

Abstract
Hyperglycemia has a profound effect on gastric motility. However, little is known about site and mechanism that sense alteration in blood glucose level. The identification of glucose-sensing neurons in the nodose ganglia led us to hypothesize that hyperglycemia acts through vagal afferent pathways to inhibit gastric motility. With the use of a glucose clamp rat model, we showed that glucose decreased intragastric pressure in a dose-dependent manner. In contrast to intravenous infusion of glucose, intracisternal injection of glucose at 250 and 500 mg dL−1 had little effect on intragastric pressure. Pretreatment with hexamethonium, as well as truncal vagotomy, abolished the gastric motor responses to hyperglycemia (250 mg dL−1), and perivagal and gastroduodenal applications of capsaicin significantly reduced the gastric responses to hyperglycemia. In contrast, hyperglycemia had no effect on the gastric contraction induced by electrical field stimulation or carbachol (10−5 M). To rule out involvement of serotonergic pathways, we showed that neither granisetron (5-HT3 antagonist, 0.5 g kg−1) nor pharmacological depletion of 5-HT using p-chlorophenylalanine (5-HT synthesis inhibitor) affected gastric relaxation induced by hyperglycemia. Lastly, NG-nitro-L-arginine methyl ester (l-NAME) and a VIP antagonist each partially reduced gastric relaxation induced by hyperglycemia, and in combination, completely abolished gastric responses. In conclusion, hyperglycemia inhibits gastric motility through a capsaicin-sensitive vagal afferent pathway originating from the gastroduodenal mucosa. Hyperglycemia stimulates vagal afferents, which, in turn, activate vagal efferent cholinergic pathways synapsing with intragastric nitric oxide- and VIP-containing neurons to mediate gastric relaxation.
Keywords: nitric oxide, peptide VIP, hyperglycemia
Introduction
Hyperglycemia has a wide range of effects on gastrointestinal functions. The inhibitory effect of hyperglycemia on gastric motility is well known. Bulato and Carlson (8) were the first to report that hyperglycemia inhibits hunger contractions in the fasted dog, and conversely, insulin-induced hypoglycemia produces gastric hypermotility that can be inhibited by I.V. glucose administration. Similarly, in healthy human subjects, glucagon-induced hyperglycemia abolishes hunger contractions, as measured with an intragastric balloon, and when blood glucose levels return to control levels, gastric contractions are restored (36). Aylett (2) showed that blood glucose elevation slows gastric emptying. Barnett and Owyang (4) used a glucose clamp study to demonstrate that acute hyperglycemia inhibits the occurrence of the gastric interdigestive migrating motor complex in healthy volunteers. During the 3-hour hyperglycemic period, gastric contractions were significantly reduced at 140 and 175 mg/ dL−1 and were almost completely absent at a blood glucose level of 250 mg dL−1. This observation has important clinical implications for patients with diabetic gastroparesis, as it may explain the common observation that stable gastric neuropathies often exhibit wide day-to-day fluctuations in gastric emptying rates and symptoms of nausea and vomiting (12,14,16,17).
Little is known about the site and mechanism that sense alteration in blood glucose level. The vagovagal reflex plays an important role in the mediation of many digestive functions, including gastrointestinal motility (23,25,37,38). The primary vagal afferents transmit sensory information about the physiological status of the gastrointestinal tract, including mechanical and chemical stimulation and energy metabolism. The identification of glucose-sensing neurons in the nodose ganglia (13,46) led us to hypothesize that hyperglycemia acts through vagal afferent pathways to inhibit gastric motility.
In this study, we examined the hypothesis that hyperglycemia stimulates vagal afferents that act by way of the brainstem to stimulate the vagal efferent cholinergic pathway synapsing with intragastric nitric oxide (NO)- and peptide VIP-containing neurons to mediate gastric relaxation. Pharmacological studies, surgical resection, and chemical ablation of vagal afferent fibers were performed with an in vivo rat model.
Materials and Methods
Ethical Approval
All experiments involving animals were approved by the University Committee on Use and Care of Animals at the University of Michigan.
Materials
The following materials were purchased: NG-nitro-L-arginine methyl ester (l-NAME) and VIP antagonist (P-chloro-d-Phe6, Leu17)-VIP from Bachem (Torrance, CA); capsaicin, atropine sulfate, carbachol, p-chlorophenylalanine (PCPA), and hexamethonium bromide from Sigma-Aldrich (St. Louis, MO). Drugs were dissolved in physiological saline.
Animal preparation
Male Sprague-Dawley rats weighing between 250–300 g were fasted with water available ad libitum. The rats were anaesthetised with urethane (1.0–1.5 g kg−1 I.P.). A tracheotomy was performed and a tracheal tube was inserted through which the animals breathed room air spontaneously. Through a midline incision, a catheter with an attached rubber balloon was inserted into the stomach through an incision in the duodenum, as described in the next section. Cannulation of the jugular veins with polyethylene tubing (PE 50, BD Diagnostics, Sparks, MD) was performed in each rat.
Measurement of intragastric pressure
Intragastric pressure was measured with the use of a rubber balloon that was tied around a polyethylene tube (PE 160) and inserted into the body of the stomach through a small incision in the duodenum, as previously described (25). The balloon was secured at the pylorus with a suture to avoid movement, and the tube was connected to a pressure transducer (World Precision Instruments, Sarasota, FL), which was connected to a transducer amplifier (TBM4M, World Precision Instruments). The balloon was filled with water at 37°C (1.2–2.0 mL, the volume determined to be the level necessary to induce an intragastric pressure of 5–10 cm H2O). Pressures were recorded and analyzed by Spike2, the data acquisition system for online analysis (Cambridge Electronic Design, Cambridge, England). The exact location of the balloon was verified after each experiment.
Hyperglycemia clamp studies
Hyperglycemia was achieved with the use of a hyperglycemic clamp, as described by Ishiguchi et al (18), who adapted the method from previous studies in humans (10). The clamp facilitates obtaining blood glucose concentrations at preset hyperglycemic levels up to 300 mg dL−1 and maintaining them for at least 30 min. The rats were anesthetized with urethane (1.0–1.5 g kg−1, i.p.). The right jugular vein was exposed and a polyethylene catheter (PE 50) was surgically placed for glucose infusion. The animals were randomly divided into 2 groups: one group was given a saline infusion (control) and the other, a 20% dextrose infusion. Glucose concentrations in blood obtained from the tail were measured every 5–10 min with a glucose meter (Accu-Check, Roche, Mannheim, Germany). For blood sampling, rat was held in a restrainer and its tail was cleaned and poked with 26G 1/2 syringe needle. A drop of blood was collected and placed on glucose test strip. Blood glucose levels were raised stepwise to preset concentrations by infusing a priming dose of 20% dextrose in the first 10 min with an infusion pump (SP 100i syringe pump, World Precision Instruments) at the rate of 100 μL min−1. After achieving hyperglycemia, the blood glucose concentration was maintained by adjusting the rate of the glucose infusion according to the blood glucose concentration measured every 5–10 min. Intragastric pressure was measured as described in the previous section.
Bilateral subdiaphragmatic vagotomy
To demonstrate that hyperglycemia acts by way of stimulation of the vagal pathways, acute bilateral subdiaphragmatic vagotomy was performed as previously described (25). A midline incision was made in the abdominal wall and the stomach was carefully manipulated to expose the esophagus. The subdiaphragmatic vagal trunks were exposed halfway between the diaphragm and the gastric cardia. Both anterior and posterior trunks of the vagal nerves were transected. For the control experiments, the abdominal vagal nerves were exposed but not cut. Hyperglycemia studies were performed as described in the previous section. To demonstrate the completeness of vagotomy, the gastric response to electrical stimulation of the vagus nerve was tested at the end of the experiments, as described in the next section.
Nerve stimulation and carbachol studies
Through a midline incision on the anterior surface of the neck, the right cervical vagus nerve was dissected free. The peripheral cut end of the cervical vagus nerve was placed on an electrode and covered with liquid paraffin. The nerve was stimulated with a Grass stimulator (10 V; 1.25, 2.5, or 5 Hz; and 2 ms for 30 s) at 30 min before and 10 min after hyperglycemia was established. To determine if hyperglycemia affects the muscle response to cholinergic stimulation, intragastric pressure response to carbachol (10−5 M, 0.1 ml given intravenously) was studied in the presence of hexamethonium (10 mg kg −1 iv). The study was repeated with intravenous infusion of glucose to induce hyperglycemia (250 mg dL−1)
Perivagal application of capsaicin
To investigate the role of the vagal afferent pathway in the mediation of the effect of hyperglycemia, we examined the effect of perivagal application of capsaicin (22,25). Following anesthetization with sodium pentobarbital (50 mg/kg ip), an upper midline laparotomy was performed and the abdominal vagal nerve trunks were exposed and isolated with a piece of parafilm. A small piece of gauze soaked in 1% capsaicin solution (0.2 mL per rat) was applied to the vagal trunks for 30 min. After capsaicin treatment, the gauze was removed. The nerve trunks were rinsed with warm saline and then the parafilm was removed. Vehicle alone was applied to the vagal trunks of the control rats. Hyperglycemia studies as described in the previous section were performed 5 days after surgery in the capsaicin-treated and control rats.
Gastroduodenal mucosal application of capsaicin
To determine if the glucose-sensitive afferent nerve endings originate from the gastroduodenal mucosa, we examined the effects of the mucosal application of capsaicin in the stomach and duodenum (25). Rats were anesthetized with sodium pentobarbital (50 mg/kg ip). After laparotomy, the stomach and duodenum were isolated and temporarily ligated at both ends. 2 mL capsaicin (6 mg mL−1 dissolved in 10% Tween 80 in olive oil) was applied topically to the gastroduodenal mucosa for 30 min. A saline application was used in control rats. Hyperglycemia studies were performed 7 days after local capsaicin application. Rats were checked for normal eye wiping movement, which is an indication that local mucosal treatment of capsaicin has no systemic effect (41).
Effects of 5-HT3 receptor antagonist and PCPA treatments
Our previous studies indicated that luminal glucose stimulates the vagal afferent pathway through the release of intestinal 5-HT, which acts as a paracrine substance to mediate vagal signal transduction by way of the 5-HT3 receptor (24,47). To rule out involvement of the 5-HT3 pathway in the mediation of the gastric motility response to hyperglycemia, we measured the effect of the 5-HT3 receptor antagonist granisetron (15). Gastric motility studies in response to hyperglycemia (250 mg dL−1) were performed 15 min after I.V. infusion of granisetron (0.5 mg kg−1). To further exclude any role for endogenous 5-HT in the mediation of gastric motility responses to hyperglycemia, we examined the effects of PCPA, a 5-HT synthesis inhibitor. PCPA inhibits tryptophan hydroxylase, the enzyme acting at the rate-limiting step of 5-HT synthesis. Depletion of 5-HT stores in the brain, intestinal tissue, and blood occurs after PCPA treatment (20,42,47). PCPA (500 mg kg−1) suspended in 2 mL gum arabic solution was administered I.P. on 2 consecutive days. Controls received only gum arabic. Studies of the gastric motility response to hyperglycemia (250 mg dL−1) were conducted as described in the previous section.
Antagonist studies
To determine the role of nicotinic receptors in the mediation of gastric relaxation induced by hyperglycemia, hexamethonium (20 mg kg−1 bolus) was injected and continuously infused at a rate of 10 mg kg−1 h−1 for 20 min before the administration of glucose, as previously described (37). To determine the role of NO and VIP, L-NAME (10 mg kg−1) and a VIP antagonist (30 nmol kg−1) were injected 10 min before the infusion of glucose. These specific doses of l-NAME and the VIP antagonist have been shown to abolish rapid and prolonged relaxation, respectively, in response to vagal stimulation in vivo (37,47).
Effect of central hyperglycemia on gastric motility
To rule out the possibility that hyperglycemia may act at a central site to induce gastric relaxation, we examined the effect of intracisternal injection of glucose on gastric motility. We adapted the method from previous study by Adelson et al. (1). Briefly, the rats were anaesthetized and the head of each rat was fixed in a stereotactic holder. A midline incision was made along the back of the head and neck, and the underlying muscles were separated and retracted. The atlantooccipital membrane was carefully exposed. A small incision was made in the membrane with a 25-gauge needle. A PE 10 polyethylene tube (BD) filled with saline was inserted through the incision into the cisterna magna. The cannula was secured with a drop of cyanoacrylate glue and the incision was closed.
Gastric motility studies were performed as described in the previous section. After a 30-min recording, 100 μL of glucose (250 or 500 mg dL−1) dissolved in 1000 μL saline was administered with a Hamilton syringe into the cisterna magna over 30 s, followed by a 5-μL saline flush. Intragastric pressure was monitored for 20 min after each injection.
Analysis of data
Results were expressed as means ± SE. Statistical analysis was performed using 1-way ANOVA followed by the Kruskal-Wallis test (a multiple comparisons procedure) or the t test, depending on the particular study design. Significance was accepted at the level of P < 0.05.
Results
Glucose Clamp Studies
After an overnight fast, the basal blood glucose level was 90 ± 7 mg dL−1 (n = 10). The intragastric pressure was set at 5–10 cm H2O with balloon distension, and this level remained stable for at least 60 min before the infusion of glucose. i.v. infusion of dextrose (20%) produced a dose-dependent inhibition of gastric motility (Fig. 1). At blood glucose concentrations of 150, 200, 250, 300, and 350 g dL−1, intragastric pressure decreased by −0.76 ± 0.34, −1.85 ± 0.38, −1.91 ± 0.28, −2.31 ± 0.23, and −2.59 ± 0.38 cm H2O, respectively. As shown in Fig. 2, increasing blood glucose levels reduced stomach muscle tone and progressively inhibited phasic gastric activities. Decreasing blood glucose levels from 300 to 150 mg dL−1 partially restored phasic gastric activities. Subsequent studies were performed with hyperglycemia maintained at 250–300 mg dL−1.
Figure 1. Dose-dependent gastric relaxation after glucose infusion.

Bar chart shows dose-related responses of acute hyperglycemia-induced gastric relaxation. Note that gastric relaxation occurred at blood glucose levels ≥100 mg dL−1 but was not statistically significant compared to baseline when the blood glucose level was between 70–100 mg dL−1. Mean ± SE of 7–10 rats is shown for each glucose level. *P < 0.05, **P < 0.01, and ***P < 0.001 compared to intragastric pressure (IGP) when the blood glucose level <100 mg dL−1. P < 0.001 based on 1-way ANOVA followed by the Kruskal-Wallis test.
Figure 2. Hyperglycemia induced gastric relaxation.

A representative tracing shows a decrease in intragastric pressure (IGP) induced by acute hyperglycemia secondary to i.v. infusion of 20% dextrose in a rat. The dextrose infusion produced inhibition of gastric tone and phasic activities. When blood glucose levels were decreased, gastric tone and phasic activities partially recovered.
Effects of hexamethonium and vagotomy
Administration of hexamethonium (10 mg kg−1) reduced basal intragastric pressure by 25 ± 3%; intragastric pressure returned to basal level within 30 min. Hexamethonium markedly reduced the inhibitory action of hyperglycemia maintained at 250 mg dL−1 (P < 0.05) (Fig. 3). This observation indicates that hyperglycemia acts at a presynaptic site of the cholinergic pathway.
Figure 3. Effects of vagotomy, i.v. administration of hexamethonium, and perivagal or gastroduodenal application of capsaicin on hyperglycemia-induced gastric relaxation.

Each procedure significantly reduced hyperglycemia-induced gastric relaxation. n = 6 for each group. *P < 0.05 compared to control on the basis of a t test. VAG, vagotomy; HEX, hexamethonium; PVcap, perivagal capsaiscin; GDcap, gastroduodenal capsaiscin.
Immediately after truncal vagotomy, basal intragastric pressure was reduced by 26 ± 4%. The intragastric pressure returned to basal level within 30 min, and the glucose infusion experiment was started. Similar to hexamethonium, truncal vagotomy markedly inhibited the gastric response to hyperglycemia at 250 mg dL−1 (P < 0.05) (Fig. 3). This suggests that the vagal pathway is the primary site of action of glucose to inhibit gastric motility.
Effect of perivagal and gastroduodenal mucosal applications of capsaicin
Perivagal application of capsaicin markedly reduced gastric relaxation in response to hyperglycemia (250 mg dL−1) (P < 0.05) (Fig. 3). Similar to perivagal application of capsaicin, gastroduodenal application of capsaicin also significantly reduced the gastric responses to hyperglycemia (P < 0.05) (Fig. 3). In separate studies, we showed that neither perivagal nor gastroduodenal mucosal applications of capsaicin affected gastric responses to electrical vagal stimulation (n = 6) (10 V, 5 Hz, 2 ms for 30 s) (data not shown), indicating that administration of capsaicin does not affect efferent vagal transmission.
Effect of hyperglycemia on gastric contraction induced by electrical vagal stimulation and intravenous administration of carbachol
Electrical vagal stimulation (10 V; 1.25, 2.5, or 5 Hz; 2 ms for 30 s) induced a frequency-dependent increase in gastric pressure. This contraction was completely abolished by atropine (100 μg kg−1 h−1). Electrical stimulation (10 V, 5Hz, 2 ms for 30 s) of the peripheral cut end of the vagus nerve produced an increase in intragastric pressure (3.32±0.33 cm H2O) (n=4). Infusion of d-glucose to achieve a blood level of 250 mg dL−1 failed to affect the gastric contraction induced by vagal stimulation (3.26±0.39 cm H2O) (n=4, P>0.05).
To determine if hyperglycemia affects the muscle response to cholinergic stimulation, we showed that carbachol (10−5 M, 0.1 ml, iv) produced an increase in intragastric pressure (6.97±0.91 cm H2O) (n = 4) in the presence of hexamethonium (10 mg kg−1, iv). Infusion of glucose to induce hyperglycemia (250 mg dL−1) failed to inhibit the contractile response induced by carbachol (6.62±1.36 cm H2O) (n=4, p>0.05).
Effects of 5-HT3 receptor antagonist and PCPA treatments
Intravenous infusion of granisetron (0.5 g kg−1) had no effect on intragastric pressure. 15 min after the administration of granisetron, i.v. infusion of 20% dextrose to achieve a blood glucose level of 250 mg dL−1 produced gastric relaxation similar to that observed under control conditions without granisetron (2.90±0.41 vs 3.21±0.65 cm H2O, n=4, P>0.05).
PCPA (500 mg kg−1) was injected I.P. into 6 rats on 2 consecutive days and 4 rats were pretreated with vehicle (gum arabic) solution. Hyperglycemia (250 mg dL−1) induced by glucose infusion stimulated a similar degree of gastric relaxation in both PCPA- and vehicle-treated rats (3.18±0.45 vs 2.94±0.54 cm H2O, n=6, P>0.05).
Effects of l-NAME and VIP antagonist
To determine if NO and VIP play a role in mediating gastric relaxation induced by hyperglycemia, we examined the effects of L-NAME and a potent VIP antagonist. NO and VIP have been shown to mediate vagally induced gastric relaxation (28). Administration of l-NAME (10 mg kg−1), which abolished the rapid transient relaxation induced by electrical vagal stimulation (28), reduced the gastric relaxation induced by hyperglycemia (250 mg dL−1) by 76 ± 9% (P < 0.05) (Fig. 4). i.v. administration of the VIP antagonist (30 nmol kg−1) also decreased gastric relaxation in response to hyperglycemia (250 mg dL−1) by 53 ± 12% (P < 0.05) (Fig. 4). A combination of l-NAME and the VIP antagonist completely abolished the gastric relaxation stimulated by hyperglycemia (250 mg dL−1) (Fig. 4).
Figure 4. Effects of l-NAME and VIP antagonist on hyperglycemia-induced gastric relaxation.

To determine if non-adrenergic, non-cholinergic pathways are involved in hyperglycemia-induced gastric relaxation, we compared gastric motility induced by hyperglycemia in animals treated with saline to that in animals treated with l-NAME (10 mgkg−1) or VIP antagonist (30 nmol kg−1), or a combination of l-NAME and VIP antagonist. l-NAME (P < 0.05, n = 6) and VIP antagonist (VIPantag) (P < 0.05, n = 8) each significantly reduced gastric relaxation after glucose infusion. The combination of l-NAME and VIP antagonist completely blocked the gastric relaxation induced by hyperglycemia (P < 0.05, n = 8).
Effect of intracisternal injection of glucose
In contrast to i.v. infusion of glucose, intracisternal injection of glucose at 250 mg dL−1 or 500 mg dL−1 did not significantly affect intragastric pressure. A tracing from one gastric pressure recording is shown in Fig. 5. Blood glucose remained eugylcemic after intracisternal injection of glucose.
Figure 5. Effects of intracisternal injection of glucose on intragastric pressure.
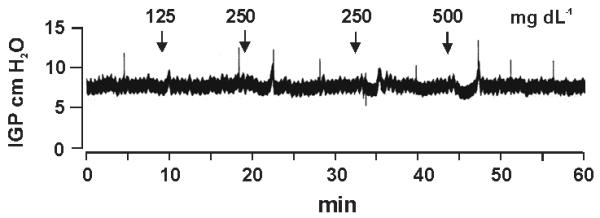
Representative recording shows the effects of intracisternal injection of glucose up to 500 mg dL−1 on gastric motility. Note the transient depression (<3 min) in intragastric pressure (IGP) at glucose doses of 250 and 500 mg dL−1.
Discussion
This study showed that hyperglycemia reduced gastric motility in a dose-dependent manner. This effect was abolished by perivagal or gastroduodenal mucosal application of capsaicin. Furthermore, we provided evidence that hyperglycemia stimulated vagal afferent pathways, which, in turn, activated vagal efferent cholinergic pathways synapsing with intragastric NO and VIP neurons to mediate gastric relaxation. This is the first demonstration that hyperglycemia inhibits gastric motility by activating the vagal afferent pathways innervating the gastroduodenal mucosa.
The infusion of concentrated dextrose as performed in the current study caused an elevation in blood insulin levels. It has been shown that insulin injection activated neurons in the enteric plexus in the stomach. The action is vagally-mediated since vagotomy abolished the c-fos expression in the stomach in response to insulin-induced hypoglycemia (45). Although it is conceivable that hyperinsulinism secondary to hyperglycemia is responsible for the inhibition of gastric motility, it is unlikely because previous work indicates a stimulatory role for insulin on gastrointestinal tract motility. Prasad and Sarna (29) reported that administration of insulin to fasted dogs results in premature migrating myoelectric complex phase III activity. Other investigators observed disruption of the migrating myoelectric complex and development of intestinal hyperactivity characteristic of a fed-like pattern during insulin infusion (7).
Although the presence of glucose-sensing neurons in the hypothalamus was first reported in 1953 (27), little is know about the site and mechanism that sense alteration in blood glucose level and modulate gastric motility. In general, the glucose-sensing neurons located in the brain are involved in the control of neuroendocrine function, nutrient metabolism, and energy homeostasis (21). These central nervous system neurons are unlikely to play a major role in mediating digestive functions in response to changes in circulating glucose under physiological conditions because the glucose level in the cerebrospinal fluid ranges between 10%–30% of blood glucose levels, and hence, these neurons are too insensitive to detect physiological changes in blood glucose concentrations (34). In this study, we further demonstrated that intracisternal injection of glucose into the cisterna magna failed to affect gastric motility, thus ruling out a central site of action of glucose to inhibit gastric motility.
However it should be noted that in contrast to peripheral hyperglycemia, insulin hypoglycemia on GI functions appears to be centrally mediated (45). These investigators showed that hypoglycemia induced by insulin induced neuronal activation in the brain vagal-regulatory nuclei including the para-ventricular nucleus (PVN) of the hypothalamus, locus coeruleus, dorsal motor nucleus of the vagus (DMN) and nucleus tractus solitarii (NTS). These in turn altered GI functions by activating the vagal efferent pathways. This action of hypoglycemia is independent of the vagal afferents (45) as bilateral cervical vagotomy did not influence insulin-induced Fos expression in the brain. Thus it appears that both glucose-sensitive neurons and glucose sensors are located both centrally and peripherally. Our current studies indicate that peripheral hyperglycemia causes gastric relaxation via its action on vagal afferents. Others show that insulin hypoglycemia exerts its action mainly in the CNS to alter gastric functions. These suggest that central and peripheral glucose sensitive neurons may have differential sensitivity to hypo- and hyper-glycemia.
Recent studies suggest that acute hyperglycemia affects a subpopulation of neurons in the nucleus tractus solitarius and the dorsal motor nucleus of the vagus (3,11,19,28,43). These investigators demonstrated a prominent action of glucose to increase vagal afferent excitatory synaptic transmission to NTS neurons. Sakaguchi et al. (31,32) reported that glucose injection into the dorsal motor nucleus of the vagus of anesthetized rats decreases gastric motility. In addition, Ferreira et al. (11) demonstrated that administration of glucose into the nucleus tractus solitarius modulates gastric motor and secretory functions. The physiological relevance of these observations is unknown; these investigators did not demonstrate that neurons in the nucleus tractus solitarius or the dorsal motor nucleus of the vagus are true primary sensors of changes in peripheral glucose levels.
Our study showed that the nicotinic receptor antagonist hexamethonium markedly reduced the inhibitory effect of hyperglycemia on gastric motility, suggesting that hyperglycemia is acting on a presynaptic site along the cholinergic pathway. To further identify the location of action, we examined the effect of bilateral subdiaphragmatic vagotomy. Similar to hexamethonium, vagotomy also markedly reduced the inhibitory action of hyperglycemia, suggesting that hyperglycemia inhibits gastric motility via a vagal pathway. To determine if hyperglycemia exerts its action via an afferent or an efferent vagal pathway, we examined the effect of perivagal treatment with the sensory neurotoxin capsaicin. Capsaicin has been widely used as a tool to investigate the role of afferent C fibers in many physiological processes (5,6,25,30,35). Systemic administration of capsaicin affects neurotransmission in all somatic and visceral capsaicin-sensitive fibers. In this study we applied capsaicin directly to the vagal trunks to avoid any damage to the afferent nerve terminals in the peripheral and central nervous systems, which has been observed with systemic administration of capsaicin. Previous studies have shown that perivagal capsaicin treatment interrupts the vagal afferent pathways that mediate CCK’s action on satiety (35), gastric motility and emptying (30), and pancreatic enzyme secretion (22). Our studies showed that perivagal pretreatment with capsaicin markedly reduced the gastric response to hyperglycemia, an effect similar to that observed with vagotomy. Similar observations have been made with secretin, which, at physiological doses, inhibits gastric motility by way of a vagal afferent pathway (25).
To further localize the site of action of hyperglycemia on the vagal afferent pathway, we examined the effect of the mucosal application of capsaicin in the gastroduodenal region. This technique has been used to demonstrate chemically the sensory fibers in the duodenal mucosa (41). We showed that, similar to perivagal capsaicin treatment, gastroduodenal application of capsaicin markedly reduced the inhibitory effect of hyperglycemia, indicating that glucose-sensitive afferent fibers originate from the vagal branch in the mucosa of the stomach and duodenum. These observations were further confirmed with the use of an in vitro vagus stomach preparation that completely eliminated any influence of the central nervous system and/or systemic responses (44). We have previously identified subsets of gastric vagal afferents that are glucose responsive (46).
Hyperglycemia reportedly inhibits cholinergic transmission (40), which may contribute to a delay in gastric emptying (9). To rule out this possibility, we showed that hyperglycemia fails to inhibit gastric contraction induced by electrical field stimulation. This suggests that hyperglycemia does not have a direct inhibitory effect on the vagal release of acetylcholine, which is the principal postsynaptic neuronal neurotransmitter mediating muscle contraction induced by electrical vagus stimulation. Hence, it argues against hyperglycemia-induced inhibition of the vagal efferent pathways to mediate gastric relaxation. Moreover, the failure of hyperglycemia to inhibit gastric contraction induced by carbachol indicates that hyperglycemia does not have a direct inhibitory effect on gastric smooth muscle cells.
Luminal stimuli, such as glucose or maltose, induce 5-HT release from mucosal enterochromaffin cells, and this, in turn, activates 5-HT3 receptors on mucosal vagal afferent terminals (47). In this manner, 5-HT acts as a paracrine substance to inhibit gastric motility through a vagal cholinergic pathway. To rule out involvement of the 5-HT3 pathway in the mediation of the gastric motility response to hyperglycemia, we showed that the 5-HT3 antagonist granisetron failed to affect gastric relaxation induced by hyperglycemia. In separate studies, we showed that pharmacological depletion of 5-HT stores with the use of a 5-HT synthesis inhibitor p-chlorophenylalanine, which markedly reduces duodenal mucosal 5-HT levels and abolishes pancreatic secretion induced by luminal administration of glucose (23), also had no affect on gastric relaxation induced by hyperglycemia (250 mg dL−1). This suggests that hyperglycemia inhibits gastric motility by way of a 5-HT-independent pathway.
Vagal stimulation produces 2 modes of relaxation in the rat stomach: rapid relaxation followed by prolonged relaxation (39). Rapid relaxation is antagonized by a NO inhibitor and prolonged relaxation is blocked by a VIP antagonist (39), which suggests that different neurotransmitters mediate different modes of relaxation. In rats, gastric distension induces a vagovagal reflex that stimulates the NO-containing gastric myenteric plexus (37). On the other hand, secretin-induced gastric relaxation is mediated by a VIP pathway (26). In this study, we showed that L-NAME and the VIP antagonist each partially reduced gastric relaxation induced by hyperglycemia. A combination of the 2 antagonists completely abolished hyperglycemia-induced gastric relaxation. These observations indicate that activation of vagal afferents by hyperglycemia stimulates vagal efferent cholinergic pathways synapsing with intragastric NO- and VIP-containing neurons to mediate gastric relaxation.
Our finding that acute hyperglycemia inhibits gastric motility differs from that reported by Shi et al. (33), who showed that hyperglycemia failed to inhibit spontaneous or bethanechol-induced gastric contractions in rats, as measured by a strain-gauge force transducer sutured to the antrum. On the other hand, I.V. infusion of glucose inhibited the antral contractions stimulated by insulin-induced hypoglycemia. This inhibitory action of glucose was not affected by sectioning the hepatic branch of the vagus nerve nor by capsaicin treatment. These differences in findings may be related to the methods used to stimulate and record gastric pressure. We measured gastric pressure with a water-filled balloon inserted into the body of the stomach, whereas Shi et al. (33) measured antral motility using a strain-gauge transducer sutured to the antrum. It is not surprising that the inhibitory effect of glucose on antral contractions induced by hyperglycemia was not sensitive to capsaicin treatment. Since insulin-induced hypoglycemia acts centrally to stimulate gastric contractions, one would not expect this action to be affected by glucose acting on the vagal afferent fibers. In fact, we also observed that hyperglycemia did not affect gastric contractions induced by direct electrical stimulation of the vagus nerve. This further emphasizes the importance of the methods used to stimulate stomach contraction because they will determine the sensitivity of gastric motor function to changes in blood glucose levels.
The demonstration that hyperglycemia inhibits gastric motility has obvious clinical importance. It suggests that hyperglycemia alone, in the absence of underlying neuropathy or myopathy, can alter gastric motor function. This may explain the common clinical observation that diabetic patients with stable motor defects often exhibit wide day-to-day variations in the severity of their symptoms depending on blood glucose control (14). In type I and type II diabetes, there is a strong correlation between delayed gastric emptying of liquids and blood glucose levels >270 mg dL−1 (16,17). Similarly, delays in solid emptying occur during periods of hyperglycemia in type I diabetes, which improve during periods of euglycemia (12). This study identifies the site and neural pathways that sense alteration in blood glucose level and modulate gastric motility.
Acknowledgments
The authors wish to thank Mr. Tin Ming Mok for technical assistance. This study was supported by the American Diabetes Association Grant 1-06-JF-58 to SY Zhou and the National Institute of Diabetes and Digestive and Kidney Diseases Grants P30-DK34933, DK-48419, DK-58913, and DK-061423 to C Owyang.
References
- 1.Adelson DW, Wei JY, Yashar M, O-Lee TJ, Taché Y. Central autonomic activation by intracisternal TRH analogue excites gastric splanchnic afferent neurons. J Neurophysiol. 1999;81:682–691. doi: 10.1152/jn.1999.81.2.682. [DOI] [PubMed] [Google Scholar]
- 2.Aylett P. Gastric emptying and change of blood glucose level as affected by glucagon and insulin. Clin Sci. 1962;22:171–178. [PubMed] [Google Scholar]
- 3.Balfour RH, Hansen AM, Trapp S. Neuronal responses to transient hypoglycemia in the dorsal vagal complex of the rat brainstem. J Physiol. 2006;570:469–484. doi: 10.1113/jphysiol.2005.098822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barnett JL, Owyang C. Serum glucose concentration as a modulator of interdigestive gastric motility. Gastroenterology. 1988;94:739–744. doi: 10.1016/0016-5085(88)90248-x. [DOI] [PubMed] [Google Scholar]
- 5.Browning KN, Zheng Z, Gettys TW, Travagli RA. Vagal afferent control of opioidergic effects in rat brain stem circuits. J Physiol. 2006;575:761–776. doi: 10.1113/jphysiol.2006.111104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Buck SH, Burks TF. The neuropharmacology of capsaicin: review of some recent observations. Pharmacol Rev. 1986;38:179–226. [PubMed] [Google Scholar]
- 7.Bueno L, Ruckebusch M. Insulin and jejunal electrical activity in dogs and sheep. Am J Physiol. 1976;230:1538–1544. doi: 10.1152/ajplegacy.1976.230.6.1538. [DOI] [PubMed] [Google Scholar]
- 8.Bulato E, Carlson AJ. The relation of the blood sugar to the hunger contractions. Am J Physiol. 1924;68:A148. [Google Scholar]
- 9.de Boer SY, Masclee AA, Lamers CB. Effect of hyperglycemia on gastrointestinal and gallbladder motility. Scand J Gastroenterol Suppl. 1992;194:13–18. doi: 10.3109/00365529209096020. [DOI] [PubMed] [Google Scholar]
- 10.DeFronzo RA, Tobin JD, Andres R. Glucose clamp technique: a method for quantifying insulin secretion and resistance. Am J Physiol. 1979;237:E214–E223. doi: 10.1152/ajpendo.1979.237.3.E214. [DOI] [PubMed] [Google Scholar]
- 11.Ferreira M, Browning KN, Sahibzada N, Verbalis JG, Gillis RA, Travagli RA. Glucose effects on gastric motility and tone evoked from the rat dorsal vagal complex. J Physiol. 2001;536:141–152. doi: 10.1111/j.1469-7793.2001.t01-1-00141.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fraser R, Horowitz M, Maddox AF, Harding PE, Chatterton BE, Dent J. Hyperglycemia slows gastric emptying in type I diabetes mellitus. Diabetologia. 1990;33:675–680. doi: 10.1007/BF00400569. [DOI] [PubMed] [Google Scholar]
- 13.Grabauskas G, Zhou SY, Song I, Owyang C. Nodose ganglia neurons demonstrate gluco-responsiveness mediated via voltage-dependent potassium channels. Gastroenterology. 2006;130:A74. [Google Scholar]
- 14.Hasler W. Disorder of gastric emptying. In: Yamada T, editor. Textbook of Gastroenterology. 3rd edn Lippincott Williams & Wilkins; Philadelphia: 1999. pp. 1341–1369. [Google Scholar]
- 15.Hillshey K, Kirkup AJ, Grundy D. Direct and indirect actions of 5-hydroxytryptamine on the discharge of mesenteric afferent fibers innervating the rat jejunum. J Physiol. 1998;506:551–564. doi: 10.1111/j.1469-7793.1998.551bw.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Horowitz M, Harding PE, Maddox AF, Wishart JM, Akkermans LM, Chatterton BE, Shearman DJ. Gastric and esophageal emptying in patients with type II (non-insulin dependent) diabetes mellitus. Diabetologia. 1989;32:151–159. doi: 10.1007/BF00265086. [DOI] [PubMed] [Google Scholar]
- 17.Horowitz M, Maddox A, Harding PE, Maddern GJ, Chatterton BE, Wishart J, Shearman DJ. Gastric and esophageal emptying in insulin-dependent diabetes mellitus. J Gastroenterol Hepatol. 1986;92:1899–1907. doi: 10.1016/0016-5085(87)90622-6. [DOI] [PubMed] [Google Scholar]
- 18.Ishiguchi T, Nakajima M, Sone H, Teda H, Kumgai AK, Takahashi T. Gastric distension-induced pyloric relaxation: central nervous system regulation and effects of acute hyperglycemia in the rat. J Physiol. 2001;533:801–813. doi: 10.1111/j.1469-7793.2001.t01-1-00801.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kobashi M, Adachi A. Effect of topical administration of glucose on neurons innervating abdominal viscera in dorsal motor nucleus of vagus in rats. J Physiol. 1994;44:729–734. doi: 10.2170/jjphysiol.44.729. [DOI] [PubMed] [Google Scholar]
- 20.Koe BK, Weissman A. p-chlorophenylalanine: a specific depletory of brain serotonin. J Pharmacol Exp Ther. 1996;154:499–516. [PubMed] [Google Scholar]
- 21.Levin BE, Routh VH, Kang L, Sanders NM, Dunn-Meynell AA. Neuronal glucosensing: what do we know after 50 years? Diabetes. 2004;53:2521–2528. doi: 10.2337/diabetes.53.10.2521. [DOI] [PubMed] [Google Scholar]
- 22.Li Y, Owyang C. Vagal afferent pathway mediates physiological action of cholecystokinin on pancreatic enzyme secretion. J Clin Invest. 1993;92:418–424. doi: 10.1172/JCI116583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li Y, Hao YB, Zhu JX, Owyang C. Serotonin released from intestinal enterochromaffin cells mediates luminal non-CCK-stimulated pancreatic secretion in rats. Gastroenterology. 2000;118:1197–1207. doi: 10.1016/s0016-5085(00)70373-8. [DOI] [PubMed] [Google Scholar]
- 24.Li Y, Wu XY, Zhu JX, Owyang C. Intestinal serotonin acts as paracrine substance to mediate pancreatic secretion stimulated by luminal factors. Am J Physiol Gastrointest Liver Physiol. 2001;281:G916–G923. doi: 10.1152/ajpgi.2001.281.4.G916. [DOI] [PubMed] [Google Scholar]
- 25.Lu Y, Owyang C. Secretin at physiological doses inhibits gastric motility via a vagal afferent pathway. Am J Physiol. 1995;268:G1012–G1016. doi: 10.1152/ajpgi.1995.268.6.G1012. [DOI] [PubMed] [Google Scholar]
- 26.Lu Y, Li Y, Owyang C. Secretin stimulates vagal afferent pathway to inhibit gastric motility: mediation via postganglionic release of vasoactive intestinal polypeptide (VIP) Gastroenterology. 1993;104:A838. [Google Scholar]
- 27.Mayer J. Glucostatic mechanism of regulation of food intake. N Engl J Med. 1953;249:13–16. doi: 10.1056/NEJM195307022490104. [DOI] [PubMed] [Google Scholar]
- 28.Mizuno Y, Oomura O. Glucose responding neurons in the nucleus tractus solitarius of the rat: in vitro study. Brain Res. 1984;307:109–116. doi: 10.1016/0006-8993(84)90466-9. [DOI] [PubMed] [Google Scholar]
- 29.Prasad KR, Sarna SK. The central and peripheral effects of insulin on migrating myoelectric complexes. Gastroenterology. 1986;90:A1589. [Google Scholar]
- 30.Raybould H, Taché Y. Cholecystokinin inhibits gastric motility and emptying via capsaicin-sensitive vagal pathways in rats. Am J Physiol. 1988;255:G242–G246. doi: 10.1152/ajpgi.1988.255.2.G242. [DOI] [PubMed] [Google Scholar]
- 31.Sakaguchi T, Ohtake M, Yamazaki M. d-glucose anomers in the nucleus of the vagus nerve can depress gastric motility of rats. Brain Res. 1985;332:390–393. doi: 10.1016/0006-8993(85)90611-0. [DOI] [PubMed] [Google Scholar]
- 32.Sakaguchi T, Sandon N, Aono T. Glucose signal in the nucleus of the vagus nerve modulates the cyclicity of gastric motility in rats. Brain Res. 1994;641:163–166. doi: 10.1016/0006-8993(94)91832-5. [DOI] [PubMed] [Google Scholar]
- 33.Shi M, Jones AR, Niedringhaus MS, Pearson RJ, Biehl AM, Ferreira M, Sahibzada N, Verbalis JG, Gillis RA. Glucose acts in the CNS to regulate gastric motility during hypoglycemia. Am J Physiol Regul Integr Comp Physiol. 2003;285:R1102–R1202. doi: 10.1152/ajpregu.00179.2003. [DOI] [PubMed] [Google Scholar]
- 34.Silver IA, Ericińska M. Extracellular glucose concentration in mammalian brain: continuous monitoring of changes during increased neuronal activity and upon limitation in oxygen supply in normo-, hypo-, and hyperglycemic animals. J Neurosci. 1994;14:5068–5076. doi: 10.1523/JNEUROSCI.14-08-05068.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.South EH, Ritter RC. Capsaicin application to central or peripheral vagal fibers attenuates CCK satiety. Peptides. 1988;9:601–612. doi: 10.1016/0196-9781(88)90171-4. [DOI] [PubMed] [Google Scholar]
- 36.Stunkard AJ, Van Itallie TB, Reiss BB. The mechanism of satiety: effect of glucagons on gastric hunger contractions in man. Proc Soc Exp Biol Med. 1955;89:258–261. doi: 10.3181/00379727-89-21776. [DOI] [PubMed] [Google Scholar]
- 37.Takahashi T, Owyang C. Characterization of vagal pathways mediating gastric accommodation reflex in rats. J Physiol. 1997;504:479–488. doi: 10.1111/j.1469-7793.1997.479be.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Takahashi T, Owyang C. Mechanism of cholecystokinin-induced relaxation of the rat stomach. J Auton Nerv Syst. 1999;75:123–130. doi: 10.1016/s0165-1838(98)00181-7. [DOI] [PubMed] [Google Scholar]
- 39.Takahashi T, Owyang C. Vagal control of nitric oxide and vasoactive intestinal polypeptide release in the regulation of gastric relaxation in rat. J Physiol. 1995;484:481–492. doi: 10.1113/jphysiol.1995.sp020680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Takahashi T, Matsuda K, Kono T, Pappas TN. Inhibitory effects of hyperglycemia on neural activity of the vagus in rats. Intensive Care Med. 2003;29:309–311. doi: 10.1007/s00134-002-1580-3. [DOI] [PubMed] [Google Scholar]
- 41.Takeuchi J, Matsumoto J, Ueshima K, Okabe S. Role of capsaicin-sensitive afferent neurons in alkaline secretory response to luminal acid in rat duodenum. Gastroenterology. 1991;101:954–961. doi: 10.1016/0016-5085(91)90721-v. [DOI] [PubMed] [Google Scholar]
- 42.Van de Kar LD. Neuroendocrine pharmacology of serotonergic (5-HT) neurons. Annu Rev Pharmacol Toxicol. 1991;31:289–320. doi: 10.1146/annurev.pa.31.040191.001445. [DOI] [PubMed] [Google Scholar]
- 43.Wan S, Browning KN. D-glucose modulates synaptic transmission from the central terminals of vagal afferent fibers. Am J Physiol Gastrointest Liver Physiol. 2008;294:G757–G763. doi: 10.1152/ajpgi.00576.2007. [DOI] [PubMed] [Google Scholar]
- 44.Wang YH, Taché Y, Sheibel AB, Go VL, Wei JY. Two types of leptin-responsive gastric vagal afferent terminals: an in vitro single unit study in rats. Am J Physiol. 1997;273:R833–R837. doi: 10.1152/ajpregu.1997.273.2.R833. [DOI] [PubMed] [Google Scholar]
- 45.Yuan PQ, Yang H. Neuronal activation of brain vagal-regulatory pathways and upper gut enteric plexuses by insulin hypoglycemia. Am J Physiol Endocrinol Metab. 2002;283:E436–E448. doi: 10.1152/ajpendo.00538.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhou SY, Lu Y, Owyang C. Evidence for glucoresponsiveness of gastric vagal afferents mediated for KATP channels: immunocytochemistry and electrophysiology studies. Gastroenterology. 2006;130:A248. [Google Scholar]
- 47.Zhu JX, Wu XY, Owyang C, Li Y. Intestinal serotonin acts as a paracrine substance to mediate vagal signal transmission evoked by luminal factors in the rat. J Physiol. 2001;530:431–442. doi: 10.1111/j.1469-7793.2001.0431k.x. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]