

A genome-wide analysis of the acquisition of stress cross-tolerance shows that reduction of growth rate is an important determinant of severe stress survival. Cellular functions important for the coupling of growth rate to stress resistance are identified, as are those required for cross-tolerance acquisition independent of growth rate reduction.
Abstract
All organisms have evolved to cope with changes in environmental conditions, ensuring the optimal combination of proliferation and survival. In yeast, exposure to a mild stress leads to an increased tolerance for other stresses. This suggests that yeast uses information from the environment to prepare for future threats. We used the yeast knockout collection to systematically investigate the genes and functions involved in severe stress survival and in the acquisition of stress (cross-) tolerance. Besides genes and functions relevant for survival of heat, acid, and oxidative stress, we found an inverse correlation between mutant growth rate and stress survival. Using chemostat cultures, we confirmed that growth rate governs stress tolerance, with higher growth efficiency at low growth rates liberating the energy for these investments. Cellular functions required for stress tolerance acquisition, independent of the reduction in growth rate, were involved in vesicular transport, the Rpd3 histone deacetylase complex, and the mitotic cell cycle. Stress resistance and acquired stress tolerance in Saccharomyces cerevisiae are governed by a combination of stress-specific and general processes. The reduction of growth rate, irrespective of the cause of this reduction, leads to redistribution of resources toward stress tolerance functions, thus preparing the cells for impending change.
INTRODUCTION
To survive in a changing external environment, microbes have to rapidly sense, respond to, and adapt their physiology to new conditions. The conditional changes often occur simultaneously, or in a recurring order (Mitchell et al., 2009). After any such change, several phases can be recognized. First are the direct physicochemical effects of the change, which either do or do not kill the cell. If the initial shock is survived, there is a phase of response and phenotypic adaptation, or acclimatization, and finally a phase of growth (Hohmann and Mager, 2003; Smits and Brul, 2005). The responses are, to some extent, specific, so that cells can withstand heat, cold, nutrient depletion, acidic or oxidative environments, changes in osmolarity, or attacks by other microbes. These specific responses are revealed in the differences in the transcriptional and protein synthesis profiles upon exposure to the various stresses. However, many different nonlethal stresses were shown to induce a common transient coordinated transcriptional response of some 900 genes; this was named the environmental stress response (ESR; Gasch et al., 2000; Gasch, 2003; Causton et al., 2001). The ESR entails the down-regulation of many genes involved in ribosome biogenesis, translation, and transcription and the up-regulation of genes controlled by the general stress transcription factors Msn2 and Msn4 (Gasch et al., 2000). On the basis of these transcriptional data, a strong overlap between growth-regulated and stress-regulated genes was noted (Regenberg et al., 2006; Castrillo et al., 2007; Brauer et al., 2008).
For many specific stress responses, components of signaling pathways, as well as the effectors of these pathways, mediating the functional acclimatization to environmental changes have been identified. In recent years, the functional requirements for growth in a plethora of conditions have been studied in a genome-wide manner, with the help of genomic deletion, overexpression, and haploinsufficiency screenings (Giaever et al., 2002; Parsons et al., 2006; Hillenmeyer et al., 2008). Here too it has become apparent that several cellular functions alter tolerance for many conditions (Dudley et al., 2005; Parsons et al., 2006; Delneri et al., 2008; Hillenmeyer et al., 2008), as well as condition specific genetic requirements. In deletion mutants of genes whose products function in groups down-regulated in the ESR—those regulating ribosome biogenesis, transcription, and translation—the growth-reducing effect of stress is often much less than on the wild type, making the mutants appear stress resistant (Giaever et al., 2002; Parsons et al., 2006; Hillenmeyer et al., 2008; Zakrzewska et al., 2010). Mutation of a number of genes and functions involved in endosomal or vacuolar sorting, or more generally vesicular trafficking, and actin cytoskeleton polarization result in reduced growth in many different nonoptimal conditions (Dudley et al., 2005; Parsons et al., 2006; Hillenmeyer et al., 2008).
The existence of the ESR and the general stress sensitivity of sets of mutants support the notion that yeast, like many other microbes, possesses a general stress response (GSR), a combination of changes in gene/protein expression, protein modification, metabolism, and structural rearrangements common to multiple stresses, in addition to stress-specific mechanisms. The functionality of such a GSR can be recognized through the capability of yeast cultures to enhance stress survival with respect to a certain stress upon exposure to either a similar stress at a mild level or to a seemingly unrelated stress (Sanchez et al., 1992; Berry and Gasch, 2008; Mitchell et al., 2009). For instance, preexposure to heat was shown to lead to increased tolerance for heat but also to freezing and salt stresses (Hall, 1983; Lindquist, 1986; Lewis et al., 1995). Similarly, cell wall perturbation or copper stress induces not only resistance to cell wall and copper stress, but also thermotolerance (de Nobel et al., 2000). The induction of heat-shock proteins, cross-talk between signal transduction pathways, the presence of the disaccharide trehalose, and other processes have been implicated in the induction of stress cross-protection (McAlister and Finkelstein, 1980; Thevelein, 1984; Lindquist, 1986; Attfield, 1987; De Virgilio et al., 1994; Lewis et al., 1995; Versele et al., 2004). The observation of the overlap between the ESR and growth-regulated genes together has led to the suggestion that growth down-regulation is an aspect of the GSR (López-Maury et al., 2008), and, indeed, thermotolerance correlates inversely with growth rate in nitrogen-limited chemostats (Lu et al., 2009 )
So far, most genome-wide studies have focused on growth only and do not measure other properties critical to population survival, such as time to phenotypic adaptation, growth yield (Warringer et al., 2008), or the survival (Lu et al., 2009) of individual cells. To understand the functional requirements for stress tolerance, we analyzed survival upon severe stress exposure. To analyze the acquisition of stress tolerance as a quantifiable parameter of the efficacy of the adaptive response, we looked at survival after a period of mild stress exposure. Genome-wide analysis of the genetic requirements for survival and tolerance acquisition revealed that growth rate reduction, whether by mutation, environmental change, or nutrient control, is a major part of the GSR and is associated with increased survival. Mathematically correcting the survival of mutants for the growth rate of that mutant, we could identify cellular functions involved in survival and the acquisition of tolerance toward severe oxidative, acid, and heat stresses. We conclude that stress survival depends on a combination of stress-specific functions and a GSR that is tightly coupled to growth rate (Castrillo et al., 2007). These closely intertwined functions reflect the strategy of survival of microbial populations in a changing environment, which is based on the evolutionary optimized intrinsic trade-off between cellular robustness and reproduction rate.
RESULTS
Stress survival depends on stress type–specific and general parameters
The obvious requirement to be able to adapt to a changed environment is survival when the change happens. It has already been well established that survival of individual yeast cells to severe stress conditions can be increased by preexposure of the population to a less severe stress. Such a pretreatment protects yeast cells not only from lethal levels of the same stress but also of other, apparently unrelated, stresses, a phenomenon called acquired stress (cross-) tolerance or resistance (McAlister and Finkelstein, 1980). This phenomenon can be interpreted as the existence of a GSR in yeast. We chose to study yeast survival after high doses of various types of stress (defined as a condition causing at least 40% loss of viability within a 10-min treatment) to determine the deletants' intrinsic stress tolerance (Figure 1). Next we determined the protective effect of various mild stress conditions (defined as a stress causing a significantly increased severe stress survival upon exposure to severe dose of a related stress without in itself causing loss of viability in a 24-h period) on survival of high levels of the same stresses. We determined the tolerance to severe oxidative, weak acid, and heat stress treatments in carefully controlled batch cultures that had been preexposed for 2 h to control or to mild heat, cold, acid, and oxidative stress conditions (Figure 1). We chose a pretreatment time of 2 h since all mild stress–severe stress combinations showed maximal survival after this period (although in several cases the maximum had been reached sooner). We observed that preexposure to any of the mild stress conditions led to an increased survival under three of the severe stress conditions, the exception being the heat shock after the cold stress.
FIGURE 1:

Severe stress survival before and after mild-stress treatment. Cultures of BY4741 were pregrown in batch fermentors at 30°C to an OD600 of ∼0.1. Independent cultures were exposed to a nonlethal mild stress treatment of 39°C, 10°C, 0.15 mM H2O2, or 1.2 mM sorbic acid at pH 5.0 for 2 h, whereas a control culture continued growth at 30°C. From each fermentor, triplicate samples were taken and exposed to severe stress treatments of 327 mM acetic acid at pH 3.0 (white bars), 10 mM H2O2 (gray bars), or 48°C (black bars), during 10 min. These samples, as well as triplicate nontreated control samples, were diluted and plated. CFUs were determined after 3 d at 30°C. Bars represent averages and standard deviations of biological quadruplicates; symbols indicate significance of the difference with the untreated samples (**p < 0.001; *p < 0.05; in a one-tailed t test assuming unequal variance).
Measurement of the extent of the increase in survival generated by the four pretreatments revealed that the three high-level stresses did not increase survival proportionally, which would be expected if one general stress response underlies the acquisition of tolerance; one general mechanism activated to various extents could increase, for example, heat resistance more strongly than oxidative stress resistance but should then always do so. This was not the case, although there were clear similarities in the induction of stress survival among acid-, oxidative-, and high temperature–induced tolerances (Supplemental Figure S1). Cold stress seems to induce tolerance by a deviating mechanism. Therefore, although induction of stress cross-tolerance occurred with all conditions tested, the underlying mechanisms generating the tolerance are likely to represent a combination of stress-specific and general processes.
A general stress response increases survival after stress pretreatment
To identify the processes required for stress survival and for acquisition of stress cross-tolerance, we performed a genome-wide analysis using a pooled yeast knockout collection. We cultivated this pooled collection in batch fermentors to early exponential growth. We sampled three times to determine the growth rates of the individual mutants. The culture was divided in two, and one-half was exposed to a mild heat stress (3 h at 38°C, time approximating one generation time for most of the deletion collection in these conditions, leading to induction of maximal severe stress survival of the total pooled collection), whereas the other half was maintained at the normal growth temperature of 30°C. Subsequently, aliquots of both cultures were exposed to severe stress treatments (10-min exposure to 10 mM H2O2, 327 mM acetic acid at pH 3.0, or 48°C; Figure 2A). The treated cells were analyzed for survival (Figure 2B). We determined the fraction of the population represented by individual deletant strains using Tag3 microarrays. Thus we determined growth rates, stress survival, and acquired stress tolerance for 4066 single-gene deletants (Supplemental Data File 1). The pretreatment increased survival of the mutant population of all three severe stresses to similar levels (Figure 2, C–E), whereas control resistance differed by orders of magnitude.
FIGURE 2:

Genome-wide analysis of the impact of single-gene deletions on stress survival and acquired stress tolerance. (A) Design of the genome-wide analysis of stress survival and acquisition of stress tolerance. (B) Survival of 10-min treatments of high levels of oxidative, acid, or heat stress of pooled deletion collection samples (± SD) of five independent replicates. (C–E) Distribution of deletant stress survival percentages within 4067 deletants analyzed, with (black bars) and without (gray bars) a 3-h pretreatment at 38°C. Stress survival was measured after 10 min of (C) oxidative stress (10 mM H2O2), (D) acid stress (327 mM acetic acid), or (E) heat stress (48°C).
We performed pairwise correlation analysis of the survival of all mutants toward oxidative, acid, and heat stress (Figure 3), both with and without the pretreatment, to understand whether the tolerance toward the three stresses depended on shared or on unrelated sets of genes. There was a mild but significant correlation in mutant survival after exposure to the three different severe stresses (r = 0.32–0.39, p < 0.0001). Mechanistic overlap can be estimated from the r2 value, and therefore the comparison of mutant survival from one stress to another suggests that the mechanistic overlap would be between 10 and 15% with the stresses we used. To understand the similarity of the mechanisms of initial survival and induced survival, we analyzed the correlation of mutant survival upon each severe stress before and after pretreatment at 38°C. Here r ranging from 0.33 for acid stress to a fully uncorrelated −0.04 for heat stress suggests that the mechanisms responsible for stress survival in cells growing in relatively optimal conditions are different from those causing the (increased) stress resistance after a mild stress pretreatment. Finally, tolerance toward the three different severe stresses of all mutants became more strongly correlated with each other after the 38°C pretreatment (r = 0.43–0.49, or estimated mechanistic overlap between 18 and 24%) than before (p values for increase <0.001 in all three cases). This suggests that the response that led to the increased stress survival of all three stresses depends at least in part on common mechanisms. Together, these observations imply that the acclimatory response leading to increased stress survival has a significant general aspect and that this is unrelated to the mechanisms responsible for the inherent tolerance to the individual stresses in unstressed exponential-phase batch cultures.
FIGURE 3:

Mild stress pretreatment causes an increase in the correlated stress survival of the deletant collection. (A) Stress survival of all individual deletants under high acid stress (327 mM acetic acid) vs. their stress survival of high oxidative stress (10 mM H2O2). The correlation between the survival was higher after 3 h of growth at 38°C (black symbols) than without this pretreatment (gray symbols), as indicated by their r (p < 0.0001). (B) Pearson correlation coefficients (p for all correlations <0.0001) of the comparison of survival percentages of all mutants upon a high-dose stress pulse of one stress compared with survival upon a pulse of another stress, either with or without a 3-h acclimatory period at 38°C, or comparison of the survival of one single pulse stress after the acclimation period with survival of the same pulse stress before the acclimation period. aPretreatment of 3 h at 38°C. bSevere stress treatment of 10 min with 10 mM H2O2 (H2O2), 327 mM acetic acid at pH 3.0 (HAc), or 48°C (48°C).
Slow growth increases stress survival
The correlations of mutant survival of one stress versus survival of another suggest that only 10–25% of the results can be explained by mechanistic overlap, whereas the mechanisms of mutant survival of a single stress before and after the 38°C pretreatment underlies overlap even less. However, in the course of the experiment we also determined the growth rates of the individual mutants. Remarkably, there was a mild to strong correlation of mutant stress survival with growth rate, revealing a significant inverse relation (Figure 4, white bars at 30°C). This means that slow-growing mutants have increased changes of surviving strong oxidative, acid, or heat stress. On average, the correlations with growth rate were stronger than those of survival of one stress with survival of another (Figure 3). Thus, based on the correlation, differences of growth rate in the mutants could explain up to half of the differences in severe stress survival (for heat stress r = −0.70, and therefore r2 = 0.5). In addition, the survival of severe stress after the 38°C pretreatment showed an inverse correlation with the growth rate at 38°C, although not as strong (Figure 4, gray bars at 38°C), suggesting again that if mutants grow more slowly at 38°C, they will be more tolerant toward severe stress. Finally, the increase in stress survival for all mutants upon mild heat pretreatment showed a clear correlation with their decrease in growth rate after the switch to 38°C (Figure 4, black bars). Thus mutants that had a stronger growth rate response upon temperature shift also had the strongest stress tolerance acquisition. This strongly suggests that mutant growth rate is one of the important determinants of stress tolerance.
FIGURE 4:

Inverse correlation of stress survival with growth rate. (A) Survival percentages of individual mutants after 10 min at 48°C plotted against their specific growth rate just before this treatment reveal an inverse correlation between the two parameters. (B) Correlation coefficients of pairwise comparison of severe stress survival and growth rate of the mutant collection. Consistent inverse correlation of survival upon severe stress is observed with the growth rate at the moment of exposure, so of survival percentages without pretreatment with growth rate at 30°C (white bars), of survival upon preexposure to 38°C with the growth rate at 38°C (gray bars), and of tolerance acquisition with the change in growth rate (μ38°C/μ30°C) upon a shift from 30 to 38°C (black bars). Survival percentages were calculated from the normalized signal intensities of all probes for one ORF before and after severe stress treatment, correcting for the viable cell counts of the total pooled deletion collection (see Materials and Methods).
To determine whether this relation between growth rate and stress survival is a general trait of yeast cultures and not just of mutants, we performed chemostat cultures of wild-type yeast at different dilution rates and compared survival after high-dose stress treatment (Figure 5, A–C). For all three severe stresses tested, we observed an inverse relation between growth rate and stress survival, showing that in wild type, a decrease in growth rate by itself already leads to an increased stress resistance. Thus slow growth, whether induced by mutation during exponential growth on glucose or induced by nutrient-limited growth in chemostats, led to an increased tolerance for all three severe stresses tested. The effect was strongest for heat stress survival, corroborating the finding that the strongest correlation observed in the mutant analysis was that between growth rate and heat-stress survival. This suggests that the GSR may well be closely related to processes involved in the modulation of growth rate.
FIGURE 5:

Growth rate affects severe stress tolerance. Culture samples from glucose-limited chemostats grown at different dilution rates were exposed to 10-min treatments of the different stressors. (A) Survival after exposure to 20 mM H2O2, 327 mM acetic acid at pH 3.0, 48°C; survival percentages were determined. (B) Biomass yield on ATP at the different dilution rates. Bars, average and SE of duplicate experiments.
One would expect that a greater investment in stress resistance activities would be costly for a cell in terms of resources and energy. Analyzing the steady-state carbon fluxes, we found that the yeast cultures were fully respiratory at dilution rates of 0.1, 0.21, and 0.25 h−1, whereas they had a mixed respirofermentative metabolism at the highest growth rate of 0.35 h−1 (Supplemental Table S1). Of interest, assuming similar biomass compositions and a constant respiratory efficiency, it can be deduced that the cultures grow more efficiently at lower dilution rates, with a higher biomass yield on ATP (Figure 5D) This suggests that scarcity of glucose leads not only to a lower growth rate, but also to a more-than-proportional saving of energy. This would liberate energy to be invested in other processes, such as those involved in stress survival.
Functional requirements for growth rate–coupled stress survival
Alteration of growth rate caused either by a mutation or by nutrient limitation in a chemostat affected high-dose stress survival. We asked which cellular functions affected both growth rate and stress sensitivity. This question can be answered by looking at significantly affected mutants and determining the enrichment for particular functional groups, but we can also directly assess whether a priori–defined functional groups are deviating from the rest of the population as a group. Such an approach is robust, as its reliability does not depend on error-prone determinations for a single mutant values (see Supplemental Figure S2) and no cutoffs for the significance of individual mutant traits have to be made. In addition, the distribution of stress survival was far from normal and usually showed strong tailing in one direction. Multiple sampling from such distributions inherently corrects for this (Kim and Volsky, 2005), such that simpler statistics can be used (Boorsma et al., 2005). We therefore determined the group-average growth rate or stress survival percentages for all Gene Ontology (GO) groups with six or more mutants measured in our screen. Next we calculated the distribution of group averages for 2000 drawings of random groups of the same number of mutants. We then determined whether the group average of a GO group was significantly different from the expected value for a group of the same size (two-tailed p of 0.05 after correction for multiple testing; Supplemental Data File 2). Mutant groups that lead to slow growth at 30°C are, as expected, generally involved in ribosome biogenesis and translation, as well as general transcription (Supplemental Table S2). At 38°C, we additionally observed groups involved in vesicular transport within the secretory pathway, as well as GTPase activities, largely involved in vesicle fusion. These activities are relevant for cellular remodeling in response to the temperature shift.
Mathematical uncoupling of mutant growth rate from gene-specific effects on stress survival
Because growth rate and stress survival were so strongly coupled, it was difficult to point out the functional groups that couple growth to survival. Knowing that growth rate directly affects stress survival, we corrected for this contribution to severe stress survival in each mutant using linear regression (see Materials and Methods). To identify those functional groups that are important for the coupling of growth rate and stress survival, we looked for those groups for which an observed survival phenotype was in fact mainly caused by a growth phenotype and therefore lose their significant phenotype upon growth rate correction (Supplemental Table S3). These groups were usually composed of slow growers, which are intrinsically stress resistant, and the growth rate correction led to decreased resistance. In general, the groups functioned in general (RNA polymerase II mediated) transcription and translation and in DNA and chromosome organization. A number of groups were involved in biogenesis and maintenance of mitochondria. Mutants in these groups were slow growers, notwithstanding the presence of high glucose concentrations, and correction for growth rate often revealed significant sensitivity, mostly for severe oxidative and acid stress. It seems therefore that the general regulation of ribosome biogenesis, RNA polymerase II–based transcription, and mitochondrial functions is the most prominent component that coordinates the direct coupling between growth rate and stress survival. This suggests that the coupling has integrated at a very basic level of cellular growth control.
Functional requirements for survival of specific stresses
After correction of mutant survival for growth rate, the phenotypes of mutants before and after the 38°C treatment became more similar (Table 1; p < 0.0001 in all cases). This revealed that the contribution of stress-specific survival mechanisms has become more prominent upon growth rate correction. However, the correlation between any two severe stresses, either before or after the 38°C treatment, decreased after this correction, which confirmed that the general aspect of the acclimatization—growth rate—which had now been now corrected for, was a major determinant of the cross-tolerance. The groups that remain affected in stress survival after the growth rate correction allow us to analyze the specific functions required for the survival of the three severe stresses individually (Supplemental Table S4). Indeed, in the mutant pools growing at 30°C, we no longer found groups that were significantly sensitive to multiple stresses, suggesting that growth rate was the most prominent general aspect of stress survival. After the 38°C pretreatment, we did find general stress sensitivity of mutant groups involved in vesicle-mediated transport and cytoskeleton organization. These groups are likely important for cellular remodeling upon environmental change (Hillenmeyer et al., 2008; Mousley et al., 2008), and in our experiments they appear to be generally important for the response to a nonlethal stress, leading to increased survival. In general, we conclude that there is a difference in the groups of genes responsible for survival of severe stress and those involved in growth in the presence of and phenotypic adaptation to chemical or physical stress.
TABLE 1:
Correlation coefficients of severe stress survival of all deletants after growth rate correction.
| H2O2 | HAc | 48°C | |||
|---|---|---|---|---|---|
| Before pretreatment | |||||
| H2O2 | — | 0.28 | 0.35 | ||
| HAc | — | 0.22 | |||
| 48°C | — | ||||
| After pretreatment of 3 h at 38°C | |||||
| H2O2 | — | 0.43 | 0.41 | ||
| HAc | — | 0.39 | |||
| 48°C | — | ||||
| Before vs. after | |||||
| H2O2 | 0.35 | ||||
| HAc | 0.40 | ||||
| 48°C | 0.25 | ||||
Severe stress treatment of 10 min with 10 mM H2O2 (H2O2), 327 mM acetic acid at pH 3.0 (HAc), or 48°C (48°C).
Pearson correlation coefficients (p for all correlations <0.0001) of the comparison of survival percentages of all mutants corrected for their growth rate at the time of pulse stress application. Mutant survival was correlated on a high-dose stress pulse of one stress compared with survival on a pulse of another stress, either with or without a 3-h acclimatory period at 38°C, or comparison of the survival of one single pulse stress after the acclimation period with survival of the same pulse stress before the acclimation period.
Stress tolerance acquisition
The study was designed to identify processes that play a role in the acquisition of stress (cross-) tolerance. We identified the reduction in growth rate to be an important general factor in the induction of stress survival. We know, however, that most stresses lead to the down-regulation of transcription of mRNA involved in translation and ribosome biogenesis but also the up-regulation of stress-related genes by Msn2p and Msn4p (Gasch et al., 2000; Causton et al., 2001). No indication of a role for these genes in the acquisition of stress tolerance could be found in our screen, and we decided to test the involvement of the Msn2/4p–regulated genes directly, using a double knockout for these transcription factors. The msn2 msn4 double-deletion mutant had much longer lag phases upon inoculation in fresh medium, but the maximal growth rate was not significantly different from that of the wild-type strain (0.38 ± 0.01 for wild-type and 0.36 ± 0.05 for the double-deletion strain). Remarkably, although higher numbers of nonviable cells were present during both normal exponential growth and during the 38°C treatment, both stress survival and stress cross-tolerance acquisition were largely unaffected (Figure 6). These results were confirmed in the BY4741 background (unpublished data). Only the acquisition of acid-induced heat tolerance was much lower than in the isogenic wild-type strain. These data strongly suggest that Msn2p and Msn4p are largely dispensable for the acquisition of stress tolerance
FIGURE 6:

Msn2p and Msn4p are not required for stress tolerance acquisition. Stress survival after 10-min exposure to 10 mM H2O2 (white bars), 327 mM acetic acid (gray bars), or 48°C (black bars) of (A) wild type (W303-1a) and (B) the isogenic msn2 msn4 double-deletion strain in control conditions or after a 3-h treatment with 38°C or 1.2 mM sorbic acid. Bars, average and SD of biological triplicates.
Deficiency of stress tolerance acquisition relied on roughly three major cellular functions (Figure 7 and Supplemental Table S5). Deletants of genes in several groups involved in vesicular protein transport and the actin cytoskeleton were defective in severe stress tolerance acquisition, as well as those in several groups involved in transcription, mRNA modification, and translation, in addition to import and export from the nucleus. Of interest, deletion of genes in quite a number of groups involved in chromatin remodeling, notably with the Swr1 and Rpd3 histone deacetylase complexes, resulted in a stress tolerance acquisition deficiency toward acid and heat stress but not oxidative stress. These groups were previously shown to be relevant for the transient up- and down-regulation of transcription in response to environmental change (Alejandro-Osorio et al., 2009; Ruiz-Roig et al., 2010). Although the functions that were required for tolerance acquisition toward three severe stresses were related, the exact aspects of these functions differed among the individual stresses.
FIGURE 7:

Functional groups involved in tolerance acquisition to different stresses are related but not identical. Mutant groups were selected if tolerance acquisition for at least one severe stress was significantly reduced (Bonferroni-corrected p <0.05, indicated with an asterisk) after correcting for growth rate–associated stress tolerance acquisition. A cutoff line designates Z values of 2, indicating noncorrected significance. White bars, tolerance acquisition for 10 mM H2O2; gray bars, tolerance acquisition for 327 mM acetic acid at pH 3.0; black bars, tolerance acquisition for 48°C.
DISCUSSION
Response versus tolerance
Microbes live in a changing environment. These changes are, to a certain extent, uncoordinated (in both timing and amplitude) but, to some extent, predictable because such changes have been occurring in a coordinated manner during many generations of the organisms' evolution, either in the wild or under conditions imposed by humans, as in, for instance, wine fermentation and the baking of bread (Mitchell et al., 2009). To cope with change, the cell should have accurate and fast mechanisms for sensing both the nature and strength of the change. We already know that for yeast and many other microbes the transcriptional responses to different stresses show a high degree of commonality (Gasch et al., 2000; Causton et al., 2001), and a part of this common response seems to be important in preparing for changes that are particularly likely to occur (Berry and Gasch, 2008; Mitchell et al., 2009) and lead to stress (cross-) tolerance.
In a changing environment, the initial selection is for survival, and this is subject to the tolerance to the stress of the population on average, as well as to the variation in tolerance among the individuals in the population (Attfield et al., 2001). The surviving individuals must sense the change and respond accordingly to finally allow adapted growth (Smits and Brul, 2005). To identify the genes and processes important for the initial survival and next for the acquisition of tolerance, we analyzed survival of the yeast haploid deletion collection after doses of heat, oxidative, and acid stress that were lethal to a large fraction of wild-type cells. Then, to find genes and functions involved in sensing and acclimatization, we analyzed the acquisition of stress tolerance after mild heat exposure of the wild type to heat, oxidative, and acid stresses.
In previous studies analyzing growth in many environmental conditions, mutation of genes involved in vesicular trafficking, notably endosome transport and vacuolar degradation, and transcription was found to lead to multistress sensitivity (Dudley et al., 2005; Parsons et al., 2006; Hillenmeyer et al., 2008), whereas mutants involved in ribosome biogenesis and translation were less affected than most. One of the difficulties in the interpretation of the results of such screens, ours included, is that we are not simply studying the effect of the absence of a single gene on stress tolerance but the complex phenotypic behavior of a strain that has in fact responded and adapted to the absence of this single gene (de Nobel et al., 2000; van Wageningen et al., 2010). In addition, the previous studies were designed to analyze the combined phases of acclimatization and growth rather than to study initial stress tolerance and then the phase of phenotypic adaptation. Indeed, we found stress sensitivity of deletants for genes in groups involved in vesicular transport and cytoskeletal reorganization, but only after mild stress pretreatment (Supplemental Table S4), suggesting that these functions are more important for the acclimatory phase than for actual tolerance. For initial tolerance, we identified stress-specific rather than general functions.
The GSR: growth rate reduction?
Besides cellular processes and functions that help yeast cope with acute changes, we aimed to identify those processes that constitute the GSR responsible for the general increase in stress tolerance upon exposure to nonlethal stresses. Consistent with other studies, we found general stress sensitivity after mild stress pretreatment of mutants involved in secretion and endocytosis and the actin cytoskeleton (Dudley et al., 2005; Brown et al., 2006; Parsons et al., 2006; Hillenmeyer et al., 2008). The Rpd3 histone deacetylase complex, previously shown to be important both for the transient up- and down-regulation of transcription in response to stress (Alejandro-Osorio et al., 2009), was revealed in our analyses to be important for the acquisition of tolerance. This same Rpd3 histone deacetylase complex was previously shown to be required for osmotolerance (De Nadal et al., 2004) and multidrug resistance (Borecka-Melkusova et al., 2008). This suggests a functional role in stress tolerance and tolerance acquisition for both the up- and down-regulated parts of the ESR.
Most important, however, we identified growth rate as a determinant for stress tolerance and growth rate reduction as a determinant for stress tolerance acquisition. Recently several studies were performed that analyzed gene expression in a growth rate–dependent manner, using chemostats with various growth rate–limiting conditions (Regenberg et al., 2006; Castrillo et al., 2007; Brauer et al., 2008; Fazio et al., 2008). In all these studies, a remarkable correlation was observed between the growth rate–dependent genes and those transcribed in the ESR (Gasch et al., 2000). We propose that a large part of the ESR expression changes reflect the lowered growth rate that almost invariably occurs in response to environmental change. Of interest, cold stress induction of severe stress survival seems to depend on different mechanisms. Cold stress did reduce growth rate but did not induce heat stress survival, the severe stress for which survival correlated most with growth rate in our experiments. Indeed, the response to low temperatures appears to be markedly different from that for many other stresses, since lowering of temperature from 30 to 12°C without affecting growth rate (in chemostat cultures) revealed a response almost opposite of the ESR, apparently counteracting the transcriptional response to low growth rate (Tai et al., 2007). Processes that are affected by the lowering of growth rate, either because of mutations in genes involved in the regulation of growth, such as ribosome biogenesis, general translation, or RNA polymerase II–mediated transcription, or because of environmental changes, all lead to an increased stress resistance. We assert that this lowering of growth rate itself is a central aspect of the GSR and is strongly coupled to increased tolerance (Lu et al., 2009).
Coordinated regulation of growth rate and stress tolerance
To identify the ways in which growth rate and stress tolerance are coupled, we need to identify those groups and processes that lead to a stress sensitivity of resistance that is significantly stronger than the growth rates of their deletants would indicate. We expected to find that the transcription factors Msn2p and Msn4p would be involved, as they are responsible for the up-regulation of transcription of ∼300 yeast genes in response to a plethora of environmental conditions (Gasch et al., 2000). When we tested the importance of MSN2 and MSN4 for severe stress survival and stress cross-tolerance acquisition, we found that a double deletion (msn2 msn4) strain was as sensitive to three severe stress treatments as the wild type and was not deficient in the acquisition of stress tolerance. We interpret this to mean that Msn2p and Msn4p are not functional in stress tolerance acquisition, which is in apparent contradiction with other observations that show that these two transcription factors play an important role in preparing for expected stresses (Berry and Gasch, 2008). Activity and localization of Msn2p and Msn4p are controlled by the protein kinase A and TOR signaling pathways (Gorner et al., 1998; Smith et al., 1998; Beck and Hall, 1999). These pathways together integrate nutrient and stress signals to control growth (Schmelzle and Hall, 2000; Jorgensen et al., 2004; De Virgilio and Loewith, 2006; Castrillo et al., 2007; Slattery et al., 2008) and have been implicated in the GSR (Park et al., 1997; Cameron et al., 1988; Smith et al., 1998; Jones et al., 2004; Shen et al., 2007; Schonbrun et al., 2009; Deveau et al., 2010; Robbins et al., 2010). Of interest, it was shown that activation of PKA was able to activate the growth-related transcriptional growth program (the inverse of the ESR) without the concomitant increase in growth (Zaman et al., 2009). Conversely, in a mutant that grows faster on nonfermentable carbon sources than on fermentable carbon sources, the growth-related gene expression was not higher on the former than on the latter. This suggests that external nutrient signals rather than growth rate itself cue the transcriptional program. Msn2p and Msn4p are crucial to this transcriptional response but not to the growth rate–related phenotypic alterations. Therefore, although the transcriptional regulation of some 1000 genes is tightly correlated to the growth rate in wild-type cells, growth rate reduction but not the transcriptional response is crucial to the acquisition of severe stress tolerance.
Growth rate and stress tolerance, or population robustness of microbes
In many detailed studies of yeast fitness, fitness is usually defined as rapid growth (Giaever et al., 2002; Parsons et al., 2006; Hillenmeyer et al., 2008), which leads to a benefit compared with other strains in a laboratory setting. In ecology, the term phenotypic plasticity is used to define the developmental choices available to an organism with one genotype, which are selected based on different life histories. In essence, cellular decision making is based on the economics of nutrition and competition, and any organism has to find the most efficient way of colonizing its niche. In natural and artificially selected yeasts, growth rate and robustness are related properties that show remarkable inverse correlations, which suggests that they cannot be optimized independently (Spor et al., 2008). Therefore the physiology of a unicellular organism in a changing environment is not optimized solely for growth rate but for a combination of growth as fast as possible while still allowing for changed requirements for population or individual robustness (Levy et al., 2007).
The basis for such a trade-off is economic; resources can only be used once and have to be distributed optimally. In physiology, the investment of energy, or ATP, is described as a choice between growth and maintenance, where growth is the increase of biomass and maintenance seemingly a measure for the inefficiency of an organism (Pirt, 1982). However, expanding our understanding of metabolism with an understanding of the cost of biosynthesis of cellular components, such as ribosomes or mitochondria, reveals that optimization of just growth rate in different nutritional environments already necessitates major changes in cellular makeup (Molenaar et al., 2009). In this study, we determined that the inherent trade-off between stress survival and growth rate is integrated already at these same basic levels of cellular functioning and thus are likely to have evolved in parallel with such optimization of yeast in its changing environment.
During evolution, the exact nature of the investments has been fine tuned, with partially overlapping pathways generating similar functions, leading to a simultaneous and interdependent optimization of growth and robustness (Papp et al., 2004; Deutscher et al., 2006; Vilaprinyo et al., 2006). The minimization of the cost of preparedness for, and acclimation to, stressful environments and nutritional changes simultaneously is solved by a combination of stochastic phenotypic variation (Balaban et al., 2004; Perkins and Swain, 2009), specific responses (such as the developmental programs of mating and sporulation [ Fischer and Sauer, 2005; Lang et al., 2009]) to “expected” changes, which include combined or related stresses (Mitchell et al., 2009), and a general stress response consisting of a growth rate reduction (López-Maury et al., 2008; Lu et al., 2009). This last response is directly coupled to increased stress tolerance and frees energy that may be diverted to cellular remodeling and specific processes required for acclimatization (Vilaprinyo et al., 2010).
The net result of the evolutionary selection for microorganisms to survive in a constantly changing environment is the trade-off between growth rate and stress tolerance; fast growth in nutrient-abundant, stress-free environments automatically leads to the selection of individuals with reduced robustness, whereas growth rate reduction leads to the generation of phenotypically and genotypically robust individuals.
MATERIALS AND METHODS
Strains, growth conditions, and genetic procedures
For batch cultivation, Saccharomyces cerevisiae strains (Table 2) were cultivated in 500-ml custom-made (in-house) batch fermentors with water jackets for temperature control. Stirring rate was set to 600 rpm and aeration rate was a fermentor volume of air per minute. The culture was kept at pH 5.0 by automatic titration with 0.1 M KOH using an Applikon ADI 1030 Controller (Applikon, Schiedam, Netherlands). The medium used for cultivation was based on a previously described mineral medium (Verduyn et al., 1992), custom made to allow changes in elemental sources and chemostat nutrient limitation, supplemented with 20 g/l glucose and any amino acid or nucleic acid bases required to satisfy the strains' auxotrophic requirements.
TABLE 2:
Strains used in this study.
| Strain | Genotype | Source or reference |
|---|---|---|
| BY4741 | MATahis3Δ1 leu2Δ0 met15Δ0 ura3Δ0 | Euroscarf |
| BY4741 pooled knockout collection | MATahis3Δ1 leu2Δ0 met15Δ0 ura3Δ0 yfgX::KanMX | Euroscarf, and this study |
| CEN.PK 113-7D | MATaMAL2-8cSUC2 | P. Kötter, personal communication |
| W303.1A | MATaleu2-3/112 ura3-1 trp1-1 his3-11/15 ade2-1 can1-100 | Thomas and Rothstein (1989) |
| W303.1A msn2Δ msn4Δ | MATaleu2-3/112 ura3-1 trp1-1 his3-11/15 ade2-1 can1-100 msn2-Δ3::HIS3 msn4-1::TRP1 | Estruch and Carlson (1993) |
| BY4741 msn2Δ msn4Δ | MATa. his3Δ1 leu2Δ2 met15Δ0 ura3Δ0 msn2Δ::KanMX msn4Δ::HIS3MX6 | This study |
Euroscarf, European Saccharomyces cerevisiae Archive for Functional Analysis (euroscarf@em.uni-frankfurt.de).
For cross-tolerance development, batch cultures in mid-exponential phase (∼2 × 106 cells/ml or an OD600 of ∼0.1–0.2) were treated with mild heat (38 or 39°C), a low concentration of weak acid (1.2 mM sorbic acid), or an oxidizing agent (0.3 mM H2O2) in batch fermentation conditions (see earlier discussion) for the times indicated. In these conditions, growth rate was affected, but no loss of viability was observed.
In chemostat experiments, strain CEN.PK113-7D was grown in aerobic, carbon-limited 2-l chemostats (Applikon) with a working volume of 1 l at a dilution rate of 0.1 h−1. Precultures were grown overnight at 30°C in mineral medium with 20 g/l glucose in shake flasks (200 rpm). The medium used for chemostat cultivation was the same mineral medium as used in the batch fermentors, but with 7.5 g/l glucose. The stirrer speed was set to 800 rpm, and the pH was set to 5.0 and kept constant by automatic titration with 1 M KOH. The temperature of the chemostat was controlled with a heat jacket and a temperature probe. The stirring rate, pH, and temperature were kept constant using an Applikon ADI 1010 Biocontroller. The chemostat was aerated by flushing air through the culture at 30 l/h. Steady states were verified by off-gas analysis for oxygen and carbon dioxide and by dry weight measurements. Steady-state flux determinations (Supplemental Table S1) were performed by analysis of culture supernatant and gas-composition analysis as described in Postmus et al. (2008).
For stress survival analysis, triplicate samples from steady-state chemostat cultures or mid-exponential–phase batch cultures (∼107 cells/ml) were taken for cell counts and severe stress treatments. Severe stress treatments (exposure to 47 or 48°C, 10 or 20 mM H2O2, or 327 mM of acetic acid at pH 3.0 for 10 min) were performed in 1.5-ml Eppendorf tubes. Tenfold serial dilutions were made in Eppendorf tubes in Verduyn medium (Verduyn et al., 1992), and 100-μl samples were plated on yeast extract/peptone/dextrose to determine the number of colony-forming units (CFUs) after 3 d of incubation at 30°C.
Strain AMY203 was generated in the BY4741 background starting with the msn2Δ strain from the deletion collection. An deletion cassette containing 50–base pair flanks for homologous recombination at the MSN4 locus with the His3MX gene was generated using forward primer 5′-CAAGTTTTCGGAGAAATAATATTCTTTCTTCCTCAGTTTACAAAACAAG TCGGATCCCCGGGTTAATTAA-3′ and reverse primer 5′-ACTTGTTTTGTAAACTGAGGAAGAAAGAATATTATTTCTCCGAAAACTTGGAATTCGAGCTCGTTTAAAC-3′ with plasmid pFA6a-His3MX6 (Longtine et al., 1998), creating 50–base pair flanks (underscored) identical to the MSN4 flanking regions. On transformation and selection for complementation of the histidine auxotrophy, gene deletion was confirmed with PCR.
Sample preparation for microarray analysis.
A total of 4825 nonessential haploid deletion strains were grown overnight in 96-well microtiter plates in synthetic complete medium, and equal culture volumes were combined and mixed. Glycerol was added to the pooled cultures to 15% wt/vol final concentration, and aliquots were frozen and stored at −80°C. Aliquots of pooled deletion collection were inoculated into Verduyn medium and cultured to a cell density of (5–8) × 106 cells/ml, corresponding to exponential growth phase. Samples of equal volumes of this culture were briefly centrifuged, and the cell pellets were resuspended in 1 ml of nonbuffered medium and then inoculated into a batch fermentor at 30°C to obtain a cell density of 2 × 105 per ml. The batch cultures were grown for 4–5 h, to a cell density of ∼1.5 × 106 cells/ml, when samples were taken for DNA analysis, cell counts, and CFU counts (t = −6 h). This was repeated after an additional 3 h of growth (t = −3 h). Now the temperature in one of the fermentors was switched from 30 to 38°C, and incubation was continued for another 3 h at both temperatures, after which samples for DNA analysis, cell counts, and CFU counts were taken (t = 0 h). Thus we also sampled at three time points during exponential growth, which allowed us to determine growth rates. Separate samples from both cultures were subjected to 10-min severe stress treatments in multiple 2-ml Eppendorf tubes, such that the total number of viable cells after treatment amounted to ∼2 × 105. Actual viability was monitored by plating samples for CFU counts. For DNA analysis, from each sample 5 × 104 viable cells (as determined in test runs of the experiment and verified in the actual experiment) were reinoculated into 100 ml of Verduyn medium and cultured for 24 h, ∼10 generations, to select live cells. Actual propagation was determined with CFU counts. Culture samples were centrifuged at 4000 rpm for 5 min, and the cell pellets were stored at −20°C. Experiments were performed in duplicate.
PCR amplification, hybridization, and data acquisition.
Yeast cells were lysed at 37°C with 0.5 mg/ml Zymolyase 100T for 60 min in an osmotically stabilized solution consisting of 1 M sorbitol, 100 mM sodium EDTA, and 14 mM β-mercaptoethanol. Genomic DNA was isolated with the Qiagen (Valencia, CA) DNeasy kit. The PCR amplification of the TAGs was performed on 200 ng of genomic DNA template. Both UPTAGs and DOWNTAGs were amplified in separate reactions using biotinylated PCR primers complementary to common regions in the replacement cassette. PCR conditions and primers used were as described previously (Delneri et al., 2008). The final, labeled UPTAGs and DOWNTAGs were purified on a YM10 Microcon column, combined, and hybridized to custom-made oligonucleotide microarrays (DNA TAG3; Affymetrix, Santa Clara, CA) as described previously (Zakrzewska et al., 2007), with the exception that four blocking primers were used in the hybridization mixture. After 16 h of hybridization at 42°C, the arrays were stained with streptavidin–phycoerythrin (Molecular Probes, Invitrogen, Carlsbad, CA) and scanned with an Affymetrix GeneChip Scanner. The hybridization intensities for each probe were determined using Affymetrix GeneChip Operating Software (GCOS).
Data processing
The majority of knock-out strains are represented by four values of signal intensity (sense and antisense array elements for each UPTAG and DOWNTAG). We calculated the mean of the signal intensities of tags representing each open reading frame (ORF) on the basis of two biological replicates of the experiment. The signal intensities of the UPTAGs and DOWNTAGs were calculated separately. We defined the background as the average of all signal intensities of nonhybridizing spots. Next the ORFs whose averaged signal intensity was below the background level in the control experiment were discarded from further analysis. We calculated growth rates for individual mutants (νi in h−1) as follows: we defined fractional intensities for each spot i (Si) within a TAG set as
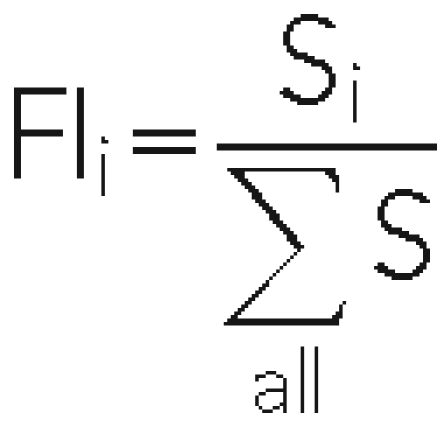 |
within that TAG set. A relative cell number was calculated for C−6, C−3, and C0 by multiplying the FIi with the culture cell density at sampling time. From these data, νi for the mutants i were calculated from the slope of a line through points −6, −3, and 0, using linear regression on the combined data from the duplicate experiments and different probes for each mutant hybridizing above background (two to eight time series per mutant). The 95% confidence intervals (CI95) for the average were calculated. Only those mutants whose νi CI95 was >0 were considered in the analysis. Growth rates at 38°C were determined from the relative cell numbers at t = 0 from the 38°C cultures compared with the 30°C cultures, multiplied by the growth rate at 30°C.
Mutant viability upon treatment was calculated as follows: for each sample, we estimated viable mutant distributions at sampling time based on the distribution after 24 h of recovery and cell division, using the growth rates calculated earlier. We calculated culture growth time from the known culture growth rate and the viable population increase over 24 h from cell counts and viable cell counts. We then calculated the best approximation FIi of each mutant at sampling time from the FIi after 24 h of recovery using the calculated νi and culture growth time, for each spot on each slide (UPTAG and DOWNTAG, forward and complementing sequence), and normalized to culture viable cell counts. Viability upon stress for each mutant is then the ratio of stress-treated sample/nontreated sample of the culture × 100%. Reproducibility between tags for one replicate and between biological replicates was similar, and therefore the aggregate average was used (Supplemental Figure S1). Averages and CI99.9 of these percentages for all individual probes belonging to one mutant were calculated. Single values above and below the CI99.9 were discarded. The resulting percentages were log transformed for correlation analyses. Full tables of raw data, growth rates, viability, and log transformations can be found at http://home.medewerker.uva.nl/g.j.smits/page1.html.
To calculate the growth rate–independent resistance of the mutants, we determined the linear relationship between log-transformed survival for each severe stress treatment of all mutants with the growth rate of the corresponding control (μ30°C, μ38°C, or the change in growth rate μ38°C/μ30°C). The prediction for survival for each individual mutant based on this regression was subtracted from the true survival.
GO analysis
For biological interpretation, we determined which functions and processes were involved in stress tolerance. Because errors in the analysis of groups of mutants are much smaller than those in individual mutants (Kim and Volsky, 2005), we analyzed the behavior of predefined groups. We used a random sampling approach: the distributions of average values for a priori–defined groups (defined by GO ontology, lists downloaded 5 December 2009) with more than six measured mutants were calculated by randomly drawing without replacement a group of identical size within each condition data set. Each of the 2000 drawings was averaged, and the distribution of such averages approximates normal (Kim and Volsky, 2005). The variation of the averages allows for calculating the Z value and associated p value for being resistant or sensitive in each condition. The p values derived from the Z value were multiplied by the number of groups tested simultaneously (Bonferroni correction), and groups whose corrected p value was <0.05 were regarded as significantly sensitive or resistant. Full tables of Z values are given in Supplemental Data File 2.
Supplementary Material
Acknowledgments
We thank L. Vogelpoel for experimental assistance. M. Versele is acknowledged for donation of strains. We thank F. M. Klis for critical reading of the manuscript. The Meer Vrouwelijke Onderzoekers als UD, Nederlandse Organisatie voor Wetenschappelijk Onderzoek (G.J.S.), Unilever (G.J.S.), and the UK Natural Environment Research Council (S.G.O.) contributed to the funding of this project.
Abbreviations used:
- CFU
colony-forming unit
- CI
confidence interval
- ESR
environmental stress response
- GSR
general stress response
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E10-08-0721) on September 30, 2011.
REFERENCES
- Alejandro-Osorio AL, Huebert DJ, Porcaro DT, Sonntag ME, Nillasithanukroh S, Will JL, Gasch AP. The histone deacetylase Rpd3p is required for transient changes in genomic expression in response to stress. Genome Biol. 2009;10:R57. doi: 10.1186/gb-2009-10-5-r57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attfield PV. Trehalose accumulates in Saccharomyces cerevisiae during exposure to agents that induce heat shock response. FEBS Lett. 1987;225:259–263. doi: 10.1016/0014-5793(87)81170-5. [DOI] [PubMed] [Google Scholar]
- Attfield PV, Choi HY, Veal DA, Bell PJ. Heterogeneity of stress gene expression and stress resistance among individual cells of Saccharomyces cerevisiae. Mol Microbiol. 2001;40:1000–1008. doi: 10.1046/j.1365-2958.2001.02444.x. [DOI] [PubMed] [Google Scholar]
- Balaban NQ, Merrin J, Chait R, Kowalik L, Leibler S. Bacterial persistence as a phenotypic switch. Science. 2004;305:1622–1625. doi: 10.1126/science.1099390. [DOI] [PubMed] [Google Scholar]
- Beck T, Hall MN. The TOR signalling pathway controls nuclear localization of nutrient-regulated transcription factors. Nature. 1999;402:689–692. doi: 10.1038/45287. [DOI] [PubMed] [Google Scholar]
- Berry DB, Gasch AP. Stress-activated genomic expression changes serve a preparative role for impending stress in yeast. Mol Biol Cell. 2008;19:4580–4587. doi: 10.1091/mbc.E07-07-0680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boorsma A, Foat BC, Vis D, Klis F, Bussemaker HJ. T-profiler: scoring the activity of predefined groups of genes using gene expression data. Nucleic Acids Res. 2005;33:W592–W595. doi: 10.1093/nar/gki484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borecka-Melkusova S, Kozovska Z, Hikkel I, Dzugasova V, Subik J. RPD3 and ROM2 are required for multidrug resistance in Saccharomyces cerevisiae. FEMS Yeast Res. 2008;8:414–424. doi: 10.1111/j.1567-1364.2007.00352.x. [DOI] [PubMed] [Google Scholar]
- Brauer MJ, Huttenhower C, Airoldi EM, Rosenstein R, Matese JC, Gresham D, Boer VM, Troyanskaya OG, Botstein D. Coordination of growth rate, cell cycle, stress response, and metabolic activity in yeast. Mol Biol Cell. 2008;19:352–367. doi: 10.1091/mbc.E07-08-0779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JA, Sherlock G, Myers CL, Burrows NM, Deng C, Wu HI, McCann KE, Troyanskaya OG, Brown JM. Global analysis of gene function in yeast by quantitative phenotypic profiling. Mol Syst Biol. 2006;2:2006–0001. doi: 10.1038/msb4100043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron S, Levin L, Zoller M, Wigler M. cAMP-independent control of sporulation, glycogen metabolism, and heat shock resistance in S. cerevisiae. Cell. 1988;53:555–566. doi: 10.1016/0092-8674(88)90572-7. [DOI] [PubMed] [Google Scholar]
- Castrillo JI, et al. Growth control of the eukaryote cell: a systems biology study in yeast. J Biol. 2007;6:4. doi: 10.1186/jbiol54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Causton HC, Ren B, Koh SS, Harbison CT, Kanin E, Jennings EG, Lee TI, True HL, Lander ES, Young RA. Remodeling of yeast genome expression in response to environmental changes. Mol Biol Cell. 2001;12:323–337. doi: 10.1091/mbc.12.2.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Nadal E, Zapater M, Alepuz PM, Sumoy L, Mas G, Posas F. The MAPK Hog1 recruits Rpd3 histone deacetylase to activate osmoresponsive genes. Nature. 2004;427:370–374. doi: 10.1038/nature02258. [DOI] [PubMed] [Google Scholar]
- de Nobel H, Ruiz C, Martin H, Morris W, Brul S, Molina M, Klis FM. Cell wall perturbation in yeast results in dual phosphorylation of the Slt2/Mpk1 MAP kinase and in an Slt2-mediated increase in FKS2-lacZ expression, glucanase resistance and thermotolerance. Microbiology. 2000;146:2121–2132. doi: 10.1099/00221287-146-9-2121. [DOI] [PubMed] [Google Scholar]
- De Virgilio C, Hottiger T, Dominguez J, Boller T, Wiemken A. The role of trehalose synthesis for the acquisition of thermotolerance in yeastIGenetic evidence that trehalose is a thermoprotectant. Eur J Biochem. 1994;219:179–186. doi: 10.1111/j.1432-1033.1994.tb19928.x. [DOI] [PubMed] [Google Scholar]
- De Virgilio C, Loewith R. The TOR signalling network from yeast to man. Int J Biochem Cell Biol. 2006;38:1476–1481. doi: 10.1016/j.biocel.2006.02.013. [DOI] [PubMed] [Google Scholar]
- Delneri D, et al. Identification and characterization of high-flux-control genes of yeast through competition analyses in continuous cultures. Nat Genet. 2008;40:113–117. doi: 10.1038/ng.2007.49. [DOI] [PubMed] [Google Scholar]
- Deutscher D, Meilijson I, Kupiec M, Ruppin E. Multiple knockout analysis of genetic robustness in the yeast metabolic network. Nat Genet. 2006;38:993–998. doi: 10.1038/ng1856. [DOI] [PubMed] [Google Scholar]
- Deveau A, Piispanen AE, Jackson AA, Hogan DA. Farnesol induces hydrogen peroxide resistance in Candida albicans yeast by inhibiting the Ras-cAMP signaling pathway. Eukaryot Cell. 2010;9:569–577. doi: 10.1128/EC.00321-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudley AM, Janse DM, Tanay A, Shamir R, Church GM. A global view of pleiotropy and phenotypically derived gene function in yeast. Mol Syst Biol. 2005;1:2005.0001. doi: 10.1038/msb4100004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estruch F, Carlson M. Two homologous zinc finger genes identified by multicopy suppression in a SNF1 protein kinase mutant of Saccharomyces cerevisiae. Mol Cell Biol. 1993;13:3872–3881. doi: 10.1128/mcb.13.7.3872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fazio A, Jewett MC, Daran-Lapujade P, Mustacchi R, Usaite R, Pronk JT, Workman CT, Nielsen J. Transcription factor control of growth rate dependent genes in Saccharomyces cerevisiae: a three factor design. BMC Genomics. 2008;9:341. doi: 10.1186/1471-2164-9-341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer E, Sauer U. Large-scale in vivo flux analysis shows rigidity and suboptimal performance of Bacillus subtilis metabolism. Nat Genet. 2005;37:636–640. doi: 10.1038/ng1555. [DOI] [PubMed] [Google Scholar]
- Gasch AP. The environmental stress response: a common yeast response to diverse environmental stresses. In: Hohmann S, Mager WH, editors. In: Yeast Stress Responses. Berlin: Springer-Verlag; 2003. pp. 11–57. [Google Scholar]
- Gasch AP, Spellman PT, Kao CM, Carmel-Harel O, Eisen MB, Storz G, Botstein D, Brown PO. Genomic expression programs in the response of yeast cells to environmental changes. Mol Biol Cell. 2000;11:4241–4257. doi: 10.1091/mbc.11.12.4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giaever G, et al. Functional profiling of the Saccharomyces cerevisiae genome. Nature. 2002;418:387–391. doi: 10.1038/nature00935. [DOI] [PubMed] [Google Scholar]
- Gorner W, Durchschlag E, Martinez-Pastor MT, Estruch F, Ammerer G, Hamilton B, Ruis H, Schuller C. Nuclear localization of the C2H2 zinc finger protein Msn2p is regulated by stress and protein kinase A activity. Genes Dev. 1998;12:586–597. doi: 10.1101/gad.12.4.586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall BG. Yeast thermotolerance does not require protein synthesis. J Bacteriol. 1983;156:1363–1365. doi: 10.1128/jb.156.3.1363-1365.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillenmeyer ME, et al. The chemical genomic portrait of yeast: uncovering a phenotype for all genes. Science. 2008;320:362–365. doi: 10.1126/science.1150021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohmann S, Mager WH. Yeast Stress Responses. Berlin: Springer-Verlag; 2003. [Google Scholar]
- Jones DL, Petty J, Hoyle DC, Hayes A, Oliver SG, Riba-Garcia I, Gaskell SJ, Stateva L. Genome-wide analysis of the effects of heat shock on a Saccharomyces cerevisiae mutant with a constitutively activated cAMP-dependent pathway. Comp Funct Genomics. 2004;5:419–431. doi: 10.1002/cfg.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen P, Rupes I, Sharom JR, Schneper L, Broach JR, Tyers M. A dynamic transcriptional network communicates growth potential to ribosome synthesis and critical cell size. Genes Dev. 2004;18:2491–2505. doi: 10.1101/gad.1228804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SY, Volsky DJ. PAGE: parametric analysis of gene set enrichment. BMC Bioinformatics. 2005;6:144. doi: 10.1186/1471-2105-6-144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang GI, Murray AW, Botstein D. The cost of gene expression underlies a fitness trade-off in yeast. Proc Natl Acad Sci USA. 2009;106:5755–5760. doi: 10.1073/pnas.0901620106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy S, Ihmels J, Carmi M, Weinberger A, Friedlander G, Barkai N. Strategy of transcription regulation in the budding yeast. Plos. 2007;ONE 2:e250. doi: 10.1371/journal.pone.0000250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis JG, Learmonth RP, Watson K. Induction of heat, freezing and salt tolerance by heat and salt shock in Saccharomyces cerevisiae. Microbiology. 1995;141:687–694. doi: 10.1099/13500872-141-3-687. [DOI] [PubMed] [Google Scholar]
- Lindquist S. The heat-shock response. Annu Rev Biochem. 1986;55:1151–1191. doi: 10.1146/annurev.bi.55.070186.005443. [DOI] [PubMed] [Google Scholar]
- Longtine MS, McKenzie A, DeMarini DJ, Shah NG, Wach A, Bracha A, Philippsen P, Pringle JR. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast. 1998;14:943–951. doi: 10.1002/(SICI)1097-0061(199807)14:10<953::AID-YEA293>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- López-Maury L, Marguerat S, Bähler J. Tuning gene expression to changing environments: from rapid responses to evolutionary adaptation. Nat Rev Genet. 2008;9:583–593. doi: 10.1038/nrg2398. [DOI] [PubMed] [Google Scholar]
- Lu C, Brauer MJ, Botstein D. Slow growth induces heat-shock resistance in normal and respiratory-deficient yeast. Mol Biol Cell. 2009;20:891–903. doi: 10.1091/mbc.E08-08-0852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAlister L, Finkelstein DB. Heat shock proteins and thermal resistance in yeast. Biochem Biophys Res Commun. 1980;93:819–824. doi: 10.1016/0006-291x(80)91150-x. [DOI] [PubMed] [Google Scholar]
- Mitchell A, Romano GH, Groisman B, Yona A, Dekel E, Kupiec M, Dahan O, Pilpel Y. Adaptive prediction of environmental changes by microorganisms. Nature. 2009;460:220–224. doi: 10.1038/nature08112. [DOI] [PubMed] [Google Scholar]
- Molenaar D, van Berlo R, de Ridder D, Teusink B. Shifts in growth strategies reflect tradeoffs in cellular economics. Mol Syst Biol. 2009;5:323. doi: 10.1038/msb.2009.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mousley CJ, Tyeryar K, Ile KE, Schaaf G, Brost RL, Boone C, Guan X, Wenk MR, Bankaitis VA. Trans-Golgi network and endosome dynamics connect ceramide homeostasis with regulation of the unfolded protein response and TOR signaling in yeast. Mol Biol Cell. 2008;19:4785–4803. doi: 10.1091/mbc.E08-04-0426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papp B, Pal C, Hurst LD. Metabolic network analysis of the causes and evolution of enzyme dispensability in yeast. Nature. 2004;429:661–664. doi: 10.1038/nature02636. [DOI] [PubMed] [Google Scholar]
- Park JI, Grant CM, Attfield PV, Dawes IW. The freeze-thaw stress response of the yeast Saccharomyces cerevisiae is growth phase specific and is controlled by nutritional state via the RAS-cyclic AMP signal transduction pathway. Appl Environ Microbiol. 1997;63:3818–3824. doi: 10.1128/aem.63.10.3818-3824.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons AB, et al. Exploring the mode-of-action of bioactive compounds by chemical-genetic profiling in yeast. Cell. 2006;126:611–625. doi: 10.1016/j.cell.2006.06.040. [DOI] [PubMed] [Google Scholar]
- Perkins TJ, Swain PS. Strategies for cellular decision-making. Mol Syst Biol. 2009;5:326. doi: 10.1038/msb.2009.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirt SJ. Maintenance energy: a general model for energy-limited and energy-sufficient growth. Arch Microbiol. 1982;133:300–302. doi: 10.1007/BF00521294. [DOI] [PubMed] [Google Scholar]
- Postmus J, Canelas AB, Bouwman J, Bakker BM, van Gulik W, de Mattos MJ, Brul S, Smits GJ. Quantitative analysis of the high temperature-induced glycolytic flux increase in Saccharomyces cerevisiae reveals dominant metabolic regulation. J Biol Chem. 2008;283:23524–23532. doi: 10.1074/jbc.M802908200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regenberg B, Grotkjaer T, Winther O, Fausboll A, Akesson M, Bro C, Hansen LK, Brunak S, Nielsen J. Growth-rate regulated genes have profound impact on interpretation of transcriptome profiling in Saccharomyces cerevisiae. Genome Biol. 2006;7:R107. doi: 10.1186/gb-2006-7-11-r107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins N, Collins C, Morhayim J, Cowen LE. Metabolic control of antifungal drug resistance. Fungal Genet Biol. 2010;47:81–93. doi: 10.1016/j.fgb.2009.07.004. [DOI] [PubMed] [Google Scholar]
- Ruiz-Roig C, Viéitez C, Posas F, de Nadal E. The Rpd3L HDAC complex is essential for the heat stress response in yeast. Mol Microbiol. 2010;76:1049–1062. doi: 10.1111/j.1365-2958.2010.07167.x. [DOI] [PubMed] [Google Scholar]
- Sanchez Y, Taulien J, Borkovich KA, Lindquist S. Hsp104 is required for tolerance to many forms of stress. EMBO J. 1992;11:2357–2364. doi: 10.1002/j.1460-2075.1992.tb05295.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmelzle T, Hall MN. TOR, a central controller of cell growth. Cell. 2000;103:253–262. doi: 10.1016/s0092-8674(00)00117-3. [DOI] [PubMed] [Google Scholar]
- Schonbrun M, Laor D, López-Maury L, Bahler J, Kupiec M, Weisman R. TOR complex 2 controls gene silencing, telomere length maintenance, and survival under DNA-damaging conditions. Mol Cell Biol. 2009;29:4584–4594. doi: 10.1128/MCB.01879-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen C, Lancaster CS, Shi B, Guo H, Thimmaiah P, Bjornsti MA. TOR signaling is a determinant of cell survival in response to DNA damage. Mol Cell Biol. 2007;27:7007–7017. doi: 10.1128/MCB.00290-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slattery MG, Liko D, Heideman W. Protein kinase A, TOR, and glucose transport control the response to nutrient repletion in Saccharomyces cerevisiae. Eukaryot Cell. 2008;7:358–367. doi: 10.1128/EC.00334-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith A, Ward MP, Garrett S. Yeast PKA represses Msn2p/Msn4p-dependent gene expression to regulate growth, stress response and glycogen accumulation. EMBO J. 1998;17:3556–3564. doi: 10.1093/emboj/17.13.3556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smits GJ, Brul S. Stress tolerance in fungi—to kill a spoilage yeast. Curr Opin Biotechnol. 2005;16:225–230. doi: 10.1016/j.copbio.2005.02.005. [DOI] [PubMed] [Google Scholar]
- Spor A, Wang S, Dillmann C, de Vienne D, Sicard D. “Ant” and “grasshopper” life-history strategies in Saccharomyces cerevisiae. PLoS One. 2008;3:e1579. doi: 10.1371/journal.pone.0001579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai SL, Daran-Lapujade P, Walsh MC, Pronk JT, Daran JM. Acclimation of Saccharomyces cerevisiae to low temperature: a chemostat-based transcriptome analysis. Mol Biol Cell. 2007;18:5100–5112. doi: 10.1091/mbc.E07-02-0131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas BJ, Rothstein R. Elevated recombination rates in transcriptionally active DNA. Cell. 1989;56:619–630. doi: 10.1016/0092-8674(89)90584-9. [DOI] [PubMed] [Google Scholar]
- Thevelein JM. Regulation of trehalose mobilization in fungi. Microbiol Rev. 1984;48:42–59. doi: 10.1128/mr.48.1.42-59.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Wageningen S, et al. Functional overlap and regulatory links shape genetic interactions between signaling pathways. Cell. 2010;143:991–1004. doi: 10.1016/j.cell.2010.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verduyn C, Postma E, Scheffers WA, Van Dijken JP. Effect of benzoic acid on metabolic fluxes in yeasts: a continuous-culture study on the regulation of respiration and alcoholic fermentation. Yeast. 1992;8:501–517. doi: 10.1002/yea.320080703. [DOI] [PubMed] [Google Scholar]
- Versele M, Thevelein JM, Van Dijck P. The high general stress resistance of the Saccharomyces cerevisiae fil1 adenylate cyclase mutant (Cyr1Lys1682) is only partially dependent on trehalose, Hsp104 and overexpression of Msn2/4-regulated genes. Yeast. 2004;21:75–86. doi: 10.1002/yea.1065. [DOI] [PubMed] [Google Scholar]
- Vilaprinyo E, Alves R, Sorribas A. Use of physiological constraints to identify quantitative design principles for gene expression in yeast adaptation to heat shock. BMC Bioinformatics. 2006;7:184. doi: 10.1186/1471-2105-7-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilaprinyo E, Alves R, Sorribas A. Minimization of biosynthetic costs in adaptive gene expression responses of yeast environmental changes. PLoS Comput Biol. 2010;6:e1000674. doi: 10.1371/journal.pcbi.1000674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warringer J, Anevski D, Liu B, Blomberg A. Chemogenetic fingerprinting by analysis of cellular growth dynamics. BMC Chem Biol. 2008;8:3. doi: 10.1186/1472-6769-8-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakrzewska A, Boorsma A, Delneri D, Brul S, Oliver SG, Klis FM. Cellular processes and pathways that protect Saccharomyces cerevisiae cells against the plasma membrane-perturbing compound chitosan. Eukaryot Cell. 2007;6:600–608. doi: 10.1128/EC.00355-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakrzewska A, Boorsma A, Ter Beek AS, Hageman JA, Westerhuis JA, Heelingwerk KJ, Brul S, Klis FM, Smits GJ. Comparative analysis of transcriptome and fitness profiles reveals general and condition specific cellular functions involved in adaptation to environmental change in Saccharomyces cerevisiae. OMICS. 2010;14:350–360. doi: 10.1089/omi.2010.0049. [DOI] [PubMed] [Google Scholar]
- Zaman S, Lippman SI, Schneper L, Slonim N, Broach JR. Glucose regulates transcription in yeast through a network of signaling pathways. Mol Syst Biol. 2009;5:245. doi: 10.1038/msb.2009.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.