

Abstract
Type 2 diabetes, a rapidly growing disease of modern aetiology, has a profound impact on morbidity and mortality. Explosions in the understanding of the underlying cellular mechanisms which lead to type 2 diabetes have recently been elucidated. In particular, the central role of endoplasmic reticulum stress (ER stress) and the unfolding protein response (UPR) in insulin resistance in type 2 diabetes has recently been discovered. We hypothesize that ER stress and UPR are not only central for type 2 diabetes but also for stress-induced diabetes. We review here the evidence that post-burn insulin resistance and hyperglycaemia have pathophysiologic mechanisms in common with type 2 diabetes. These recent discoveries not only highlight the importance of ER stress in the post-burn patient recovery, but furthermore enable new models to study fundamental and interventional aspects of type 2 diabetes.
Keywords: burn, trauma, ER stress, insulin resistance, hyperglycaemia, diabetes, calcium
Introduction
A burn injury represents one of the most severe forms of trauma, and occurs in more than two to three million people in North America each year [1]. According to the World Health Organization, an estimated 330,000 deaths per year worldwide are related to thermal injury [2]. A severe burn represents a devastating injury affecting nearly every organ system and leading to significant morbidity and mortality [3]. Stress-induced diabetes with hyperglycaemia and insulin resistance during acute hospitalization is a hallmark of severely burned patients and directly effects patient morbidity and mortality [4]. Various studies demonstrated that hyperglycaemia and insulin resistance are associated with a significantly increased incidence of infection, sepsis, ventilator-associated pneumonia, delay in wound healing and increased death [5-7]. Thus, to improve patient’s outcome it is important to understand the molecular mechanisms which lead to post-burn insulin resistance.
Insulin resistance and hyperglycaemia in burn patients
Based on recent findings we propose that post-burn insulin resistance is similar in pathophysiology to type 2 diabetes, differing only in its acute onset and severity. Similar to type 2 diabetes, in the clinical scenario stress-induced diabetes with insulin resistance and hyperglycaemia can be overcome by exogenous insulin administration, which normalizes glucose levels and improves muscle protein synthesis, accelerates donor site healing time, and attenuates lean body mass loss and the acute phase response [8-15]. We and others hypothesize that these vast and detrimental responses are driven by cytokines and stress hormones, which are the primary mediators of the hypermetabolic response to trauma or burn [16, 17]. We have shown that stress hormones are increased 10- to 40-fold after severe burn and remain elevated for up to 2 years post-injury [18, 19]. Of special interest are the effects of stress hormones on glucose metabolism and insulin resistance [20]. During the early phases of post-burn, hyperglycaemia is due to an increased rate of glucose appearance along with an impaired tissue extraction of glucose leading to an increase of glucose and lactate [21, 22].
Euglycaemic control in critically ill and burn patients
The clinical implications of tight euglycaemic control as published by van den Berghe and colleagues significantly and rapidly changed intensive care unit (ICU) practice [23]. They showed that insulin administered to maintain glucose at levels below 110 mg/dl decreased mortality, the incidence of infections, sepsis and sepsis-associated multi-organ failure (MOF) in surgical patients, reduced kidney injury, and accelerated weaning from mechanical ventilation and discharge from the ICU in medical patients [24]. In several follow-up studies, the authors confirmed the advantageous effects of tight euglycaemic control. The authors showed recently that tight euglycaemic control improved mortality in paediatric ICU patients [25]. Insulin given during the acute phase not only improved acute hospital outcomes, but also improved long-term rehabilitation of critical ill patients over a period of 1 year [26, 27], indicating the advantage of insulin therapy. However, all studies presented by the Leuven group were unicentre trials, so various unicentre and multicentre studies followed the Leuven trials to determine whether tight euglycaemic control improves outcomes in a different setting. The results of these trials were mixed, with some showing benefits with the use of euglycaemic control [28, 29]. Other studies, however, failed to show improved outcomes. In contrast, some of these studies even demonstrated detrimental effects associated with tight euglycaemic control and a dramatic increase in the incidence of hypoglycaemia [30]. Intensive discussion thus arose as to whether tight euglycaemic control is beneficial. To end this discussion, a large multicenter trial was initiated: the NICE SUGAR trial. This trial enrolled more than 6,000 patients and failed to show beneficial outcome for critically ill patients with intensive insulin therapy [31], and delineated the risks and problems associated with this therapy. Therefore, many ICUs have now changed their tight euglycaemic protocols to be less strict. Despite the aforementioned studies, none of the trials investigated whether tight euglycaemic control is beneficial in severely thermally injured patients. Hyperglycaemia is a hallmark of burned patients [4]. The clinical relevance of hyperglycaemia after a severe burn was shown in several studies in which the authors demonstrated that burn patients with poor glucose control had a significantly higher incidence of bacteremia/fungemia and mortality [5-7], indicating that hyperglycaemia represents a significant clinical problem in burn patients. We conducted a prospective randomized controlled trial in severely paediatric burn patients and found that tight glycaemia control significantly improves post-burn morbidity [32] indicating the benefit of insulin administration post-burn and overcoming post-burn insulin resistance.
Molecular mechanisms associated with post-burn insulin resistance: endoplasmic reticulum stress and unfolded protein response
The metabolic alterations post-burn are well-defined by increased glucose production along with decreased glucose uptake in the liver and periphery. However, the molecular changes lacked understanding for a long time. Recently, changes in insulin receptor substrate-1 (IRS-1) and other downstream insulin receptor signalling pathways have been a focus of intense research. A cell organelle that has gained substantial attention in the diabetes literature is the ER. The ER is the site of protein synthesis and folding of secreted and membrane-bound proteins [33]. The presence of an excess of misfolded proteins results in the activation of signalling pathways to restore homeostasis (Fig. 1; Ref. [33]). Calcium depletion from ER stores leads to increased unfolded and/or misfolded proteins in the ER lumen. This results in the activation of the UPR [33]. The accumulation of unfolded proteins is detected by the cell via three key ER transmembrane receptors, protein kinase RNA (PKR)-like ER kinase (PERK), inositol-requiring enzyme 1 (IRE1) and activating transcription factor 6 (ATF6). IRE1 is an ER-resident transmembrane protein with a luminal domain and a cytoplasmic kinase domain [34, 35]. The luminal domain of IRE1 is bound to the ER Hsp70 homologue BiP in unstressed cells. In response to unfolded proteins, IRE1 dissociates from BiP and oligomerizes in the plane of the membrane and trans-autophosphorylates juxtaposed kinase domains, which leads to an IRE1-mediated sequence-specific splicing of the mRNA transcript for X-box binding protein-1 (XBP1). The protein product of spliced XBP1 mRNA (XBP1s) induces the gene transcription of chaperones and ER-associated degradation proteins to increase the ER folding capacity [36]. IRE1 can also act by recruitment of TRAF2 [tumour necrosis factor receptor (TNFR)-associated factor-2]. TRAF2 signals and activates Jun N-terminal kinase (JNK) [37]. Ozcan et al. showed that ER stress leads to suppression of insulin receptor signalling through hyperactivation of JNK and subsequent serine phosphorylation of insulin receptor substrate 1 (IRS-1) [38]. They further showed that mice deficient in XBP-1 developed insulin resistance. The authors conclude that ER stress is a central contributing feature of peripheral insulin resistance and type 2 diabetes in vivo [38]. Insulin resistance is a known pathophysiologic occurrence post-burn, therefore IRE1 signalling may contribute to insulin resistance post-burn.
Fig 1.
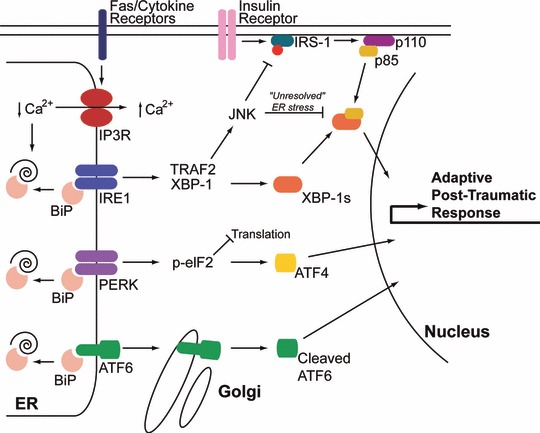
ER stress pathways activated post-burn are similar to those mediating type 2 diabetes. Burn injury causes multi-organ ER stress characterized by activation of all three arms of the ER stress response. In animal burn models, hepatic ER calcium stores are depleted most likely by activation of IP3R calcium channels leading to accumulation of unfolded polypeptides and activation of IRE1, PERK and ATF6 [53]. The mechanism by which the IP3R is activated is still unclear, but may be mediated by Fas receptor activation or calcium mobilizing cytokines (see text for details). Activation of JNK is prominent post-burn, leading to IRS-1 phosphorylation and suppression of PI3K activation (schematically represented by the p110 and p85 subunits) and ultimately insulin resistance [54, 55]. Insulin resistance in burn patients persists for an extended period of time (‘unresolved’ ER stress) similar to type 2 diabetes [78]. This may be mediated by suppression of XBP-1s/p85 transcriptional activity by JNK [60].
The second ER stress transducer is ATF6, a founding member of a novel class of metazoan-specific ER stress transducers [39]. ATF6 is a family member of the cyclic AMP response element binding protein (CREB) family of transcription factors. In unstressed cells, ATF6 resides in the ER membrane. ATF6 trafficking is hindered by binding to BiP at the luminal domain of ATF6 [36]. ER stress disrupts BiP binding and ATF6 is subsequently transported to the Golgi apparatus [40], where it is cleaved by Golgi-resident proteases: first by site 1 protease (S1P) and then in an intra-membrane region by site 2 protease (S2P) to release the cytosolic DNA-binding domain. ATF6 then translocates to the nucleus to activate gene expression of UPR target genes [39].
The third ER stress transducer, PERK, is also an ER-localized type I transmembrane protein with luminal stress-sensing domains that is similar in structure and function to IRE1 [41]. Like IRE1 and ATF6, the luminal domain of PERK is bound to BiP in unstressed cells. The cytoplasmic portion of PERK contains a protein kinase domain, which undergoes activation by oligomerization in ER-stressed cells. Activated PERK phosphorylates the α-subunit of eukaryotic translation initiation factor-2 (eIF2α) at serine 51, leading to derepression of translational inhibition by elongation factor 2B (eIF2B) [36]. The resulting reduced activities of eIF2α account for all of the important consequences of PERK activity. Because other eIF2 kinases can activate this pathway independently of ER stress, this portion of the unfolded protein response is termed the integrated stress response [42, 43]. The function of PERK activation is to lower global protein synthesis to reduce the ER unfolded protein load and to alter gene transcription. For example, phospho-PERK increases transcription of the activating transcription factor-4 (ATF4) and stimulates nuclear factor κB (NF-κB) activation [44, 45]. The transcription factor C/EBP-homologous protein (CHOP) is also activated transcriptionally by ATF4, and its target genes include DNA damage-inducible protein-34 (GADD34), a regulatory subunit of phosphatase PP1 that dephosphorylates eIF2α and terminates PERK signalling [46].
Induction of the ER stress response and intracellular calcium stores
The concentration of calcium in various intracellular compartments are exquisitely regulated by various ATPases and exchangers. Levels in the cytosol are maintained in the nanomolar range, whereas ER, mitochondrial and extracellular levels are in the millimolar range. The cell expends a significant amount of energy to maintain these steep gradients, and this is required for maintaining normal physiologic function of these organelles. Maintaining a steep calcium gradient also allows this ion to function as a signalling molecule. In the ER, millimolar calcium is required to maintain the protein folding capacity, because critical molecular chaperones that participate in the folding, assembly and maturation of secretory proteins, such as calnexin, calreticulin, PDI and BiP, require calcium to function properly [47]. One of the classic and most widely used in vitro models of ER stress is treatment with the sarco-endoplasmic reticulum ATPase (SERCA) poison thapsigargin. Inhibition of SERCA activity uncovers a passive leak of calcium from the ER, and leads to rapid and profound reductions in ER luminal calcium levels. This leads to protein misfolding in the ER and induction of the ER stress response.
We found in a rat burn model that thermal injury has dramatic effects on intracellular calcium levels in the liver (Fig. 1). Burn injury resulted in marked reduction in ER calcium levels, with a concomitant increase in cytoplasmic calcium levels [48]. This was associated with decreased responsiveness to vasopressin and the purinergic agonist ATP. Furthermore, the depletion of ER calcium stores was associated with marked activation of the ER stress response, including up-regulation of pro-apoptotic ER stress signalling through JNK and Bim. We concluded that hepatic ER stress and apoptosis post-burn is mediated primarily by depletion of intracellular calcium stores [48]. A question remains as to the mechanism of ER calcium store depletion. It is possible that traumatic injury results in dramatic up-regulation of calcium mobilizing agonists in the liver and elsewhere, including pro-inflammatory cytokines such as IL-1 [49] or members of the TNF-α receptor family [50]. Indeed, we have found in unpublished observations that Fas receptor levels and mRNA are dramatically up-regulated in the liver after burn injury. As Fas receptor is coupled to ER calcium release [50, 51], this could be a possible mechanism for ER calcium store depletion and cell death (Fig. 1). A different model was recently proposed by Fu et al., where they showed that hepatic ER lipid synthesis was greatly increased in an obese mouse model resulting in reduced SERCA pump activity [52]. Reduced SERCA activity led to depleted ER calcium stores and chronic ER stress. Strikingly, SERCA overexpression reversed these effects and improved ER stress and insulin sensitivity. Interestingly, in our rat burn model SERCA expression is up-regulated 24 and 48 hrs after burn, which we interpreted to be reflective of a compensatory mechanism in response to depleted ER calcium stores [53]. It will be interesting to investigate if hepatic SERCA activity is inhibited post-burn.
Similar to our studies in the rat burn model, we found that the molecular chaperones GRP78/BiP and PDI were significantly increased in the liver up to 21 days post-burn in mice [54]. The expression of phosphorylated PERK increased within 1 day post-burn and then dropped to normal levels by day 14. Phospho-IRE-1was increased at 1 day post-burn, exhibiting a peak at 7 days post-burn. The cleavage of ATF-6 increased significantly at the late time point of 21 days post-burn. The expression of calnexin and calreticulin were dramatically increased at 1 day post-burn in mice, and remained elevated 21 days later, indicative of a compensatory mechanism in response to calcium store depletion. Downstream activation of CHOP was seen at 7 and 14 days post-burn. The aforementioned changes all indicate that burn induces marked ER stress/UPR not only in rats but also in mice and that ER stress/UPR persists over a prolonged period of time, indicating the cellular, metabolic and inflammatory alterations persisting longer than previously thought.
Type 2 diabetes in animal models and links to the ER stress response
As mentioned earlier, ER stress activates JNK resulting in phosphorylation of insulin receptor substrate-1 [38]. This prevent tyrosine phosphorylation of JNK by the insulin receptor, inhibiting downstream signalling through the PI3K/Akt pathway and transport of GLUT4 to the plasma membrane. We have found similar findings in the liver in a rat burn model which accurately recapitulates what is seen clinically. In particular, burn injury results in serine phosphorylation of IRS-1 resulting in decreased sensitivity to exogenously administered insulin [55]. Furthermore, insulin administration post-burn protected against ER stress and associated hepatic damage and insulin resistance [56]. Thus, at least in the liver (and possibly skeletal muscle [57, 58]), similar pathways are activated in animal models of type 2 diabetes and burn injury.
In order for the liver to meet metabolic demands under high glucose conditions, there is a transient activation of the unfolded protein response to increase the folding capacity in the ER lumen due to increased translation of metabolic proteins [59]. Thus, ER stress is a ‘normal’ response in the liver to increased metabolic demands. Two recent reports have highlighted a direct link between insulin receptor pathway and ER stress, with direct implications for insulin resistance [60, 61]. It was found in wild-type mice that after a meal ER stress was activated by binding of the catalytic subunit of PI3K (p85) to the protein product of XBP-1 (XBP-1s), and this was required for the stabilization and nuclear translocation of XBP-1s (Fig. 1). It is important to appreciate that the p85 subunit is normally heterodimerized with the p110 catalytic subunit under resting conditions, and is released after insulin receptor activation. Importantly, in ob/ob mice there was not sufficient free p85 subunit to promote the downstream activation of XBP-1s target genes critical for restoring the folding capacity of the ER, leading to increased unfolded protein burden [60, 61]. As the effects of p85 are independent of the other arms of the ER stress response, continued ER stress would ultimately result in activation of the pro-apoptotic and pro-inflammatory arms of the ER stress response. Similar to animal models of obesity and insulin resistance, we have found dramatically increased splicing of XBP-1 post-burn (unpublished), and this is associated with suppressed PI3K/Akt signalling [55].
Clinical relevance of insulin resistance and ER stress in the post-burn response
Burn injury produces a profound hypermetabolic stress response characterized by increased glucose production, lipolysis and protein catabolism [3, 23, 62]. The hypermetabolic stress response is driven by the inflammatory response, which encompasses hormones, cytokines and acute phase proteins [63-65]. Clinical studies have shown that a sustained or increased inflammatory and acute phase response can be life threatening with the uncontrolled and prolonged action of pro-inflammatory cytokines and acute phase proteins contributing to multi-organ failure, hypermetabolism, morbidity and mortality [64-66]. We hypothesize that the underlying mechanism is an increased inflammatory response associated with depletion of ER calcium stores and induction of ER stress and apoptotic pathways.
To validate our studies in animal models, we investigated whether a severe injury induces ER stress in severely burned patients. Twenty severely burned paediatric patients were compared to 36 non-burned children. Clinical markers, protein analysis and transcriptome analysis was used to identify transcriptional changes in ER stress and insulin resistance-related signalling cascades in peripheral blood leukocytes, fat and muscle at admission up to 466 days post-burn. We found in severely burned patients that a burn injury induces a vast inflammatory response which leads to cellular stress responses in various tissues associated with profound insulin resistance and hyperglycaemia. We found that these changes were associated with the induction of systemic ER stress (unpublished observations). We determined the genomic changes in peripheral blood leukocytes, fat and muscle post-burn and compared these changes to the same tissues from normal, healthy, non-burned volunteers [67]. Genomic expression data collected over four time periods (0–10, 11–49, 50–250 and 251+ days post-burn) in burned patients was compared to expression data from non-burned patients. We identified the following canonical signalling pathways in Ingenuity Knowledge Base that are associated with insulin resistance and ER/SR stress: ER/SR stress, insulin receptor, phosphoinositide-3-kinase (PI3K), extracellular-regulated MAP kinase 1/2/mitogen-activated protein kinase 1 (ERK/MAPK), c-Jun N-terminal kinase (SAPK/JNK), acute phase response, calcium and apoptosis. Super-imposition of the expression data for the significantly altered genes onto the networks of known molecular interactions and canonical signalling pathways enabled identification of genes that may mediate the post-burn insulin resistance response in different tissues. Within the identified pathways, transcripts of 455 genes in peripheral blood leukocytes, 360 genes in fat and 448 genes in muscle were significantly changed in a temporal manner following a severe burn injury [67]. The majority of these genes did not return to normal expression levels by even 251+ days following the burn injury, indicating pervasive and persistent ER/SR stress, UPR and insulin resistance. Thus, the dramatic changes in gene expression in various tissues may provide the molecular basis for clinical sequela such as insulin resistance, muscle wasting, fat loss and persistent hyperinflammation during the first year following a burn injury. These findings indicate that ER stress and UPR is not limited to one tissue type but is present in many if not all organs and tissues post-burn.
Targeting the ER stress response as a therapeutic for burn injury
As mentioned earlier, ER stress is primarily triggered by an increase in the unfolded protein burden in the ER. Molecular chaperones in the ER lumen such as Bip and Grp94 maintain proteostasis in the ER by binding to unfolded/misfolded polypeptide chains. Recently, several ‘chemical chaperones’ have been identified which function in a manner analogous the endogenous molecular chaperone machinery and reduce ER stress [68-71]. The mechanisms by which these compounds favourably affect protein folding are still incompletely understood, but may be related to their function as osmolytes [72], or direct effects on the endogenous protein folding machinery [71].
In a mouse model of type 2 diabetes (ob/ob mice), Ozcan et al. demonstrated that administration of two of these compounds, 4-phenyl butyric acid (PBA) and tauroursodeoxycholic acid (TUDCA), reduced hepatic ER stress and insulin resistance in vivo [70]. Furthermore, both compounds restored systemic glucose metabolism. In this experimental paradigm, PBA was administered by gavage and TUDCA by intraperitoneal injection. A separate study demonstrated that PBA alleviates macrophage ER stress and vascular disease in a mouse model of atherosclerosis [73]. A small clinical study examining orally administered TUDCA in type 2 diabetic patients revealed improvements in hepatic and muscle insulin sensitivity and an increase in muscle IRS-1 and Akt phosphorylation, suggesting a reversal of post-receptor insulin resistance [74]. Both PBA and TUDCA are FDA approved medications for various disorders unrelated to ER stress/type 2 diabetes, and thus may be promising targets for intervention in type 2 diabetes [71, 75-77]. Thus, PBA and TUDCA may be beneficial for the treatment of post-burn ER stress and insulin resistance.
Conclusions
Traumatic injuries such as burns cause profound metabolic and hyper-inflammatory responses, which result in multi-organ dysfunction. One hallmark of burn injury is profound and prolonged insulin resistance which mimics at the molecular level the cellular changes seen in type 2 diabetes. Importantly, post-burn insulin resistance directly affects patient morbidity and mortality. Recent studies by our group and others have delineated a clear role for multi-organ ER stress as a critical mediator of post-burn insulin resistance. Thus, targeting ER stress therapeutically may have a dramatic impact on post-traumatic insulin resistance, patient recovery, and patient reintegration.
Acknowledgments
This study was supported by the National Institutes of Health Grants GM081685 and GM081685-S1 (DB) and GM087285 (MGJ), grants 8640, 8690 and 8660 from Shriners of North America, CFI Leader’s Opportunity Fund: Project #25407, and Physicians’ Services Incorporated Foundation—Health Research Grant Program.
Conflicts of interest
The authors confirm that there are no conflicts of interest.
References
- 1.Bringham PA, McLoughlin E. Burn incidence and medical care use in the United States: estimates, trends and data sources. J Burn Care Rehabil. 1996;17:95–107. doi: 10.1097/00004630-199603000-00003. [DOI] [PubMed] [Google Scholar]
- 2.Peden M, McGee K, Sharma G. The injury chart book: a graphical overview of the global burden of injuries. Geneva: World Health Organization; 2002. [Google Scholar]
- 3.Herndon DN, Tompkins RG. Support of the metabolic response to burn injury. Lancet. 2004;363:1895–902. doi: 10.1016/S0140-6736(04)16360-5. [DOI] [PubMed] [Google Scholar]
- 4.Jeschke MG, Chinkes DL, Finnerty CC, et al. Pathophysiologic response to severe burn injury. Ann Surg. 2008;248:387–401. doi: 10.1097/SLA.0b013e3181856241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gore DC, Chinkes D, Heggers J, et al. Association of hyperglycemia with increased mortality after severe burn injury. J Trauma. 2001;51:540–4. doi: 10.1097/00005373-200109000-00021. [DOI] [PubMed] [Google Scholar]
- 6.Gore DC, Chinkes DL, Hart DW, et al. Hyperglycemia exacerbates muscle protein catabolism in burn-injured patients. Crit Care Med. 2002;30:2438–42. doi: 10.1097/00003246-200211000-00006. [DOI] [PubMed] [Google Scholar]
- 7.Hemmila MR, Taddonio MA, Arbabi S, et al. Intensive insulin therapy is associated with reduced infectious complications in burn patients. Surgery. 2008;144:629–35. doi: 10.1016/j.surg.2008.07.001. ; discussion 35–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ferrando AA, Chinkes DL, Wolf SE, et al. A submaximal dose of insulin promotes net skeletal muscle protein synthesis in patients with severe burns. Ann Surg. 1999;229:11–8. doi: 10.1097/00000658-199901000-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pierre EJ, Barrow RE, Hawkins HK, et al. Effects of insulin on wound healing. J Trauma. 1998;44:342–5. doi: 10.1097/00005373-199802000-00019. [DOI] [PubMed] [Google Scholar]
- 10.Thomas SJ, Morimoto K, Herndon DN, et al. The effect of prolonged euglycemic hyperinsulinemia on lean body mass after severe burn. Surgery. 2002;132:341–7. doi: 10.1067/msy.2002.126871. [DOI] [PubMed] [Google Scholar]
- 11.Zhang XJ, Chinkes DL, Wolf SE, et al. Insulin but not growth hormone stimulates protein anabolism in skin would and muscle. Am J Physiol. 1999;276:E712–20. doi: 10.1152/ajpendo.1999.276.4.E712. [DOI] [PubMed] [Google Scholar]
- 12.Jeschke MG, Klein D, Bolder U, et al. Insulin attenuates the systemic inflammatory response in endotoxemic rats. Endocrinology. 2004;145:4084–93. doi: 10.1210/en.2004-0592. [DOI] [PubMed] [Google Scholar]
- 13.Jeschke MG, Klein D, Herndon DN. Insulin treatment improves the systemic inflammatory reaction to severe trauma. Ann Surg. 2004;239:553–60. doi: 10.1097/01.sla.0000118569.10289.ad. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jeschke MG, Rensing H, Klein D, et al. Insulin prevents liver damage and preserves liver function in lipopolysaccharide-induced endotoxemic rats. J Hepatol. 2005;42:870–9. doi: 10.1016/j.jhep.2004.12.036. [DOI] [PubMed] [Google Scholar]
- 15.Klein D, Schubert T, Horch RE, et al. Insulin treatment improves hepatic morphology and function through modulation of hepatic signals after severe trauma. Ann Surg. 2004;240:340–9. doi: 10.1097/01.sla.0000133353.57674.cd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harrison TS, Seaton JF, Feller I. Relationship of increased oxygen consumption to catecholamine excretion in thermal burns. Ann Surg. 1967;165:169–72. doi: 10.1097/00000658-196702000-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wilmore DW, Long JM, Mason AD, Jr, et al. Catecholamines: mediator of the hypermetabolic response to thermal injury. Ann Surg. 1974;180:653–69. doi: 10.1097/00000658-197410000-00031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goodall M, Stone C, Haynes BW., Jr Urinary output of adrenaline and noradrenaline in severe thermal burns. Ann Surg. 1957;145:479–87. doi: 10.1097/00000658-195704000-00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wilmore DW, Aulick LH. Metabolic changes in burned patients. Surg Clin North Am. 1978;58:1173–87. doi: 10.1016/s0039-6109(16)41685-3. [DOI] [PubMed] [Google Scholar]
- 20.Wolfe RR, Durkot MJ, Allsop JR, et al. Glucose metabolism in severely burned patients. Metabolism. 1979;28:1031–9. doi: 10.1016/0026-0495(79)90007-6. [DOI] [PubMed] [Google Scholar]
- 21.Gore DC, Ferrando A, Barnett J, et al. Influence of glucose kinetics on plasma lactate concentration and energy expenditure in severely burned patients. J Trauma. 2000;49:673–7. doi: 10.1097/00005373-200010000-00015. ; discussion 7–8. [DOI] [PubMed] [Google Scholar]
- 22.Wolfe RR, Miller HI, Spitzer JJ. Glucose and lactate kinetics in burn shock. Am J Physiol. 1977;232:E415–8. doi: 10.1152/ajpendo.1977.232.4.E415. [DOI] [PubMed] [Google Scholar]
- 23.van den Berghe G, Wouters P, Weekers F, et al. Intensive insulin therapy in the critically ill patients. N Engl J Med. 2001;345:1359–67. doi: 10.1056/NEJMoa011300. [DOI] [PubMed] [Google Scholar]
- 24.Van den Berghe G, Wilmer A, Hermans G, et al. Intensive insulin therapy in the medical ICU. N Engl J Med. 2006;354:449–61. doi: 10.1056/NEJMoa052521. [DOI] [PubMed] [Google Scholar]
- 25.Vlasselaers D, Milants I, Desmet L, et al. Intensive insulin therapy for patients in paediatric intensive care: a prospective, randomised controlled study. Lancet. 2009;373:547–56. doi: 10.1016/S0140-6736(09)60044-1. [DOI] [PubMed] [Google Scholar]
- 26.Ellger B, Debaveye Y, Vanhorebeek I, et al. Survival benefits of intensive insulin therapy in critical illness: impact of maintaining normoglycemia versus glycemia-independent actions of insulin. Diabetes. 2006;55:1096–105. doi: 10.2337/diabetes.55.04.06.db05-1434. [DOI] [PubMed] [Google Scholar]
- 27.Ingels C, Debaveye Y, Milants I, et al. Strict blood glucose control with insulin during intensive care after cardiac surgery: impact on 4-years survival, dependency on medical care, and quality-of-life. Eur Heart J. 2006;27:2716–24. doi: 10.1093/eurheartj/ehi855. [DOI] [PubMed] [Google Scholar]
- 28.Finney SJ, Zekveld C, Elia A, et al. Glucose control and mortality in critically ill patients. JAMA. 2003;290:2041–7. doi: 10.1001/jama.290.15.2041. [DOI] [PubMed] [Google Scholar]
- 29.Preiser JC, Devos P. Clinical experience with tight glucose control by intensive insulin therapy. Crit Care Med. 2007;35:S503–7. doi: 10.1097/01.CCM.0000278046.24345.C7. [DOI] [PubMed] [Google Scholar]
- 30.Brunkhorst FM, Engel C, Bloos F, et al. Intensive insulin therapy and pentastarch resuscitation in severe sepsis. N Engl J Med. 2008;358:125–39. doi: 10.1056/NEJMoa070716. [DOI] [PubMed] [Google Scholar]
- 31.Finfer S, Chittock DR, Su SY, et al. Intensive versus conventional glucose control in critically ill patients. N Engl J Med. 2009;360:1283–97. doi: 10.1056/NEJMoa0810625. [DOI] [PubMed] [Google Scholar]
- 32.Jeschke MG, Kulp GA, Kraft R, et al. Intensive insulin therapy in severely burned pediatric patients: a prospective randomized trial. Am J Respir Crit Care Med. 2010;182:351–9. doi: 10.1164/rccm.201002-0190OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Szegezdi E, Logue SE, Gorman AM, et al. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006;7:880–5. doi: 10.1038/sj.embor.7400779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cox JS, Shamu CE, Walter P. Transcriptional induction of genes encoding endoplasmic reticulum resident proteins requires a transmembrane protein kinase. Cell. 1993;73:1197–206. doi: 10.1016/0092-8674(93)90648-a. [DOI] [PubMed] [Google Scholar]
- 35.Mori K, Ma W, Gething MJ, et al. A transmembrane protein with a cdc2+/CDC28-related kinase activity is required for signaling from the ER to the nucleus. Cell. 1993;74:743–56. doi: 10.1016/0092-8674(93)90521-q. [DOI] [PubMed] [Google Scholar]
- 36.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–29. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 37.Urano F, Wang X, Bertolotti A, et al. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science. 2000;287:664–6. doi: 10.1126/science.287.5453.664. [DOI] [PubMed] [Google Scholar]
- 38.Ozcan U, Cao Q, Yilmaz E, et al. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004;306:457–61. doi: 10.1126/science.1103160. [DOI] [PubMed] [Google Scholar]
- 39.Haze K, Yoshida H, Yanagi H, et al. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol Biol Cell. 1999;10:3787–99. doi: 10.1091/mbc.10.11.3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shen J, Chen X, Hendershot L, et al. ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev Cell. 2002;3:99–111. doi: 10.1016/s1534-5807(02)00203-4. [DOI] [PubMed] [Google Scholar]
- 41.Bertolotti A, Zhang Y, Hendershot LM, et al. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat Cell Biol. 2000;2:326–32. doi: 10.1038/35014014. [DOI] [PubMed] [Google Scholar]
- 42.Harding HP, Zhang Y, Bertolotti A, et al. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol Cell. 2000;5:897–904. doi: 10.1016/s1097-2765(00)80330-5. [DOI] [PubMed] [Google Scholar]
- 43.Harding HP, Zhang Y, Zeng H, et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell. 2003;11:619–33. doi: 10.1016/s1097-2765(03)00105-9. [DOI] [PubMed] [Google Scholar]
- 44.Wek RC, Cavener DR. Translational control and the unfolded protein response. Antioxid Redox Signal. 2007;9:2357–71. doi: 10.1089/ars.2007.1764. [DOI] [PubMed] [Google Scholar]
- 45.Wek RC, Jiang HY, Anthony TG. Coping with stress: eIF2 kinases and translational control. Biochem Soc Trans. 2006;34:7–11. doi: 10.1042/BST20060007. [DOI] [PubMed] [Google Scholar]
- 46.Marciniak SJ, Yun CY, Oyadomari S, et al. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004;18:3066–77. doi: 10.1101/gad.1250704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Burdakov D, Petersen OH, Verkhratsky A. Intraluminal calcium as a primary regulator of endoplasmic reticulum function. Cell Calcium. 2005;38:303–10. doi: 10.1016/j.ceca.2005.06.010. [DOI] [PubMed] [Google Scholar]
- 48.Jeschke MG, Gauglitz GG, Song J, et al. Calcium and ER stress mediate hepatic apoptosis after burn injury. J Cell Mol Med. 2009;13:1857–65. doi: 10.1111/j.1582-4934.2008.00644.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang Q, Downey GP, Choi C, et al. IL-1 induced release of Ca2+ from internal stores is dependent on cell-matrix interactions and regulates ERK activation. FASEB J. 2003;17:1898–900. doi: 10.1096/fj.03-0069fje. [DOI] [PubMed] [Google Scholar]
- 50.Wozniak AL, Wang X, Stieren ES, et al. Requirement of biphasic calcium release from the endoplasmic reticulum for Fas-mediated apoptosis. J Cell Biol. 2006;175:709–14. doi: 10.1083/jcb.200608035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Akimzhanov AM, Wang X, Sun J, et al. T-cell receptor complex is essential for Fas signal transduction. Proc Natl Acad Sci USA. 2010;107:15105–10. doi: 10.1073/pnas.1005419107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fu S, Yang L, Li P, et al. Aberrant lipid metabolism disrupts calcium homeostasis causing liver endoplasmic reticulum stress in obesity. Nature. 2011;473:528–31. doi: 10.1038/nature09968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jeschke MG, Gauglitz GG, Song J, et al. Calcium and ER stress mediate hepatic apoptosis after burn injury. J Cell Mol Med. 2009;13:1857–65. doi: 10.1111/j.1582-4934.2008.00644.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Song J, Finnerty CC, Herndon DN, et al. Severe burn-induced endoplasmic reticulum stress and hepatic damage in mice. Mol Med. 2009;15:316–20. doi: 10.2119/molmed.2009.00048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gauglitz GG, Halder S, Boehning DF, et al. Post-burn hepatic insulin resistance is associated with endoplasmic reticulum (Er) stress. Shock. 2010;33:299–305. doi: 10.1097/SHK.0b013e3181b2f439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jeschke MG, Kraft R, Song J, et al. Insulin protects against hepatic damage post-burn. Mol Med. 2011;17:516–22. doi: 10.2119/molmed.2010.00166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang Q, Carter EA, Ma BY, et al. Molecular mechanism(s) of burn-induced insulin resistance in murine skeletal muscle: role of IRS phosphorylation. Life Sci. 2005;77:3068–77. doi: 10.1016/j.lfs.2005.02.034. [DOI] [PubMed] [Google Scholar]
- 58.Ikezu T, Okamoto T, Yonezawa K, et al. Analysis of thermal injury-induced insulin resistance in rodents. Implication of postreceptor mechanisms. J Biol Chem. 1997;272:25289–95. doi: 10.1074/jbc.272.40.25289. [DOI] [PubMed] [Google Scholar]
- 59.Wek RC, Anthony TG. Obesity: stressing about unfolded proteins. Nat Med. 2010;16:374–6. doi: 10.1038/nm0410-374. [DOI] [PubMed] [Google Scholar]
- 60.Park SW, Zhou Y, Lee J, et al. The regulatory subunits of PI3K, p85alpha and p85beta, interact with XBP-1 and increase its nuclear translocation. Nat Med. 2010;16:429–37. doi: 10.1038/nm.2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Winnay JN, Boucher J, Mori MA, et al. A regulatory subunit of phosphoinositide 3-kinase increases the nuclear accumulation of X-box-binding protein-1 to modulate the unfolded protein response. Nat Med. 2010;16:438–45. doi: 10.1038/nm.2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Herndon DN, Hart DW, Wolf SE, et al. Reversal of catabolism by beta-blockade after severe burns. N Engl J Med. 2001;345:1223–9. doi: 10.1056/NEJMoa010342. [DOI] [PubMed] [Google Scholar]
- 63.Finnerty CC, Herndon DN, Przkora R, et al. Cytokine expression profile over time in severely burned pediatric patients. Shock. 2006;26:13–9. doi: 10.1097/01.shk.0000223120.26394.7d. [DOI] [PubMed] [Google Scholar]
- 64.Tracey KJ, Fong Y, Hesse DG, et al. Anti-cachectin/TNF monoclonal antibodies prevent septic shock during lethal bacteraemia. Nature. 1987;330:662–4. doi: 10.1038/330662a0. [DOI] [PubMed] [Google Scholar]
- 65.Tracey KJ, Lowry SF, Fahey TJ, 3rd, et al. Cachectin/tumour necrosis factor induces lethal shock and stress hormone responses in the dog. Surg Gynecol Obstet. 1987;164:415–22. [PubMed] [Google Scholar]
- 66.Moshage H. Cytokines and the hepatic acute phase response. J Pathol. 1997;181:257–66. doi: 10.1002/(SICI)1096-9896(199703)181:3<257::AID-PATH756>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 67.Jeschke MG, Finnerty CC, Herndon DN, et al. Severe injury is associated with insulin resistance, er stress response, and unfolded protein response. Ann Surg. 2011 doi: 10.1097/SLA.0b013e31823e76e7. ; In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.de Almeida SF, Picarote G, Fleming JV, et al. Chemical chaperones reduce endoplasmic reticulum stress and prevent mutant HFE aggregate formation. J Biol Chem. 2007;282:27905–12. doi: 10.1074/jbc.M702672200. [DOI] [PubMed] [Google Scholar]
- 69.Gong B, Zhang LY, Pang CP, et al. Trimethylamine N-oxide alleviates the severe aggregation and ER stress caused by G98R alphaA-crystallin. Mol Vis. 2009;15:2829–40. [PMC free article] [PubMed] [Google Scholar]
- 70.Ozcan U, Yilmaz E, Ozcan L, et al. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science. 2006;313:1137–40. doi: 10.1126/science.1128294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Engin F, Hotamisligil GS. Restoring endoplasmic reticulum function by chemical chaperones: an emerging therapeutic approach for metabolic diseases. Diabetes Obes Metab. 2010;12:108–15. doi: 10.1111/j.1463-1326.2010.01282.x. [DOI] [PubMed] [Google Scholar]
- 72.Bolen DW, Baskakov IV. The osmophobic effect: natural selection of a thermodynamic force in protein folding. J Mol Biol. 2001;310:955–63. doi: 10.1006/jmbi.2001.4819. [DOI] [PubMed] [Google Scholar]
- 73.Erbay E, Babaev VR, Mayers JR, et al. Reducing endoplasmic reticulum stress through a macrophage lipid chaperone alleviates atherosclerosis. Nat Med. 2009;15:1383–91. doi: 10.1038/nm.2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kars M, Yang L, Gregor MF, et al. Tauroursodeoxycholic acid may improve liver and muscle but not adipose tissue insulin sensitivity in obese men and women. Diabetes. 2010;59:1899–905. doi: 10.2337/db10-0308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Maestri NE, Brusilow SW, Clissold DB, et al. Long-term treatment of girls with ornithine transcarbamylase deficiency. N Engl J Med. 1996;335:855–9. doi: 10.1056/NEJM199609193351204. [DOI] [PubMed] [Google Scholar]
- 76.Collins AF, Pearson HA, Giardina P, et al. Oral sodium phenylbutyrate therapy in homozygous beta thalassemia: a clinical trial. Blood. 1995;85:43–9. [PubMed] [Google Scholar]
- 77.Poupon RE, Bonnand AM, Chretien Y, et al. Ten-year survival in ursodeoxycholic acid-treated patients with primary biliary cirrhosis. The UDCA-PBC Study Group. Hepatology. 1999;29:1668–71. doi: 10.1002/hep.510290603. [DOI] [PubMed] [Google Scholar]
- 78.Gauglitz GG, Herndon DN, Kulp GA, et al. Abnormal insulin sensitivity persists up to three years in pediatric patients post-burn. J Clin Endocrinol Metab. 2009;94:1656–64. doi: 10.1210/jc.2008-1947. [DOI] [PMC free article] [PubMed] [Google Scholar]