-04-0394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klingelhöfer J, Troyanovsky RB, Laur OY, Troyanovsky SM. Exchange of catenins in cadherin-catenin complex. Oncogene. 2003;22:1181–1188. doi: 10.1038/sj.onc.1206245. [DOI] [PubMed] [Google Scholar]
- Magie CR, Pinto-Santini D, Parkhurst SM. Rho1 interacts with p120ctn and alpha-catenin, and regulates cadherin-based adherens junction components in Drosophila. Development. 2002;129:3771–3782. doi: 10.1242/dev.129.16.3771. [DOI] [PubMed] [Google Scholar]
- Miyashita Y, Ozawa M. Increased internalization of p120-uncoupled E-cadherin and a requirement for a dileucine motif in the cytoplasmic domain for endocytosis of the protein. J Biol Chem. 2007a;282:11540–11548. doi: 10.1074/jbc.M608351200. [DOI] [PubMed] [Google Scholar]
- Miyashita Y, Ozawa M. A dileucine motif in its cytoplasmic domain directs beta-catenin-uncoupled E-cadherin to the lysosome. J Cell Sci. 2007b;120:4395–4406. doi: 10.1242/jcs.03489. [DOI] [PubMed] [Google Scholar]
- Nelson WJ. Regulation of cell-cell adhesion by the cadherin-catenin complex. Biochem Soc Trans. 2008;36:149–155. doi: 10.1042/BST0360149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohkubo T, Ozawa M. p120(ctn) binds to the membrane-proximal region of the E-cadherin cytoplasmic domain and is involved in modulation of adhesion activity. J Biol Chem. 1999;274:21409–21415. doi: 10.1074/jbc.274.30.21409. [DOI] [PubMed] [Google Scholar]
- Pettitt J, Cox EA, Broadbent ID, Flett A, Hardin J. The Caenorhabditis elegans p120 catenin homologue, JAC-1, modulates cadherin-catenin function during epidermal morphogenesis. J Cell Biol. 2003;162:15–22. doi: 10.1083/jcb.200212136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pokutta S, Weis WI. Structure and mechanism of cadherins and catenins in cell-cell contacts. Annu Rev Cell Dev Biol. 2007;23:237–261. doi: 10.1146/annurev.cellbio.22.010305.104241. [DOI] [PubMed] [Google Scholar]
- Provost E, Rimm DL. Controversies at the cytoplasmic face of the cadherin-based adhesion complex. Curr Opin Cell Biol. 1999;11:567–72. doi: 10.1016/s0955-0674(99)00015-0. [DOI] [PubMed] [Google Scholar]
- Reynolds AB, Carnahan RH. Regulation of cadherin stability and turnover by p120ctn: implications in disease and cancer. Semin Cell Dev Biol. 2004;15:657–663. doi: 10.1016/j.semcdb.2004.09.003. [DOI] [PubMed] [Google Scholar]
- Shibamoto S, et al. Association of p120, a tyrosine kinase substrate, with E-cadherin/catenin complexes. J Cell Biol. 1995;128:949–957. doi: 10.1083/jcb.128.5.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoreson MA, Anastasiadis PZ, Daniel JM, Ireton RC, Wheelock MJ, Johnson KR, Hummingbird DK, Reynolds AB. Selective uncoupling of p120(ctn) from E-cadherin disrupts strong adhesion. J Cell Biol. 2000;148:189–202. doi: 10.1083/jcb.148.1.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troyanovsky RB, Laur O, Troyanovsky SM. Stable and unstable cadherin dimers: mechanisms of formation and roles in cell adhesion. Mol Biol Cell. 2007;18:4343–4352. doi: 10.1091/mbc.E07-01-0084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troyanovsky RB, Sokolov EP, Troyanovsky SM. Endocytosis of cadherin from intracellular junctions is the driving force for cadherin adhesive dimer disassembly. Mol Biol Cell. 2006;17:3484–3493. doi: 10.1091/mbc.E06-03-0190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia X, Carnahan RH, Vaughan MH, Wildenberg GA, Reynolds AB. p120 serine and threonine phosphorylation is controlled by multiple ligand-receptor pathways but not cadherin ligation. Exp Cell Res. 2006;312:3336–48. doi: 10.1016/j.yexcr.2006.07.007. [DOI] [PubMed] [Google Scholar]
- Xiao K, Allison DF, Buckley KM, Kottke MD, Vincent PA, Faundez V, Kowalczyk AP. Cellular levels of p120 catenin function as a set point for cadherin expression levels in microvascular endothelial cells. J Cell Biol. 2003;163:535–545. doi: 10.1083/jcb.200306001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao K, Garner J, Buckley KM, Vincent PA, Chiasson CM, Dejana E, Faundez V, Kowalczyk AP. p120-Catenin regulates clathrin-dependent endocytosis of VE-cadherin. Mol Biol Cell. 2005;16:5141–51. doi: 10.1091/mbc.E05-05-0440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagisawa M, Huveldt D, Kreinest P, Lohse CM, Cheville JC, Parker AS, Copland JA, Anastasiadis PZ. A p120 catenin isoform switch affects Rho activity, induces tumor cell invasion, and predicts metastatic disease. J Biol Chem. 2008;283:18344–18354. doi: 10.1074/jbc.M801192200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap AS, Crampton MS, Hardin J. Making and breaking contacts: the cellular biology of cadherin regulation. Curr Opin Cell Biol. 2007;19:508–514. doi: 10.1016/j.ceb.2007.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.