

Abstract
Recently, we reported the first total synthesis of chloropeptin II (1, complestatin), the more strained and challenging of the two naturally occurring chloropeptins. Central to the design of the approach and by virtue of a single-step, acid-catalyzed ring expansion rearrangement of chloropeptin II to chloropeptin I, the route also provided a total synthesis of chloropeptin I. Herein, we report a complementary and divergent oxidation of chloropeptin II (1, complestatin) to either complestatin A (2, neuroprotectin A) or complestatin B (3, neuroprotectin B), providing the first synthesis of the natural products and establishing their remaining stereochemical assignments. Key to the approach to complestatin A (2, neuroprotectin A) was the development of two different single-step indole oxidations (HCl–DMSO and NBS, THF–H2O) that avoid the rearrangement of chloropeptin II (1) to chloropeptin I (4), providing the 2-oxindole 2 in superb yields (93% and 82%). With a mechanistic understanding of features that impact the latter oxidation and an appreciation of the intrinsic reactivity of the chloropeptin II indole, its modification (NCS, THF–H2O; Cs2CO3, DMF–H2O) provided a two-step, single-pot oxidation of chloropeptin II (1) to afford directly the 3-hydroxy-2-oxindole complestatin B (3, neuroprotectin B). Extensive studies conducted on the fully functionalized synthetic DEF ring system of chloropeptin II were key to the unambiguous assignment of the stereochemistry as well as the exploration and subsequent development of the mild oxidation conditions used in the synthesis of complestatin A and B.
Introduction
Complestatin A and B, fermentation products isolated from Streptomyces sp. MA7234, were disclosed by Merck1 as inhibitors of HIV-1 integrase, and their structures and partial stereochemistry elucidated (Figure 1). Independently and at the same time, the structures were also disclosed as neuroprotectin A and B, isolated from the fermentation broth of Streptomyces sp. Q27107, and shown to protect cultured neurons from glutamate- and kainate-induced excitotoxicities, albeit through an unknown mechanism.2 These two natural products were isolated along with the previously disclosed chloropeptins3,4 (1 and 4) with which they share many similarities, including elements of their reported biological properties and the presence of an embedded tryptophan (Trp) residue as an integral part of the macrocyclic core.
Figure 1.

Natural products.
The chloropeptins have attracted considerable attention due to their reported anti-HIV activity, mediated through two independent sites of action (inhibit gp120-CD4 binding and HIV integrase), and their structural complexity.3-5 Although structurally similar to the glycopeptide antibiotics,6 one of the characteristic diaryl ether linkages is replaced with a biaryl linkage to C6 or C7 of the indole of a (R)-tryptophan within the macrocyclic core, adopting a single atropisomer stereochemistry incapable of interconversion. In complestatin A and B (2 and 3), the embedded tryptophan indole of chloropeptin II (1, complestatin) has been oxidized to a 2-oxindole (2, complestatin A) or 3-hydroxy-2-oxindole (3, complestatin B). Although clearly related to the chloropeptins (1 and 4, Figure 2), whose stereochemistry has been firmly established by extensive NMR spectroscopy paired with computational modeling studies7 and confirmed by subsequent total syntheses,8,9 both the atropisomer orientation of the tryptophan-derived indole and the assignment of the new stereocenter in complestatin A and B were not addressed at the time of their disclosures.
Figure 2.

Acid-catalyzed rearrangement of chloropeptin II (complestatin) to chloropeptin I.
The biaryl linkage between the central amino acid and the oxidized indole of 2 and 3 was shown to have the same C6 versus C7 connectivity as chloropeptin II (1), resulting in a more strained 16-membered versus 17-membered macrocyclic core.1,2 Complementary to the synthetic efforts of Hoveyda, Snapper, and others,8-11 we reported the first total synthesis of chloropeptin II (1), the more strained and challenging of the two chloropeptins.9 Central to the design of our approach and by virtue of a remarkable single-step, acid-catalyzed ring expansion rearrangement12 of chloropeptin II (1) to chloropeptin I (4, Figure 2), the route also provided a total synthesis of 4. Herein, we report a single-step oxidation of 1 to either 2 or 3 and the use of this divergent13 strategy and the strain inherent in chloropeptin II (1) to provide the first synthesis of both complestatin A (2) and complestatin B (3), establishing their remaining stereochemical assignments.
Results and Discussion
Based on an assessment of the spectroscopic data of 2 and 3 and data on related compounds possessing the unnatural atropisomer stereochemistry, we were confident that complestatin A and B shared the same spatial orientation of the indole-derived ring, that this atropisomer stereochemistry matched that found in the chloropeptins (1 and 4), and that the 2-oxindole C3-chiral center possessed the stereochemistry shown (Figure 1).
The 1H NMR chemical shifts of the Trp-derived subunit are most diagnostic of the atropisomer stereochemistry where Trp α-CH appears at δ 4.18 with Trp β-CH2 at δ 3.50 and δ 2.89 (DMSO-d6) in chloropeptin II (1, complestatin). In the unnatural atropisomer (5, isocomplestatin) and as first highlighted by Hoveyda and Snapper,10 Trp α-CH and Trp β-CH2 appear at δ 5.18 and at δ 3.44 and δ 3.36 (acetone-d6), respectively. In addition to the pronounced shielding of Trp α-CH in the natural atropisomer, the diastereotopic Trp β-CH2 signals exhibit large chemical shift differences in the natural configuration (Δ 0.59 ppm), whereas the difference is characteristically much smaller (Δ 0.08 ppm) with the unnatural atropisomer. The chemical shifts of complestatin A and B exhibit an upfield shielded Trp α-CH at δ 3.48 and δ 3.49 (DMSO-d6), and large Trp β-CH2 chemical shift differences (Δ 1.08 ppm and Δ 1.09 ppm), respectively, supporting the assigned stereochemistry. Conformational searches on the full structure using Monte Carlo sampling (MacroModel, OPLS 2005 forcefield) supported this assignment. Specifying the (R)-configuration for the 2-oxindole C3 stereocenter produced a family of lowest energy structures in which the molecule possessed the indole spatial orientation of chloropeptin II (Figure 3). Small calculated coupling constants (J = 2.7 and 4.4 Hz) between the Trp γ-CH and Trp β-CH2 and a predicted strong NOE signal between the amino acid D-ring to C5-H of the indole proved characteristic of the lowest energy conformation of this structure. In contrast, specifying the (S)-configuration for the new stereocenter produced a structure similar to isocomplestatin (5). Diagnostic of its lowest energy conformation, this structure was calculated to exhibit at least one large coupling constant (J = 10.7 and 1.2 Hz) between Trp γ-CH and Trp β-CH2 and would be expected to exhibit a strong NOE between amino acid D-ring to C7-H of the indole. Based on the reported coupling constants for Trp γ-CH of complestatin A (t, J = 3.0 Hz) and those calculated (J = 2.7 and 4.4 Hz), the spectroscopic evidence supporting the (R)-configuration was even more compelling (Figure 3).
Figure 3.

Calculated distances and 1H NMR coupling constants.
Our efforts to establish the stereochemistry and understand the latent oxidation reactivity of the strained indole began with the synthetic macrocyclic DEF right-hand subunits 6 and 7,9 which share key features of chloropeptin II, including the strained 16-membered macrocycle.
Conformational searches with Monte Carlo sampling on the simplified oxindoles (R)-8 and (S)-8 produced results nearly identical with those obtained with the full complestatin system, suggesting that results from this advanced model system would be of predictive value. These simplified systems, available through an atroposelective Pd(0)-mediated Larock indole macrocyclization (Scheme 1),9,14 were exposed to a wide variety of oxidants. Initially, treatment of the fully functionalized N-acetyl-2-triethylsilylindoles (R)-6 and (S)-6 with dimethyldioxirane15 (DMDO) or m-chloroperbenzoic acid16 (m-CPBA) led to complex mixtures of products. These reaction mixtures appeared to contain products bearing both natural (R) and unnatural (S) configurations, albeit in low yields combined with many over-oxidation products. In efforts to develop a more controlled oxidation, the substrate indole was further simplified by removing the 2-triethylsilyl group and hydrolyzing the indole N-acetyl group to afford the natural and unnatural indole atropisomers (R)-7 and (S)-7 in excellent yield (Scheme 1). Due to the strain inherent in the macrocycle and with an appreciation of the enhanced indole reactivity, (R)-7 and (S)-7 were subjected to a variety of oxidation conditions.
Scheme 1.

Exposure of the natural atropisomer (R)-7 to DMDO or m-CPBA at room temperature (1 h) produced the 2-oxindole (R)-8 as a single stereoisomer, but also in disappointingly low yields (∼20%), along with additional unidentified products. However, examination of an alternative oxidation involving treatment of (R)-7 with concentrated aqueous HCl in dimethyl sulfoxide (DMSO)17 overnight at room temperature produced the 2-oxindole (R)-8 as a single stereoisomer in significantly higher yield (73%) (Scheme 2). Under the modified conditions we developed using conc. HCl as a reagent versus solvent, no indole C6 to C7 ring expansion rearrangement occurred, no skeletal C3 to C2 indole substituent migration was detected, and the NHBoc group remained intact, highlighting the mild reaction conditions of the modified protocol. Without further optimization, the same conditions applied to the unnatural atropisomer (S)-7 yielded the diastereomeric 2-oxindole (S)-8 in good yield (Scheme 2), and again no competitive acid-catalyzed rearrangement products were detected. Comparison of the spectroscopic properties of the two diastereomers (R)-8 and (S)-8 provided compelling evidence supporting the stereochemical assignments of complestatin A and B. The diastereomer (R)-8 (vs (S)-8) bearing the chloropeptin II natural atropisomer stereochemistry exhibited the characteristic shielded Trp α-CH chemical shift of δ 2.84 (vs δ 3.81), the small Trp β-CH2 to Trp γ-CH coupling constants of 3.0 Hz (δ 3.60, t, J = 3.0 Hz) versus 11.0 Hz (δ 3.57, d, J = 11.0 Hz), a diagnostic NOE between the central amino acid D-ring to C5-H of the indole (vs C7-H) indicative of the spatial atropisomer orientation, and a large chemical shift difference in the diastereotopic protons of Trp β-CH2 at δ 3.04 and 1.90, Δ 1.14 ppm (vs δ 2.44 and δ 2.11, Δ 0.33 ppm), matching the signals reported for complestatin A (2). The 13C NMR spectra provided further evidence for the stereochemical assignments. According to both isolation reports, the 2-oxindole of 2 exhibited chemical shifts of δ 178.6 (indole C2), 43.8 (indole C3), and 30.0 (Trp β-CH2), comparing favorably to those found with (R)-8 in which shifts of δ 178.7 (indole C2), 43.8 (indole C3), and 30.1 (Trp β-CH2) were observed, whereas the diastereomer (S)-8 exhibited shifts of δ 178.4, 42.1, and 29.5, respectively (Figure 4).
Scheme 2.

Figure 4.

Diagnostic NMR chemical shifts and NOEs for 2-oxindoles (DMSO-d6).
Mechanistically, the stereochemistry can be rationalized by a selective indole C3 protonation occurring from the external, sterically more accessible top face of the molecule as depicted (Scheme 3). With the natural atropisomer, this results in protonation from the si-face, setting the C3 (R)-configuration, whereas the analogous top face attack on the unnatural atropisomer results in re-face protonation, producing the (S)-isomer. This is followed by DMSO addition to the indole-derived iminium ion and subsequent elimination of dimethyl sulfide with oxidation to the 2-oxindole. The use of more acidic reaction conditions (1 N HCl in AcOH with DMSO18 or 4 N HCl in dioxane with DMSO) resulted in reduced yields with the generation of additional side-products, some of which suggest that the biaryl indole C6 to C7 acid-catalyzed rearrangement becomes competitive with the DMSO-mediated oxidation.
Scheme 3.

Mechanism of the DMSO-mediated oxidation.
Confident in the stereochemical assignment and with an appreciation of the indole reactivity, the remaining challenge was the synthesis of the further oxidized 3-hydroxy-2-oxindole characteristic of complestatin B (3). Although 2-oxindoles have been converted to 3-hydroxy-2-oxindoles by a variety of oxidants,11c exposure of (R)-8 or (S)-8 to the commonly reported reagents DMDO or m-CPBA provided either recovered starting material under mild conditions or significant decomposition under more forcing conditions, suggesting that the strained substrates fail to effectively enolize. It is known19 that exposure of an indole to N-bromosuccinimide (NBS) under aqueous conditions results in generation of 3-bromo-2-oxindoles along with 2-oxindoles and we expected that such a 3-bromo-2-oxindole could be used to introduce a C3-alcohol through a subsequent displacement reaction using water as a nucleophile.19e Exposure of (R)-7 to stoichiometric NBS in aqueous tetrahydrofuran (THF) produced the 2-oxindole (R)-8 (52%) along with a smaller, but significant amount of the 3-bromo-2-oxindole (R)-9 (31%). As might be anticipated, further investigation of the reaction revealed that dilute reaction conditions (0.4 mM) and limiting the amount of NBS (1 or even 2 equiv) favored the formation of the 2-oxindole (80–90%), whereas the use of more concentrated (> 4 mM) reaction conditions and larger amounts of NBS (3 equiv), facilitating subsequent NBS-mediated oxidation of the 3-bromo-2-hydroxyindoline, led to the generation of increasing amounts of the 3-bromo-2-oxindole (20–30%). Upon detailed characterization, it was established that the 3-bromo-2-oxindole (R)-9 possessed the expected C3 configuration (to give the (S) geometry) resulting from bromination on the sterically more accessible si face, and its exposure to sodium bicarbonate (NaHCO3) in aqueous THF (48 h) provided the 3-hydroxy-2-oxindole (R)-10 in high yield (92%, Scheme 2). Comprehensive NMR characterization established that the C3 configuration was retained in the displacement reaction, indicating a stepwise elimination of HBr to provide an intermediate indol-2-one19d,e followed by nucleophilic addition of water from the sterically more accessible si face, rather than direct SN2 displacement (Eq. 1). Potential side products derived from skeleton rearrangement or aryl bromination were not observed, demonstrating the pronounced reactivity of the strained indole ring at C3.
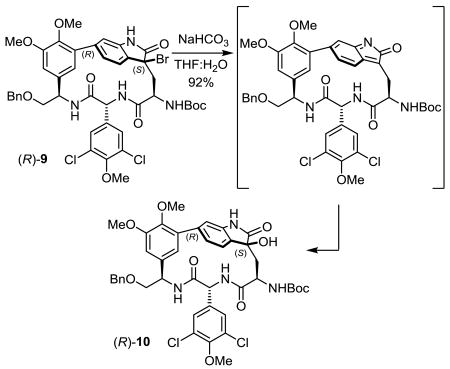 |
(1) |
The NMR chemical shifts of (R)-10 were diagnostic of the stereochemistry, especially the 13C NMR chemical shifts of the Trp-derived residue. Chemical shifts of δ 178.4 and δ 75.4 for complestatin B are reported for the newly oxidized carbons, and chemical shifts of δ 178.4 and δ 75.4 are observed for (R)-10. A diagnostic NOE between the central amino acid D-ring with C5-H of the indole-derived ring and the absence of an NOE to C7-H proved indicative of the atropisomer orientation, and the diagnostic large chemical shift distinctions in the diastereotopic protons of Trp β-CH2 at δ 2.93 and 1.85 (Δ 1.14 ppm) match the characteristic signals reported for complestatin B (3) (Figure 5).
Figure 5.

Diagnostic NMR chemical shifts and NOEs for 3-hydroxy-2-oxindoles (DMSO-d6).
Mindful that the chloropeptins and their atropisomers are incapable of thermal interconversion, equilibration studies were conducted with the 2-oxindoles (R)-8 and (S)-8 to establish whether additional atropisomers were accessible or if there was an available racemization pathway. Microwave heating solutions of pure samples of both 2-oxindoles in degassed, anhydrous o-dichlorobenzene at 160 °C led only to recovery of starting material, suggesting that these structures are incapable of atropisomer interconversion or facile racemization and reside in a stable spatial orientation relative to the C3 stereocenter (Eq. 2).
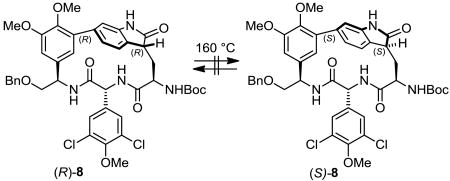 |
(2) |
With effective protocols developed on the macrocyclic DEF right-hand subunit, we turned our attention to complestatin (1) itself. Initial efforts began with room temperature exposure of 1 to concentrated HCl in DMSO, the conditions adopted for (R)-8, and the studies indicated that the reaction was slower, with only a small amount of complestatin A detected. Encouraged by this initial result and in the absence of competitive rearrangement to provide chloropeptin I, additional acid catalyst was added, and further product conversion was observed. Additional optimization demonstrated that both natural and synthetic9 chloropeptin II (1) could be converted cleanly to complestatin A (2, neuroprotectin A) in superb yield (93%) over 24–48 h in the presence of reagent amounts of concentrated HCl in DMSO, with additional HCl added after 24 h if needed (Scheme 4). The 1H NMR spectrum of synthetic 2 was in complete agreement with the published data1,2 reported for complestatin A (Supporting Information Table S1), confirming the structure and establishing the stereochemistry of the remaining indole-derived stereocenters in the natural product.
Scheme 4.

Oxidation of chloropeptin II (complestatin) to complestatin A (neuroprotectin A) or complestatin B (neuroprotectin B).
Extensions of these efforts to the further oxidized natural product complestatin B (3) proved less straightforward. Analogous to the reaction of the isolated DEF-ring system (R)-7, we expected that treatment of chloropeptin II (1) with aqueous NBS would provide workable amounts of the 3-bromo-2-oxindole that could be used to access complestatin B (3). Remarkably, treatment of 1 with aqueous NBS (1 equiv, THF–H2O, 22 °C, 0.5 h) under the conditions enlisted for (R)-7 furnished only the 2-oxindole complestatin A (2) in excellent yield (82%), but without evidence of 3-bromo-2-oxindole formation. Because chloropeptin II (1) possesses such poor solubility properties, it was not possible to increase the reaction concentration, facilitating the requisite NBS-mediated intermolecular 2-oxindole oxidation competitive with elimination of HBr, and the use of larger amounts of NBS (5 equiv) did not result in the generation of detectable amounts of 3-bromo-2-oxindole. As a result and while this provided a second effective single-step oxidation of 1 to provide complestatin A (2), we were not able to extend the aqueous NBS oxidation directly to a synthesis of the 3-hydroxy-2-oxindole complestatin B (3).
With a recognition that the analogous elimination of HCl from an initial chlorohydrin to provide a 2-hydroxy-2H-indole and its subsequent tautomerization to a 2-oxindole should be slower, the substitution of N-chlorosuccinimide (NCS) for NBS in the aqueous oxidation conditions was examined. Gratifyingly and without the opportunity for optimization because of the limiting amounts of 1 in hand, room temperature treatment of chloropeptin II (1) with NCS (3 equiv) in aqueous THF (9:1 THF/H2O, 3 mM, 22 °C, 30 min) exclusively provided the 3-chloro-2-oxindole in superb conversion (84%) without detection of the corresponding 2-oxindole, which in turn was converted to the 3-hydroxy-2-oxindole complestatin B (3) upon exposure to aqueous Cs2CO3 (20 equiv) in dimethylformamide (4:1 DMF/H2O, 22 °C, 1 h, 30%) via an intermediate indol-2-one.19d,e This two-step, single-pot oxidation using NCS was first explored with (R)-7, providing excellent overall conversions (72%) to the 3-hydroxy-2-oxindole (R)-10, where neither aqueous NaHCO3 or LiOH served to displace the chloride as effectively as aqueous Cs2CO3 (Eq. 3).19e,20 The 1H NMR spectrum of synthetic 3 was in agreement with the published data1,2 reported for complestatin B (Supporting Information Table S2), establishing the stereochemistry of the remaining indole-derived stereocenters in the natural product.
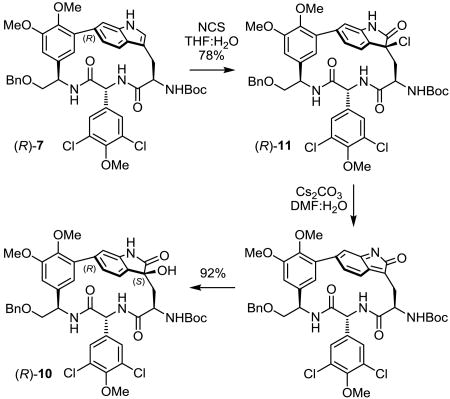 |
(3) |
Finally, a key feature of our total synthesis of chloropeptin II (1) was the development of an atroposelective Pd(0)-mediated intramolecular Larock indole annulation14 for macrocyclization to furnish a functionalized N-acetyl-2-triethylsilylindole. The N-acetyl group served to tame the inherent reactivity of the product strained indole, whereas the 2-triethylsilyl group controlled the regiochemistry of the intramolecular indole cyclization. Consequently, we explored whether this uniquely functionalized indole could serve as a suitable or alternative precursor to oxidation. Following removal of the indole N-acetyl group in order to release the indole reactivity, studies were conducted directly on (R)-12 and (S)-12 to determine if the 2-triethylsilyl group could serve as a handle for a controlled indole oxidation (Scheme 5).
Scheme 5.

Alternative synthesis of 2-oxindole.
Their treatments with concentrated HCl–DMSO (25 °C, 16 h) simply provided the 2-oxindoles 8 in conversions approaching those observed with 7 and most likely proceed by initial proto-desilylation to provide 7 itself. Remarkably, the reaction of NBS in H2O–THF (1 equiv, 0.5 h) with (R)-12 and (S)-12 led to the isolation of the surprisingly semi-stable bromine containing imines (R)-13 and (S)-13,21 respectively. This was true of both individual atropisomers, and not only provides further evidence of a facially selective electrophilic reaction occurring at the indole C3 to provide the diastereomers shown, but also highlights a reactivity overriding the a priori expected C2 reaction, with direct bromide substitution of the triethylsilyl group. These semi-stable imines could be converted to the corresponding 2-oxindoles diastereoselectively and in excellent yields by exposure to aqueous acid (TsOH, THF–H2O, 25 °C, 16 h), either by proto-desilylation or possibly by acid activation of the imine and addition of water, followed by elimination of triethylsilylbromide and a subsequent tautomerization to furnish 2-oxindoles (R)-8 and (S)-8. By contrast, their exposure to aqueous base (NaHCO3, THF–H2O, 25 °C), even for extended reaction times, provided only recovered starting material.
Conclusions
Herein we disclose details of studies that complete a divergent total synthesis of complestatin A and B (neuroprotectin A and B), the development of mild reaction conditions for two unique indole oxidations that take advantage of the strain intrinsic in the chloropeptin II (complestatin) 16-membered macrocycle and selectively provide either a 2-oxindole or 3-hydroxy-2-oxindole directly, and the definition of factors impacting the reactions and their diastereoselectivity. Key issues surrounding the structure and the remaining stereochemical assignments of complestatin A and B are discussed and were unambiguously established.
Supplementary Material
Acknowledgments
We gratefully acknowledge the financial support of the National Institutes of Health (CA041101), the Skaggs Institute for Chemical Biology, and fellowship support from the National Science Foundation (S.P.B.). We are especially grateful to Dr. S. B. Singh (Merck) for an authentic sample (5.0 mg) of complestatin (chloropeptin II) and Dr. Hiroyuki Shimamura for initial studies examining the DMDO and m-CPBA oxidation of the model DEF ring system.
Footnotes
Supporting Information Available: Full experimental details. This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.Singh SB, Jayasuriya H, Salituro GM, Zink DL, Shafiee A, Heimbuch B, Silverman KC, Lingham RB, Genilloud O, Teran A, Vilella D, Felock P, Hazuda D. J Nat Prod. 2001;64:874–882. doi: 10.1021/np000632z. [DOI] [PubMed] [Google Scholar]
- 2.(a) Kobayashi H, Shin-Ya K, Furhata K, Nagai K, Suzuki KI, Hayakawa Y, Seto H, Yun BS, Ryoo IJ, Kim JS, Kim CJ, Yoo ID. J Antibiot. 2001;54:1019–1024. doi: 10.7164/antibiotics.54.1019. [DOI] [PubMed] [Google Scholar]; (b) Kobayashi H, Shin-Ya K, Nagai K, Suzuki KI, Hayakawa Y, Seto H, Yun BS, Ryoo IJ, Kim JS, Kim CJ, Yoo ID. J Antibiot. 2001;54:1013–1018. doi: 10.7164/antibiotics.54.1013. [DOI] [PubMed] [Google Scholar]
- 3.(a) Kaneko I, Fearon DT, Austen KF. J Immunol. 1980;124:1194–1198. [PubMed] [Google Scholar]; (b) Tachikawa K, Hasumi K, Endo A. Thromb Haemost. 1997;77:137–142. [PubMed] [Google Scholar]; (c) Tachikawa K, Hasumi K, Endo A. Thrombosis Res. 1997;87:571–576. doi: 10.1016/s0049-3848(97)00186-2. [DOI] [PubMed] [Google Scholar]; (d) Kaneko I, Kamoshida K, Takahashi S. J Antibiot. 1989;42:236–241. doi: 10.7164/antibiotics.42.236. [DOI] [PubMed] [Google Scholar]; (e) Seto H, Fujioka T, Furihata K, Kaneko I, Takahashi S. Tetrahedron Lett. 1989;30:4987–4990. [Google Scholar]; (f) Momota L, Kaneko I, Kimura S, Mitamura K, Shimada K. Biochem Biophys Res Commun. 1991;179:243–250. doi: 10.1016/0006-291x(91)91361-f. [DOI] [PubMed] [Google Scholar]
- 4.(a) Matsuzaki K, Ogino T, Matsumoto A, Woodruff HB, Tanaka H, Omura S. J Antibiot. 1994;47:1173–1174. doi: 10.7164/antibiotics.47.1173. [DOI] [PubMed] [Google Scholar]; (b) Matsuzaki K, Ogino T, Matsumoto A, Woodruff HB, Tanaka H, Omura S. J Antibiot. 1997;50:58–65. doi: 10.7164/antibiotics.50.58. [DOI] [PubMed] [Google Scholar]; (c) Matsuzaki K, Ogino T, Matsumoto A, Woodruff HB, Tanaka H, Omura S. J Antibiot. 1997;50:66–69. doi: 10.7164/antibiotics.50.66. [DOI] [PubMed] [Google Scholar]
- 5.(a) Singh SB, Jayasuriya H, Hazuda DL, Felock P, Homnick CF, Sardana M, Patane MA. Tetrahedron Lett. 1998;39:8769–8770. [Google Scholar]; (b) Hedge VR, Puar MS, Dai P, Patel M, Gullo VP, Chan TM, Silver J, Pramanik BN, Jenh CH. Bioorg Med Chem Lett. 2003;13:573–575. doi: 10.1016/s0960-894x(02)00919-8. [DOI] [PubMed] [Google Scholar]; (c) Hedge VR, Puar MS, Dai P, Patel M, Gullo VP, Pramanik B, Jenh CH. Tetrahedron Lett. 2002;43:5339–5341. [Google Scholar]
- 6.Kahne D, Leimkuhler C, Lu W, Walsh CT. Chem Rev. 2005;105:425–448. doi: 10.1021/cr030103a.Boger DL. Med Res Rev. 2001;21:356–381. doi: 10.1002/med.1014.. For related ongoing synthetic studies, see: Boger DL, Miyazaki S, Kim SH, Wu JH, Loiseleur O, Castle SL. J Am Chem Soc. 1999;121:3226.Boger DL, Miyazaki S, Kim SH, Wu JH, Castle SL, Loiseleur O, Jin Q. J Am Chem Soc. 1999;121:10004.Boger DL, Kim SH, Miyazaki S, Strittmatter H, Weng JH, Mori Y, Rogel O, Castle SL, McAtee JJ. J Am Chem Soc. 2000;122:7416.Boger DL, Kim SH, Mori Y, Weng JH, Rogel O, Castle SL, McAtee JJ. J Am Chem Soc. 2001;123:1862. doi: 10.1021/ja003835i.McComas CC, Crowley BM, Boger DL. J Am Chem Soc. 2003;125:9314. doi: 10.1021/ja035901x.Crowley BM, Mori Y, McComas CC, Tang D, Boger DL. J Am Chem Soc. 2004;126:4310. doi: 10.1021/ja039795a.Crowley BM, Boger DL. J Am Chem Soc. 2006;128:2885. doi: 10.1021/ja0572912.Xie J, Pierce JG, James RC, Okano A, Boger DL. J Am Chem Soc. 2011;133:13946. doi: 10.1021/ja207142h.
- 7.Gouda H, Matsuzaki K, Tanaka H, Hirono S, Omura S, McCauley JA, Sprengeler PA, Furst GT, Smith AB. J Am Chem Soc. 1996;118:13087–13088. [Google Scholar]
- 8.Deng H, Jung JK, Liu T, Kuntz KW, Snapper ML, Hoveyda AH. J Am Chem Soc. 2003;125:9032–9034. doi: 10.1021/ja030249r. [DOI] [PubMed] [Google Scholar]
- 9.(a) Garfunkle J, Kimball FS, Trzupek JD, Takizawa S, Shimamura H, Tomishima M, Boger DL. J Am Chem Soc. 2009;131:16036–16038. doi: 10.1021/ja907193b. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Shimamura H, Breazzano SP, Garfunkle J, Kimball FS, Trzupek JD, Boger DL. J Am Chem Soc. 2010;132:7776–7783. doi: 10.1021/ja102304p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shinohara T, Deng H, Snapper ML, Hoveyda AH. J Am Chem Soc. 2005;127:7334–7336. doi: 10.1021/ja051790l. [DOI] [PubMed] [Google Scholar]
- 11.(a) Wang Z, Bois-Choussy M, Jia Y, Zhu J. Angew Chem Int Ed. 2010;49:2018–2022. doi: 10.1002/anie.200906797. [DOI] [PubMed] [Google Scholar]; (b) Yamada Y, Akiba A, Arima S, Okada C, Yoshida K, Itou F, Kai T, Satou T, Takeda K, Harigaya T. Chem Pharm Bull Jpn. 2005;53:1277–1290. doi: 10.1248/cpb.53.1277. [DOI] [PubMed] [Google Scholar]; (c) Smith AB, III, Chruma JJ, Han Q, Barbosa J. Bioorg Med Chem Lett. 2004;14:1697–1702. doi: 10.1016/j.bmcl.2004.01.056. [DOI] [PubMed] [Google Scholar]; (d) Elder AM, Rich DH. Org Lett. 1999;1:1443–1446. doi: 10.1021/ol990990x. [DOI] [PubMed] [Google Scholar]; (e) Roussi G, Zamora EG, Carbonnelle AC, Beugelmans R. Heterocycles. 1999;51:2041–2063. [Google Scholar]; (f) Kai T, Kajimoto N, Konda Y, Harigaya Y, Takayanagi H. Tetrahedron Lett. 1999;40:6289–6292. [Google Scholar]; (g) Gurjar MK, Tripathy NL. Tetrahedron Lett. 1997;38:2163–2166. [Google Scholar]
- 12.(a) Jayasuriya H, Salituro GM, Smith SK, Heck JV, Gould SJ, Singh SB. Tetrahedron Lett. 1998;39:2247–2248. [Google Scholar]; (b) Hedge VR, Dai P, Patel M, Gullo VP. Tetrahedron Lett. 1998;39:5683–5684. [Google Scholar]
- 13.Boger DL, Brotherton CE. J Org Chem. 1984;49:4050–4055. [Google Scholar]
- 14.(a) Larock RC, Yum EK. J Am Chem Soc. 1991;113:6689–6690. [Google Scholar]; (b) Larock RC, Yum EK, Refvik MD. J Org Chem. 1998;63:7652–7662. [Google Scholar]; (c) Shen M, Li G, Lu BZ, Hossain A, Roschangar F, Farina V, Senanayake CH. Org Lett. 2004;6:4129–4132. doi: 10.1021/ol048114t. [DOI] [PubMed] [Google Scholar]
- 15.(a) Adam W, Ahrweiler M, Peters K, Schmeideskamp B. J Org Chem. 1994;59:2733–2739. [Google Scholar]; (b) Zhang X, Foote CS. J Am Chem Soc. 1993;115:8867–8868. [Google Scholar]; (c) Saurez-Castillo OR, Sanchez-Zavala M, Melendez-Rodriguez M, Castelan-Duarte LE, Morales-Rios MS, Joseph-Nathan P. Tetrahedron. 2006;62:3040–3051. [Google Scholar]
- 16.(a) Alamgir M, Mitchell PSR, Bowyer PK, Kumar N, Black DStC. Tetrahedron. 2008;64:7136–7142. [Google Scholar]; (b) Hino T, Yamaguchi H, Matsuki K, Nakano K, Sodeoka M, Nakagawa M. J Chem Soc Perkin Trans I. 1983:141–146. [Google Scholar]
- 17.(a) Savige WE, Fontana A. J Chem Soc Chem Commun. 1976:599–600. [Google Scholar]; (b) Szabo-Pustay K, Szabo L. Synthesis. 1979;4:276–277. [Google Scholar]
- 18.Zhou N, Polozov AM, O'Connell M, Burgeson J, Yu P, Zeller W, Zhang J, Onua E, Ramirez J, Palsdottir GA, Halldorsdottir GV, Andresson T, Kiselyov AS, Gurney M, Singh J. Bioorg Med Chem Lett. 2010;20:2658–2664. doi: 10.1016/j.bmcl.2010.02.028. [DOI] [PubMed] [Google Scholar]
- 19.(a) Hinman RL, Bauman CP. J Org Chem. 1964;29:1206–1215. [Google Scholar]; (b) Lawson WB, Witkop R. J Org Chem. 1961;26:263–264. [Google Scholar]; (c) Lawson WB, Patchornik A, Witkop R. J Am Chem Soc. 1960;82:5918–5923. [Google Scholar]; (d) Fuchs JR, Funk RL. J Am Chem Soc. 2004;126:5068–5069. doi: 10.1021/ja049569g. [DOI] [PubMed] [Google Scholar]; (e) Fuchs JR, Funk RL. Org Lett. 2005;7:677–680. doi: 10.1021/ol047532v. [DOI] [PubMed] [Google Scholar]; (f) Hino T, Tonozuka M, Nakagawa M. Tetrahedron. 1974;30:2123–2133. [Google Scholar]; (g) Hino T, Nakamura T, Nakagawa M. Chem Pharm Bull Jpn. 1975;23:2990–2997. [Google Scholar]
- 20.Goldberg FW, Magnus PD, Turnbull R. Org Lett. 2005;7:4531–4534. doi: 10.1021/ol051943+. [DOI] [PubMed] [Google Scholar]
- 21.Similar intermediates have been reported with 2-alkylthioindoles see: Hino T, Endo M, Tonozuka M, Nakagawa M. Heterocycles. 1974;2:565–570.Hino T, Nakagawa M. Heterocycles. 1977;6:1680–1685.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.