

Abstract
Nuclear hormone receptor (NR) function relies on association of agonist-bound receptors with steroid receptor coactivator (SRC) proteins through a small pentapeptide motif (LXXLL) of the SRC that binds to a hydrophobic groove on the NR. We have synthesized a series of bicyclo[2.2.2]octanes that are close structural mimics of the two key lysine residues of this SRC sequence as bound in the hydrophobic groove of the estrogen receptor. These bicyclic systems block the NR-SRC interaction with modest potency.
Nuclear receptors (NRs) are transcription factors that control many physiological and pathological processes by directly regulating the expression of select target genes.1 Members of the NR gene superfamily have multi-domain structures, with a conserved DNA-binding domain and a ligand-binding domain (LBD). The transcriptional activity of NRs is initiated by hormone binding that stabilizes the conformation of the LBD. When an agonist ligand binds, conformational stabilization rigidifies surface features that function as docking sites for coactivator protein complexes that are recruited to modify chromatin structure and activate RNA polymerase II. The most notable of these are the p160 class of steroid receptor coactivators (SRCs).2,3
X-ray crystallography has revealed details of these NR-SRC interactions,4–9 which are largely mediated by a short, conserved, pentapeptide LXXLL (L=leucine, X=amino acid) motif of the SRC, termed the NR box. The interaction between the estrogen receptor α (ERα) LBD and a peptide corresponding to NR box 2 of SRC1 (RHKILHRLLQE) is shown in Fig. 1.5 Selected residues of the peptide interact with the LBD through a two-turn amphipathic α-helical motif that places the first and third leucine residues (L690 and L694) in a deep, solvent-exposed hydrophobic groove made up of residues from various helices of the LBD; histidine and arginine residues of the peptide (H691 and R692, removed for clarity) are solvent exposed and appear unnecessary for the interaction.5 The intrinsic dipole moment of the coactivator α-helix is aligned with charged residues on the surface of the ERα-LBD (E542 interacts with the peptide N-terminus and K362 with its C-terminus), together forming a “charge clamp”.5
Figure 1.
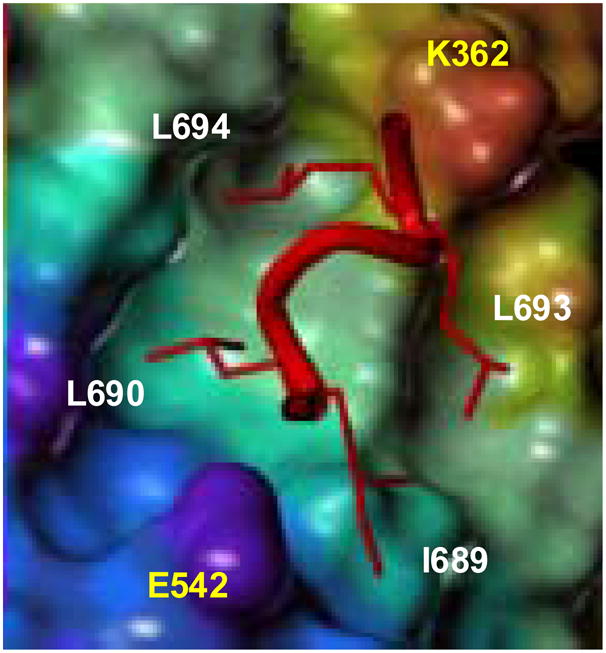
Schematic of a crystal structure of an SRC NR2 peptide bound to ERα LBD. Residues in yellow are from the LBD, white from the peptide.
The traditional strategy to block agonist signaling of NRs involves hormone antagonists (antihormones) that bind to the LBD, displacing the agonist and stabilizing alternative conformations that cannot interact with the coactivators.4–8 The effectiveness of NR antagonists can be compromised by cellular adaptations that enable coactivators to bind to NR-antihormone complexes, such as increased coactivator expression10 or covalent modifications of the interacting components.11,12 This is exemplified by the development of resistance to the antiestrogen tamoxifen in ER-based endocrine therapies for breast cancer, a phenomenon that is a serious limitation on such therapies.11,12
An alternative approach to interrupting NR signaling is the use of compounds capable of blocking the step following NR-ligand binding, namely, the interaction between the agonist-liganded ER and coactivators. Such compounds could be termed coactivator binding inhibitors (CBIs). Biochemical studies using peptides and peptide mimics of the LXXLL motifs of the SRCs have demonstrated that this alternative approach is feasible.13–16 If disruption of this helix-groove protein-protein interaction between the ERs and SRCs could be effected with small molecules, as is possible in other helix-groove interactions,17–23 such molecules would be intriguing molecular probes for studying ER signaling. They might also afford an alternative strategy for blocking estrogen action in breast cancer less likely to be compromised by the cellular adaptations responsible for tamoxifen resistance.11,12
Recently, we reported that certain small molecules, particularly 2,4,6-trisubstituted pyrimidines, are inhibitors of coactivator binding to ERα;24 since then, examples of related compounds from other functional group classes have appeared.25 These reports provide a proof-of-principle that small molecule CBIs can be developed,24 and as part of our interest in ER CBIs, we are pursuing refinements to improve their potency.
Previously, we took an “outside-in” design, in which the heterocycle (pyrimidine) functioned as a peptide scaffold mimic onto which we appended groups mimicking the key leucine residues.24 Here, we have taken an alternative “inside-out” approach, beginning with a structural surrogate of the two leucine residues in the ILHRLL sequence (L690 and L694, bolded) that extend most deeply into the hydrophobic pocket (Fig. 1) and attaching to it structural elements that mimic other residues and functional elements of the NR box helix.
Figure 2 illustrates how this can be done using a bicyclo[2.2.2]octane as the core structural mimic of the first and third leucine residues (L690 and L694) in the NR box motif. The alignment of the two isopropyl groups of these two leucine residues can be replicated almost exactly by interposing a boat cyclohexane ring (Fig. 2, purple). Two atoms of one of the isopropyl groups make a 1,4-bridge on the cyclohexane, creating the bicyclo[2.2.2]octane system; the other isopropyl group is attached to the cyclohexane by its tertiary carbon. This “inside” portion of a potential CBI can then be further adorned with functionality mimicking the N-terminal isoleucine (I689) and the second leucine (L693); appropriately charged groups could also be added to mimic the helix dipole of the NR box sequence.
Figure 2.
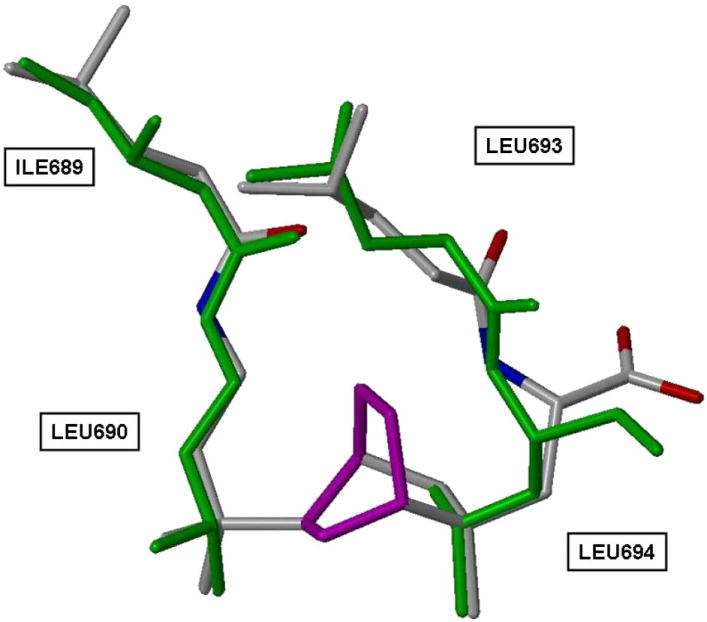
Cyclohexane connector (purple) creates a bicyclo[2.2.2]-octane CBI core from L690 and L694 of an SRC NR2 box peptide
Retrosynthetic analysis shows that this bicyclo[2.2.2]-octane system, for example, compound 1, can be assembled in two key steps: addition of a side arm and a Diels-Alder reaction of a 5-substitued 1,3-cyclohexadiene with methacrolein to form the bicyclo[2.2.2]octane core (Scheme 1). The synthesis, therefore, was divided into three parts: synthesis of the 5-substituted 1,3-cyclohexadiene, the Diels-Alder reaction, and addition of the side arms.
Scheme 1.

Retrosynthetic analysis of bicyclo[2.2.2]octane CBI 1
Preparation of the desired diene (Scheme 2) began with the cyclohexadienyl cation salt 3, prepared by abstraction of a hydride from 1,3-cyclohexadiene iron tricarbonyl 2 with triphenylcarbenium tetrafluoroborate. Nucleophilic addition of isobutyraldehyde provided aldehyde 4, in which the two methyl groups necessary to mimic L690 of the coactivator peptide have been introduced. To install one of the side arms, a Henry reaction of aldehyde 4 with nitromethane gave nitroalkene 5, which was reduced to amine 6 with lithium borohydride and Me3SiCl and converted to amide 7 with isoamyl acid chloride. We chose to make the leucine mimic instead of the isoleucine (I689) to reduce the number of potential stereoisomeric products. Trimethylamine N-oxide decomplexation of the iron tricarbonyl gave the iron-free diene 8.
Scheme 2.

Synthesis of 5-substituted 1,3-cyclohexadiene
The key step in preparing bicyclo[2.2.2]octane aldehyde 10 is a Diels-Alder reaction between diene 8 and methacrolein 9 (Scheme 3). Lewis acids are typically used in catalytic quantities to accelerate Diels-Alder reactions, and they enhance stereo- and regioselectivity. However, because of the amide function in diene 8, stoichiometric amounts of Lewis acids were needed. Ytterbium trichloride was an effective catalyst.26
Scheme 3.

Diels-Alder reaction products, relative yields, steric energies, and aldehyde 1H NMR data
Because of secondary orbital overlap, Diels-Alder reactions, when run under conditions of kinetic control, particularly with Lewis acid catalysis, favor products with endo stereochemistry.27,28 In the reaction of diene 8 with methacrolein 9, three distinct aldehyde peaks were observed between 9.25 and 9.65 ppm, indicating that three isomeric products were formed (Scheme 3). We expected that the 5-alkyl substituent on the 1,3-cyclohexadiene would control facial selectivity, directing dienophile approach from the less hindered side to give the desired stereoisomer, the one having the new dimethylene bridge anti to the C-5 group. Because alkyl substitution of the diene is at a remote position (carbon 5), there were no groups on the diene itself to strongly direct the regioselectivity of the reaction. Nevertheless, steric factors would still be expected to favor formation of regioisomers (10a and 10b), in which the carboxyaldehyde and tertiary alkyl substituents were in a 1,4- rather than a 1,3-relationship (10c) with respect to one another.
The endo and exo isomers could readily be distinguished by 1H NMR spectral comparison to reported analogs (Scheme 3), because when the double bond is syn to the aldehyde, its anisotropic effect shields the aldehyde proton, shifting its signal upfield.28,29 The structure of a minor isomer was tentatively assigned as regioisomer 10c, based on steric energy calculations (MacroModel), which predicted it to be the third most stable isomer (The steric energy values for the three isomers are given in brackets). Further synthesis was done on the major, desired isomer, 10a.
To add a second short side chain containing a carboxylic acid for interaction with the lysine charge clamp residue (K362), aldehyde 10a was converted to the unsaturated methyl ester 12 by a Horner-Wadsworth-Emmons reaction (Scheme 4). Hydrogenation gave the saturated methyl ester 14, and both esters were saponified, giving acids 13 and 15.
Scheme 4.

Synthesis of carboxylic acid side arm
Part of our original design of the bicyclo[2.2.2]octane core CBI involved addition of a more extended side chain bearing another amide group to mimic the third leucine residue, but introduction of this second amide proved surprisingly difficult. Condensations of methyl isocyanoacetate, N-(benzyloxycarbonyl)-phosphonoglycine trimethyl ester or methyl nitroacetate with aldehyde 10 proved to be unsuccessful in our hands. Aldehyde 10 did react with dimethyl malonate 16 and TiCl4 to give unsaturated diester 17 (Scheme 5). Hydrogenation gave the saturated ester 18, and saponification, the saturated monocarboxylic acid 19. Curtius rearrangement via the acyl azide gave an isocyanate, which was trapped with benzyl alcohol to give the Cbz-protected amino ester 20. Curiously, attempts to deprotect the carbobenzyloxy group of amino ester 20 under various conditions were only marginally successful; so only a trace amount of the desired amino ester 21 was obtained.
Scheme 5.

Installation of the amide side arm
We used a time-resolved fluorescence resonance energy transfer (TR-FRET) assay to evaluate the potential of these bicyclic compounds to compete with the nuclear receptor domain of SRC3 (627–829, encompassing all three NR boxes) for agonist-liganded ER (The development of this assay will be described elsewhere). Biotin-labeled ERα LBD, tagged with terbium-labeled streptavidin, was incubated with estradiol and fluorescein-labeled SRC3 NRD, labeled non-specifically through cysteine residues with iodoacetamide-fluorescein. The Fl-SRC3 binds to the estradiol-liganded Tb-ER complex, and CBI activity is assayed by the ability of increasing concentrations of compound to compete for ER-SRC binding, reducing the FRET signal. Unlabeled SRC1-NR Box2 peptide (LTERHKILHRLLQEGSPSD) and a pyrimidine prepared previously24 are used as CBI positive controls. Vehicle (DMF) and αβV peptide (that binds to tamoxifen-liganded ER) were used as negative controls.13,14 IC50 values were converted to Ki values. Representative competition binding curves for the bicyclo[2.2.2]-octanes are shown in Figure 3; additional ones are listed.
Figure 3.

TR-FRET assay of bicyclo[2.2.2]oxtane CBI and SRC1 NR2, αβV peptide and pyrimidine control compounds and vehicle activity on ERα-SRC3. Pyrimidine control is compound 12a from ref. 24.
While the control peptide and a pyridimine prepared previously show full competition curves with good potency, the bicyclo-octane CBIs have curves that are right shifted and show less complete competition. Ki values are in the range of 7–40 μM, indicating lower potency than the peptide and pyrimidine control compounds. While we are not certain how to interpret the incomplete competition curves, they could represent stabilization by these compounds of an ER complex that does not result in full displacement of the Fl-peptide but binds it with lower affinity or altered geometry, reducing but not eliminating the FRET signal. Despite the structural mimicry of the LXXLL peptide motif by these bicyclo-octanes, there is also no obvious relationship between their structures and Ki values.
Compounds that block the interaction of agonist-liganded ER with CoAs could provide unique pharmacological tools for interrupting the signal transduction of this receptor and might also allow the estrogen receptor to circumvent certain modes of cellular resistance to conventional antagonists. In this paper, we describe the structure-inspired de novo design, synthesis, and evaluation of such small molecule coactivator binding inhibitors (CBIs), based on a bicyclo[2.2.2]octane skeleton. Despite their accurate structural mimicry of the key residues involved in the ER-coactivator interaction, their potency and effectiveness is limited. Nevertheless, this work contributes to the proof-of-principle that effective small molecule CBIs for the ER can be developed.
Acknowledgments
Supported by grants from the NIH (PHS 5R37 DK15556). Funding for NMR and MS instrumentation is from the Keck Foundation, NIH and NSF. We are grateful to Dr. Sung Hoon Kim and Terry Moore for helpful comments, and Jaimin Amin for conducting the coactivator binding assays.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Katzenellenbogen BS, Katzenellenbogen JA. Breast Cancer Res. 2000;2:335. doi: 10.1186/bcr78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McKenna NJ, O’Malley BW. Endocrinology. 2002;143:2461. doi: 10.1210/endo.143.7.8892. [DOI] [PubMed] [Google Scholar]
- 3.McKenna NJ, O’Malley BW. Cell. 2002;108:465. doi: 10.1016/s0092-8674(02)00641-4. [DOI] [PubMed] [Google Scholar]
- 4.Brzozowski AM, Pike AC, Dauter Z, Hubbard RE, Bonn T, Engstrom O, Ohman L, Greene GL, Gustafsson JA, Carlquist M. Nature. 1997;389:753. doi: 10.1038/39645. [DOI] [PubMed] [Google Scholar]
- 5.Shiau AK, Barstad D, Loria PM, Cheng L, Kushner PJ, Agard DA, Greene GL. Cell. 1998;95:927. doi: 10.1016/s0092-8674(00)81717-1. [DOI] [PubMed] [Google Scholar]
- 6.Shiau AK, Barstad D, Radek JT, Meyers MJ, Nettles KW, Katzenellenbogen BS, Katzenellenbogen JA, Agard DA, Greene GL. Nat Struct Biol. 2002;9:359. doi: 10.1038/nsb787. [DOI] [PubMed] [Google Scholar]
- 7.Pike AC, Brzozowski AM, Hubbard RE, Bonn T, Thorsell AG, Engstrom O, Ljunggren J, Gustafsson JA, Carlquist M. Embo J. 1999;18:4608. doi: 10.1093/emboj/18.17.4608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pike AC, Brzozowski AM, Walton J, Hubbard RE, Thorsell AG, Li YL, Gustafsson JA, Carlquist M. Structure. 2001;9:145. doi: 10.1016/s0969-2126(01)00568-8. [DOI] [PubMed] [Google Scholar]
- 9.Darimont BD, Wagner RL, Apriletti JW, Stallcup MR, Kushner PJ, Baxter JD, Fletterick RJ, Yamamoto KR. Genes Dev. 1998;12:3343. doi: 10.1101/gad.12.21.3343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shang Y, Brown M. Science. 2002;295:2465. doi: 10.1126/science.1068537. [DOI] [PubMed] [Google Scholar]
- 11.Schiff R, Massarweh S, Shou J, Osborne CK. Clin Cancer Res. 2003;9:447S. [PubMed] [Google Scholar]
- 12.Osborne CK, Bardou V, Hopp TA, Chamness GC, Hilsenbeck SG, Fuqua SA, Wong J, Allred DC, Clark GM, Schiff R. J Natl Cancer Inst. 2003;95:353. doi: 10.1093/jnci/95.5.353. [DOI] [PubMed] [Google Scholar]
- 13.Connor CE, Norris JD, Broadwater G, Willson TM, Gottardis MM, Dewhirst MW, McDonnell DP. Cancer Res. 2001;61:2917. [PubMed] [Google Scholar]
- 14.Norris JD, Paige LA, Christensen DJ, Chang CY, Huacani MR, Fan D, Hamilton PT, Fowlkes DM, McDonnell DP. Science. 1999;285:744. doi: 10.1126/science.285.5428.744. [DOI] [PubMed] [Google Scholar]
- 15.Galande AK, Bramlett KS, Trent JO, Burris TP, Wittliff JL, Spatola AF. Chembiochem. 2005;6:1991. doi: 10.1002/cbic.200500083. [DOI] [PubMed] [Google Scholar]
- 16.Galande AK, Bramlett KS, Burris TP, Wittliff JL, Spatola AF. J Pept Res. 2004;63:297. doi: 10.1111/j.1399-3011.2004.00152.x. [DOI] [PubMed] [Google Scholar]
- 17.Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, Fotouhi N, Liu EA. Science. 2004;303:844. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 18.Wang JL, Liu D, Zhang ZJ, Shan S, Han X, Srinivasula SM, Croce CM, Alnemri ES, Huang Z. Proc Natl Acad Sci U S A. 2000;97:7124. doi: 10.1073/pnas.97.13.7124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Degterev A, Lugovskoy A, Cardone M, Mulley B, Wagner G, Mitchison T, Yuan J. Nature Cell Biology. 2001;3:173. doi: 10.1038/35055085. [DOI] [PubMed] [Google Scholar]
- 20.Issaeva N, Bozko P, Enge M, Protopopova M, Verhoef LGGC, Masucci M, Pramanik A, Selivanova G. Nature Medicine. 2004;10:1321. doi: 10.1038/nm1146. [DOI] [PubMed] [Google Scholar]
- 21.Parks DJ, LaFrance LV, Calvo RR, Milkiewicz KL, Gupta V, Lattanze J, Ramachandren K, Carver TE, Petrella EC, Cummings MD, Maguire D, Grasberger BL, Lu T. Bioorganic and Medicinal Chemistry Letters. 2005;15:765. doi: 10.1016/j.bmcl.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 22.Ernst JT, Becerril J, Park HS, Yin H, Hamilton AD. Angew Chem Int Ed Engl. 2003;42:535. doi: 10.1002/anie.200390154. [DOI] [PubMed] [Google Scholar]
- 23.Kutzki O, Park HS, Ernst JT, Orner BP, Yin H, Hamilton AD. J Am Chem Soc. 2002;124:11838. doi: 10.1021/ja026861k. [DOI] [PubMed] [Google Scholar]
- 24.Rodriguez AL, Tamrazi A, Collins ML, Katzenellenbogen JA. J Med Chem. 2004;47:600. doi: 10.1021/jm030404c. [DOI] [PubMed] [Google Scholar]
- 25.Shao D, Berrodin TJ, Manas E, Hauze D, Powers R, Bapat A, Gonder D, Winneker RC, Frail DE. J Steroid Biochem Mol Biol. 2004;88:351. doi: 10.1016/j.jsbmb.2004.01.007. [DOI] [PubMed] [Google Scholar]
- 26.Furuno H, Hanamoto T, Sugimoto Y, Inanaga J. Org Lett. 2000;2:49. doi: 10.1021/ol991189q. [DOI] [PubMed] [Google Scholar]
- 27.Liu W, You F, Mocella CJ, Harman WD. J Am Chem Soc. 2006;128:1426. doi: 10.1021/ja0553654. [DOI] [PubMed] [Google Scholar]
- 28.Corey EJ, Shibata T, Lee TW. J Am Chem Soc. 2002;124:3808. doi: 10.1021/ja025848x. [DOI] [PubMed] [Google Scholar]
- 29.Ishihara K, Kurihara H, Matsumoto M, Yamamoto H. Journal of the American Chemical Society. 1998;120:6920. [Google Scholar]