

Abstract
Autologous keratinocytes can be used to augment cutaneous repair, such as in the treatment of severe burns and recalcitrant ulcers. Such cells can be delivered to the wound bed either as a confluent sheet of cells or in single-cell suspension. The standard method for expanding primary human keratinocytes in culture uses lethally irradiated mouse 3T3 fibroblasts as feeder cells to support keratinocyte attachment and growth. In an effort to eliminate xenobiotic cells from clinical culture protocols where keratinocytes are applied to patients, we investigated whether human autologous primary fibroblasts could be used to expand keratinocytes in culture. At a defined ratio of a 6:1 excess of keratinocytes to fibroblasts, this co-culture method displayed a population doubling rate comparable to culture with lethally irradiated 3T3 cells. Furthermore, morphological and molecular analysis showed that human keratinocytes expanded in co-culture with autologous human fibroblasts were positive for proliferation markers and negative for differentiation markers. Keratinocytes expanded by this method thus retain their proliferative phenotype, an important feature in enhancing rapid wound closure. We suggest that this novel co-culture method is therefore suitable for clinical use as it dispenses with the need for lethally irradiated 3T3 cells in the rapid expansion of autologous human keratinocytes.
Keywords: Cell culture, Tissue engineering, Skin, Keratinocyte
Introduction
The use of autologous keratinocytes in the treatment of cutaneous defects, such as full thickness burns and venous leg ulcers, is established as a method for improving epithelial closure in patients suffering extensive skin damage (Boyce et al. 2002). In order to augment epithelial repair, autologous human keratinocytes have been applied to the wound bed either as a confluent sheet (CEA, cultured epithelial autograft) or in single-cell suspension (LaFrance and Armstrong 1999; Atiyeh and Costagliola 2007; Cooper and Spielvogel 1994b; Horch et al. 1998; Gustafson and Kratz 1999; Currie et al. 2003; Navarro et al. 2000).
The rapid expansion of keratinocytes from a small skin biopsy is essential in the tissue engineering of new skin, especially when large numbers of cells are necessary to repopulate and repair extensive areas of cutaneous damage. Rheinwald and Green (1975) were the first to establish the culture of keratinocytes from skin biopsies. They reported that the use of lethally irradiated mouse 3T3 cells as a feeder layer allowed keratinocytes to form colonies and proliferate. The feeder layer of fibroblastic cells were reported as being key to facilitating the attachment and proliferation of keratinocytes in culture, this being due to the secretion of extracellular matrix proteins and growth factors by these cells (Navsaria et al. 1994). The method established by Rheinwald and Green is still currently used, including in the preparation of keratinocytes for clinical use (Moustafa et al. 2007; James et al. 2010).
Due to growing concerns in relation to animal disease transmission when reapplying cells back to the patient in a clinical situation, the development of systems that dispense with xenobiotic feeder cells and materials is highly desirable. Indeed it has been shown recently that human embryonic and mesenchymal stem cells expanded in the presence of mouse feeder cells can become contaminated with xenoantigens originating from the feeder cells (Martin et al. 2005; Heiskanen et al. 2007). This indicates the use of xenobiotic tissue for clinical use could pose substantial and unacceptable risks to patients. In an effort to limit such risks, growth-arrested 3T3 feeder cells of mouse origin have been replaced either by the use of human allogeneic fibroblasts (Sun et al. 2004) or alternatively by lethally irradiated autologous fibroblasts (Mujaj et al. 2010). The use of allogeneic cells, however, poses a potential risk in terms of immunological rejection. Furthermore, the methods employed to growth-arrest feeder cells such as irradiation or chemical treatment may lead to damage and transformation of the cells and potential transmission of damaged genetic material to the keratinocytes. We have therefore devised a novel protocol utilising primary autologous non-irradiated fibroblasts to facilitate the expansion of primary autologous keratinocytes for clinical use. Our aim was to define the co-culture conditions that would support the rapid expansion of human keratinocytes in the presence of autologous fibroblasts.
Materials and methods
Primary human keratinocytes and fibroblasts were isolated and cultured as previously described (Harris et al. 2009). Briefly, skin was obtained with patient consent and full approval from the National Regional Ethics Service (REC: 06/Q1907/81) from discarded tissue during routine surgical procedures. The dermis and epidermis were separated using dispase (4 mg/mL, Invitrogen, UK), and keratinocytes were released from the epidermis by incubating in 0.5% trypsin for 30 min at 37 °C (Gibco, UK). Fibroblasts were isolated by incubation of the dermis in 0.5% collagenase for 2 h at 37 °C (Sigma, UK). Following counting using a trypan blue exclusion assay using a haemocytometer cells were seeded at a density of 2 × 104 cells/cm2 (Navsaria et al. 1994) in Rheinwald and Green medium (R&G), consisting of DMEM and Ham’s F12 medium (Invitrogen, UK) at a 3:1 ratio, supplemented with 10% FCS (Gibco, UK), 10 ng/mL human recombinant EGF (Invitrogen, UK), 10 nM cholera toxin (Sigma, UK) and 0.4 mg/mL hydrocortisone (Sigma, UK) or in defined medium EpiLife (Gibco, UK) with EDGS supplement (Gibco, UK). As controls, keratinocytes were grown on a feeder layer of lethally irradiated 3T3 cells. All cells were cultured at 37 °C with 5% CO2 and medium was changed twice weekly. Cultures were passaged by incubation in 0.05% Trysin-EDTA (Gibco, UK) and reseeding in fresh medium.
Primary keratinocyte and fibroblast co-culture
Co-culture experiments were set up at ratios ranging from 10:1 to 1:1 of keratinocytes to fibroblasts, with cells being mixed prior to seeding (Sun et al. 2004). As cultures reached a confluent state, the percentage of keratinocytes in each co-culture was assessed visually using phase contrast microscopy. Those cultures that had reached confluency and contained at least 80% keratinocytes were passaged as described and re-seeded at 2 × 104 cells/cm2 to establish their cumulative population doublings. The following formula was used to calculate the number of population doublings (n) of autologous keratinocytes (AK):
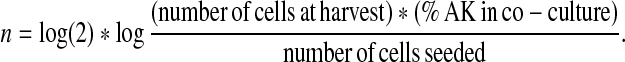 |
Indirect Immunofluorescence
Cells grown on glass coverslips were fixed in 3% paraformaldehyde (Sigma, UK), permeabilised in 0.2% saponin (Sigma, UK) and blocked in 0.2% gelatin (Sigma, UK) and 0.02% saponin (all in PBS). Primary antibody incubations were 60 min, secondary antibody incubations 30 min in the dark, both performed at room temperature in a moist chamber. Coverslips were mounted in Prolong Antifade Gold with DAPI (Invitrogen, UK). Images were acquired using a Zeiss Axioscope and analysed using Axiovision software (Zeiss, Germany). Antibodies used were cytokeratin 14 (K14, Vector Laboratories, UK, 1:200 dilution), cytokeratin 10 (K10, Abcam, UK, 1:500), vimentin (Abcam, UK, 1:100), anti-mouse FITC (Stratech Scientific, UK, 1:100) and anti-rabbit TRITC (Abcam, UK, 1:400).
Flow cytometry
Confluent cultures were harvested utilising 0.05% Trysin-EDTA (Gibco, UK) and stained according to Beckman Coulter Cell Lab pan-keratin protocol (Beckman Coulter, USA). Briefly, cells were fixed in 4% formaldehyde (Sigma, UK) and subsequently permeabilised in ice-cold 90% methanol (Sigma, UK). Blocking, staining and washing were performed in 0.5% BSA in PBS. 5 × 106 cells were used per sample. Primary antibody incubations were 1 h with cytokeratin 14 (1:100), cytokeratin 6 (1:30) (both Abcam, UK), mouse or rabbit IgG isotype controls (Vector Laboratories, UK), secondary antibody incubations were 30 min with DyLight 488 or DyLight 649 (both Vector Laboratories, UK). Cells were washed after staining, resuspended in PBS and analysis was performed using the Accuri C6 flow cytometer and Flow Plus software (Accuri, UK). Secondary antibody-only and isotype antibody staining acted as controls. Cytokeratin 14/isotype control mouse with DyLight 488 was captured in the FL-1 channel and cytokeratin 6/isotype control rabbit with DyLight 649 in the FL-4 channel on the standard Accuri C6 filter settings. Gating strategy was R1 (FSC vs. SSC) and R2 (FL1 or FL4 vs. SSC) and percentage positive events after gating were compared between triplicate samples. For statistical analysis, all measurements were compared using Anova (all pair-wise comparison) with SigmaStat software (Systat, USA).
Results
Co-culture in Rheinwald & Green medium (R&G)
In the presence of autologous fibroblasts (AF), autologous keratinocytes (AK) were able to form colonies and to proliferate in culture (Fig. 1a, b). Clear separate colonies of AK were observed by 5 days after plating of autologous keratinocytes and fibroblasts in co-culture. The polygonal AK formed mosaic-like colonies with distinct cell and colony boundaries. As the AK colonies expanded, the AF occupied increasingly smaller foci that were surrounded by keratinocytes. This is comparable to culture in the presence of lethally irradiated 3T3 cells where the latter initially occupy distinct regions surrounded by the keratinocyte colonies (Fig. 1d), although at a later stage, being lethally irradiated cells, 3T3 cells become detached from the culture flask, round up and undergo cell death.
Fig. 1.

Evaluation of cell morphology of keratinocytes in co-culture. a–b AK in R&G with AF 6:1 and (c) 10:1 or (d) with lethally irradiated 3T3 cells. (e) AK with AF in EpiLife + EDGS 6:1 and (f) AK in R&G with AF 3:1. a–e AK grew as mosaic-like colonies with AF or 3T3 cells in culture.
AK for clinical use are routinely seeded at 2 × 104 cells/cm2 in culture (Navsaria et al. 1994) and have previously been reported to grow in culture in the presence of allogeneic fibroblasts when keratinocytes were seeded in excess (Sun et al. 2004). We thus tested ratios of AK to AF ranging from 10:1 to 1:1 to establish the optimum conditions for AK expansion. A ratio of 6:1 of AK to AF resulted in a confluent culture containing 90% keratinocytes within fifteen days in culture. AF were found in small foci which were completely surrounded by keratinocytes (Fig. 1a, b). Ratios lower than 6:1 and ranging down to 1:1 were not able to support keratinocyte expansion, and in such cases the phenomenon of fibroblast overgrowth was evident in the cultures (Fig. 1f). Ratios greater than 6:1, ranging up to 10:1, were able to support keratinocyte expansion but at such ratios in our hands we found that keratinocyte colonies grew significantly slower (Fig. 1c). At seeding densities lower than 2 × 104 cells/cm2, AK colonies did not expand successfully and fibroblast overgrowth occurred in these cultures at all ratios tested. Thus, based on these results, a ratio of 6:1 of AK to AF at a seeding density of 2 × 104 cells/cm2 was chosen for subsequent experiments.
Serial expansion of keratinocytes in co-culture
Clinical application of AK in burn injuries usually occurs between 2–3 weeks after hospital admission. It is therefore important that AK are expanded rapidly during the first 3 weeks in culture to ensure their availability for re-introduction to the wound bed of the patient at the clinically appropriate time. To test the suitability of our co-culture method for the serial sub-cultivation of keratinocytes over a 3 week period, we compared the cumulative population doubling (CPD) rates of keratinocytes expanded in co-culture with autologous fibroblasts with those expanded in the presence of lethally irradiated 3T3 cells. We further included the co-culture of keratinocytes with autologous fibroblasts in the commercially available medium EpiLife supplemented with EpiLife defined growth supplement (EDGS) (EpiLife + EDGS) in order to determine the suitability of this defined medium in expanding human keratinocytes in co-culture. EpiLife with supplement EDGS was able to support the growth of AK in co-culture with AF at a 6:1 ratio (Fig. 1e). Analysis of the CPD rates showed that AK expanded in co-culture with AF in R&G displayed a nearly identical cumulative population doubling rate to AK culture with lethally irradiated 3T3 cells over 25 days, the cultures being passaged once during this time period (Fig. 2). Although AK in co-culture with AF were also able to expand to P2 when grown in EpiLife + EDGS, the rate of population doubling was significantly less compared with co-cultures grown in R&G medium. This indicates that although EpiLife + EDGS can sustain AK growth in co-culture, the use of this defined medium in expanding AK to maximum numbers for clinical use is limited. Importantly, these data showed that autologous fibroblasts were equally as effective in supporting the expansion of keratinocytes as lethally irradiated 3T3 cells, indicating that our co-culture method is suitable for producing keratinocytes in large quantities, as is required for clinical application.
Fig. 2.

Cumulative population doublings of keratinocytes in different culture conditions. (filled circles ) AK in R&G with lethally irradiated 3T3 s or (circle) with AF displayed very similar rates of population doublings. (filled inverted triangle) AK in EpiLife + EDGS with AF showed a lower population doubling rate, n = 3
Phenotypic characterisation of autologous keratinocytes in co-culture
AK grown in co-culture maintained a high level of immunoreactivity for cytokeratin 14 (K14), a marker of highly proliferative basal keratinocytes. Colonies stained positive for K14 in both R&G and EpiLife + EDGS (Fig. 3a). Furthermore, AK in co-culture remained largely negative for cytokeratin 10 (K10), a marker for supra-basal differentiating keratinocytes (Fig. 3b). The distribution patterns of both markers were comparable with AK grown in co-culture with lethally irradiated 3T3 cells (Fig. 3a, b). Flow cytometric analysis confirmed these findings. AK grown in co-culture in both R&G and EpiLife + EDGS displayed high percentages of positive events for K14 and the wound healing phenotype marker cytokeratin 6 (K6), these levels being comparable to culture in the presence of lethally irradiated 3T3 cells (Fig. 4). Although the percentage of positive events was greater in co-cultures in EpiLife + EDGS compared to culture in R&G, this difference was not statistically significant (Fig. 4b, c). These important findings show that AK retained their phenotype in co-culture with AF in both R&G and EpiLife + EDGS.
Fig. 3.

Indirect immunofluorescence of keratinocytes in different culture conditions. a Cytokeratin 14 (K14) staining showed the majority of AK were of a highly proliferative, basal phenotype. b Cytokeratin 10 (K10) staining identified a small population of differentiating AK surrounded by fibroblasts, stained with vimentin (Vim). The small areas labelled positive for the differentiation marker cytokeratin 10 (K10) show a small proportion cells that are differentiatiated. These cells were stratified in culture and more round in appearance, thus it might appear that they are smaller. The larger area stained in AK:AF is a more flattened but differentiated cell
Fig. 4.

Flow cytometry of markers for proliferation and wound healing. Representative overlay histograms showed no difference in the positive staining for (a) the proliferation marker cytokeratin 14 (K14) and (c) the wound healing marker cytokeratin 6 (K6) between AK in different culture conditions. Anova all pair-wise comparison showed no statistically significant differences in the percentage of positive events of (b) K14 and (d) K6 between the different culture conditions (n = 3, p = 0.05)
Thus, we have shown that the presence of autologous fibroblasts in co-culture facilitates the expansion of autologous keratinocytes, and the latter maintain a phenotype suitable for clinical use. This co-culture method dispenses with the need to use lethally irradiated 3T3 feeder cells. Although the defined media EpiLife + EDGS supported the expansion of highly proliferative autologous keratinocytes in co-culture, the population expansion rate was too low to provide the numbers of cells that are required and necessary to successfully augment epithelial repair in patients. However, in R&G medium, the co-culture of human keratinocytes with autologous fibroblasts is a suitable method for rapidly expanding keratinocytes for clinical use.
Discussion
The use of xenobiotic material such as irradiated cells poses a potential risk of disease transmission to the patient and is therefore undesirable in clinical practice where autologous cells are being re-introduced to the patient. Many groups still rely on the use of lethally irradiated 3T3 cells for the expansion of keratinocytes as this system reliably produces highly proliferative keratinocytes. The aim of our study was to develop a culture method that replaces lethally irradiated murine 3T3 feeder cells with autologous non-irradiated fibroblasts for the culture of autologous keratinocytes for clinical application.
Our data show that AK rapidly formed colonies and proliferated in the presence of autologous fibroblasts in both serum-containing R&G medium and defined EpiLife + EDGS medium. At a ratio of 6:1 fibroblasts did not overgrow the cultures, as was observed in ratios lower than 6:1; instead AF were able to support the attachment and growth of AK in a comparable manner to culture in the presence of lethally irradiated 3T3 cells. Furthermore, in R&G medium, the cumulative population doubling rate of the 6:1 co-culture was comparable to culture with lethally irradiated 3T3 cells, and a large number of AK was produced in a clinically relevant timeframe of 2–3 weeks. However, in the defined EpiLife + EDGS medium, the cumulative population doubling rate was significantly lower, indicating that this medium is not compatible with our co-culture method when large numbers of cells are required in a limited time period. Immunostaining and flow cytometric analysis showed that there was no difference in the distribution and levels of markers of proliferation between cells grown in co-culture with autologous fibroblasts and those grown with lethally irradiated 3T3 cells. Similarly, in the timescale of the experiment, AK had not undergone differentiation, as indicated by the lack of keratinocyte differentiation markers. It has been shown that cultured epithelial autografts (CEAs) contain a high proportion of differentiated cells that are unable to further proliferate upon transplantation, with the result that CEAs do not respond well in clinical application and often fail to “take” (Cooper and Spielvogel 1994a; Atiyeh and Costagliola 2007). It is therefore very important to use proliferating keratinocytes in the treatment of cutaneous defects as these cells are able to proliferate rapidly and re-populate the wound bed to ensure prompt epithelial closure.
We have shown the successful expansion of highly proliferative human keratinocytes in the presence of autologous fibroblasts at the same rate as current clinical protocols which use lethally irradiated 3T3 cells. Dispensing with the need for xenobiotic cells is only the first step toward a protocol that is fully defined or free of materials of animal origin. Considerable efforts have been made by other groups towards developing and testing defined medium conditions (Boyce and Ham 1983; Sun et al. 2004; Coolen et al. 2007; Mujaj et al. 2010; De Corte et al. 2011). In these experiments we did not test defined medium conditions beyond the use of EpiLife with EDGS. However, future work will assess the suitability of our co-culture method in a range of defined media, in an effort to establish a protocol for clinical expansion of autologous keratinocytes that is completely devoid of animal products.
Matrix proteins secreted by fibroblasts such as fibronectin and collagen may also be important in supporting the attachment and proliferation of keratinocytes in such a co-culture system. Further work investigating the secretion of matrix proteins, their sequestration of growth factors and the interaction of the two cell types through direct cell contact would provide additional important information. Such information would allow the further optimisation of co-culture conditions and provide information on how the two cell types may interact on the healing wound bed clinically.
Furthermore, we have recently shown an additional advantage to our co-culture system. The application of a small population of fibroblasts with the keratinocytes may have significant benefits in the clinical treatment of cutaneous wounds. When cutaneous wounds close, there is a significant amount of contraction that occurs at the wound site which results in the formation of deforming scars and contractures. These can restrict movement, particularly if they occur over a joint, and are often very painful for the patient (Billingham and Medawar 1955). It is therefore desirable to reduce contraction during the healing process. In an in vivo porcine model of cutaneous wound repair, we have recently successfully shown that contraction of wounds is significantly reduced when full thickness wounds are treated with a combination of autologous keratinocytes and fibroblasts delivered to the wound bed on microcarriers, compared with wounds that were treated either with keratinocytes alone or with an ultra-thin skin graft (Martin et al. 2011). Other groups have previously reported that full thickness wounds treated with dermal substitutes seeded with autologous fibroblasts contracted less than those treated with dermal substitutes alone (Lamme et al. 2000). We, and others, have speculated that the presence of additional cells delivered to the wound bed may reduce the need for host fibroblasts of the contractile myofibroblast phenotype to migrate into the remodelling dermis, thus reducing the contractile tension on the healing wound (Lamme et al. 2000; Martin et al. 2011).
In conclusion, we describe a novel co-culture method for the expansion of autologous keratinocytes for clinical use which replaces lethally irradiated murine 3T3 feeder cells with autologous non-irradiated fibroblasts. Our method could have great clinical benefit due to the exclusion of xenobiotic cells. It may provide the additional advantage of co-application of autologous fibroblasts during treatment with autologous keratinocytes, which could significantly reduce the contraction of wounds during the healing process.
Acknowledgments
This work was funded by the Blond McIndoe Research Foundation.
References
- Atiyeh BS, Costagliola M. Cultured epithelial autograft (CEA) in burn treatment: three decades later. Burns. 2007;33:405–413. doi: 10.1016/j.burns.2006.11.002. [DOI] [PubMed] [Google Scholar]
- Billingham RE, Medawar PB. Contracture and intussusceptive growth in the healing of extensive wounds in mammalian skin. J Anat. 1955;89:114–123. [PMC free article] [PubMed] [Google Scholar]
- Boyce ST, Ham RG. Calcium-regulated differentiation of normal human epidermal keratinocytes in chemically defined clonal culture and serum-free serial culture. J Invest Dermatol. 1983;81:33s–40s. doi: 10.1111/1523-1747.ep12540422. [DOI] [PubMed] [Google Scholar]
- Boyce ST, Kagan RJ, Yakuboff KP, Meyer NA, Rieman MT, Greenhalgh DG, Warden GD. Cultured skin substitutes reduce donor skin harvesting for closure of excised, full-thickness burns. Ann Surg. 2002;235:269–279. doi: 10.1097/00000658-200202000-00016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coolen NA, Verkerk M, Reijnen L, Vlig M, Bogaerdt AJ, Breetveld M, Gibbs S, Middelkoop E, Ulrich MM. Culture of keratinocytes for transplantation without the need of feeder layer cells. Cell Transpl. 2007;16:649–661. [PubMed] [Google Scholar]
- Cooper ML, Spielvogel RL. Artificial skin for wound healing. Clin Dermatol. 1994;12:183–191. doi: 10.1016/0738-081X(94)90268-2. [DOI] [PubMed] [Google Scholar]
- Cooper ML, Spielvogel RL. Artificial skin for wound healing. Clin Dermatol. 1994;12:183–191. doi: 10.1016/0738-081X(94)90268-2. [DOI] [PubMed] [Google Scholar]
- Currie LJ, Martin R, Sharpe JR, James SE. A comparison of keratinocyte cell sprays with and without fibrin glue. Burns. 2003;29:677–685. doi: 10.1016/S0305-4179(03)00155-4. [DOI] [PubMed] [Google Scholar]
- De Corte P, Verween G, Verbeken G, Rose T, Jennes S, De Coninck A, Roseeuw D, Vanderkelen A, Kets E, Haddow D, Pirnay JP (2011) Feeder layer- and animal product-free culture of neonatal foreskin keratinocytes: improved performance, usability, quality and safety. Cell Tissue Bank [Epub ahead of print] [DOI] [PMC free article] [PubMed]
- Gustafson CJ, Kratz G. Cultured autologous keratinocytes on a cell-free dermis in the treatment of full-thickness wounds. Burns. 1999;25:331–335. doi: 10.1016/S0305-4179(99)00004-2. [DOI] [PubMed] [Google Scholar]
- Harris KL, Bainbridge NJ, Jordan NR, Sharpe JR. The effect of topical analgesics on ex vivo skin growth and human keratinocyte and fibroblast behavior. Wound Repair Regen. 2009;17:340–346. doi: 10.1111/j.1524-475X.2009.00488.x. [DOI] [PubMed] [Google Scholar]
- Heiskanen A, Satomaa T, Tiitinen S, Laitinen A, Mannelin S, Impola U, Mikkola M, Olsson C, Miller-Podraza H, Blomqvist M, Olonen A, Salo H, Lehenkari P, Tuuri T, Otonkoski T, Natunen J, Saarinen J, Laine J. N-glycolylneuraminic acid xenoantigen contamination of human embryonic and mesenchymal stem cells is substantially reversible. Stem Cells. 2007;25:197–202. doi: 10.1634/stemcells.2006-0444. [DOI] [PubMed] [Google Scholar]
- Horch RE, Bannasch H, Kopp J, Andree C, Stark GB. Single-cell suspensions of cultured human keratinocytes in fibrin-glue reconstitute the epidermis. Cell Transpl. 1998;7:309–317. doi: 10.1016/S0963-6897(98)00005-0. [DOI] [PubMed] [Google Scholar]
- James SE, Booth S, Dheansa B, Mann DJ, Reid MJ, Shevchenko RV, Gilbert PM. Sprayed cultured autologous keratinocytes used alone or in combination with meshed autografts to accelerate wound closure in difficult-to-heal burns patients. Burns. 2010;36:e10–e20. doi: 10.1016/j.burns.2008.11.011. [DOI] [PubMed] [Google Scholar]
- LaFrance ML, Armstrong DW. Novel living skin replacement biotherapy approach for wounded skin tissues. Tissue Eng. 1999;5:153–170. doi: 10.1089/ten.1999.5.153. [DOI] [PubMed] [Google Scholar]
- Lamme EN, Leeuwen RT, Brandsma K, Marle J, Middelkoop E. Higher numbers of autologous fibroblasts in an artificial dermal substitute improve tissue regeneration and modulate scar tissue formation. J Pathol. 2000;190:595–603. doi: 10.1002/(SICI)1096-9896(200004)190:5<595::AID-PATH572>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- Martin MJ, Muotri A, Gage F, Varki A. Human embryonic stem cells express an immunogenic nonhuman sialic acid. Nat Med. 2005;11:228–232. doi: 10.1038/nm1181. [DOI] [PubMed] [Google Scholar]
- Martin Y, Eldardiri M, Lawrence-Watt DJ, Sharpe JR. Microcarriers and their potential in tissue regeneration. Tissue Eng Part B Rev. 2011;17:71–80. doi: 10.1089/ten.teb.2010.0559. [DOI] [PubMed] [Google Scholar]
- Moustafa M, Bullock AJ, Creagh FM, Heller S, Jeffcoate W, Game F, Amery C, Tesfaye S, Ince Z, Haddow DB, MacNeil S. Randomized, controlled, single-blind study on use of autologous keratinocytes on a transfer dressing to treat nonhealing diabetic ulcers. Regen Med. 2007;2:887–902. doi: 10.2217/17460751.2.6.887. [DOI] [PubMed] [Google Scholar]
- Mujaj S, Manton K, Upton Z, Richards S. Serum-free primary human fibroblast and keratinocyte coculture. Tissue Eng Part A. 2010;16:1407–1420. doi: 10.1089/ten.tea.2009.0251. [DOI] [PubMed] [Google Scholar]
- Navarro FA, Stoner ML, Park CS, Huertas JC, Lee HB, Wood FM, Orgill DP. Sprayed keratinocyte suspensions accelerate epidermal coverage in a porcine microwound model. J Burn Care Rehabil. 2000;21:513–518. doi: 10.1097/00004630-200021060-00007. [DOI] [PubMed] [Google Scholar]
- Navsaria HA, Sexton C, Bouvard V, Leigh IM. Human epidermal keratinocytes. In: Leigh IM, Watt FM, editors. Keratinocyte methods. Cambridge: Cambridge University Press; 1994. pp. 5–12. [Google Scholar]
- Rheinwald JG, Green H. Serial cultivation of strains of human epidermal keratinocytes: the formation of keratinizing colonies from single cells. Cell. 1975;6:331–343. doi: 10.1016/S0092-8674(75)80001-8. [DOI] [PubMed] [Google Scholar]
- Sun T, Higham M, Layton C, Haycock J, Short R, MacNeil S. Developments in xenobiotic-free culture of human keratinocytes for clinical use. Wound Repair Regen. 2004;12:626–634. doi: 10.1111/j.1067-1927.2004.12609.x. [DOI] [PubMed] [Google Scholar]