

Abstract
D-Xylose isomerase (XI) and triosephosphate isomerase (TIM) catalyze the aldose-ketose isomerization reactions of D-xylose and D-glyceraldehyde 3-phosphate (DGAP), respectively. D-Glyceraldehyde (DGA) is the triose fragment common to the substrates for XI and TIM. The XI-catalyzed isomerization of DGA to give dihydroxyacetone (DHA) in D2O was monitored by 1H NMR spectroscopy and kcat/Km = 0.034 M−1 s−1 was determined for this isomerization at pD 7.0. This is similar to kcat/Km = 0.017 M−1 s−1 for the TIM-catalyzed carbon deprotonation reaction of DGA in D2O at pD 7.0 [Amyes, T. L.; O’Donoghue, A. C. and Richard J. P. (2001) J. Am. Chem. Soc. 123, 11325–11326]. The much larger activation barrier for XI-catalyzed isomerization of D-xylose (kcat/Km = 490 M−1 s−1) than for the TIM-catalyzed isomerization of DGAP (kcat/Km = 9.6 x 106 M−1 s−1) is due to: (i) The larger barrier to conversion of cyclic D-xylose to the reactive linear sugar (5.4 kcal/mol) than for conversion of DGAP hydrate to the free aldehyde (1.7 kcal/mol). (ii) The smaller intrinsic binding energy [Jencks, W. P. (1975) Adv. Enzymol. Relat. Areas Mol. Biol. 43, 219–410] of the terminal ethylene glycol fragment of D-xylose (9.3 kcal/mol) than of the phosphodianion group of DGAP (ca. 12 kcal/mol). The XI-catalyzed isomerization of DGA in D2O at pD 7.0 gives a 90% yield of [1-1H]-DHA and a 10% yield of [1-2H]-DHA, the product of isomerization with deuterium incorporation from solvent D2O. By comparison, the transfer of 3H from labeled hexose substrate to solvent is observed only once in every 109 turnovers for the XI-catalyzed isomerization of [2-3H]-glucose in H2O [Allen, K. N., Lavie, A., Farber, G. K., Glasfeld, A., Petsko, G. A., and Ringe, D. (1994), Biochemistry 33, 1481–1487]. We propose that truncation of the terminal ethylene glycol fragment of D-xylose to give DGA results in a large decrease in the rate of XI-catalyzed isomerization with hydride transfer compared with that for proton transfer. An ultra-high resolution (0.97 Å) X-ray crystal structure was determined for the complex obtained by soaking crystals of XI with 50 mM DGA. The triose binds to XI as the unreactive hydrate, but ligand binding induces metal cofactor movement and conformational changes in active site residues similar to those observed for XI•sugar complexes.
INTRODUCTION
Two mechanisms have evolved for the enzyme-catalyzed isomerization of α-hydroxyaldehydes (aldoses) to give α-hydroxyketones (ketoses). Isomerization of sugar phosphates, as exemplified by conversion of D-glyceraldehyde 3-phosphate (DGAP)1 to give dihydroxyacetone phosphate catalyzed by triosephosphate isomerase (TIM, Scheme 1A) (1–8), proceeds by a proton transfer mechanism through an enediolate intermediate (8, 9), while isomerization of sugars, as exemplified by reactions catalyzed by D-xylose isomerase (XI, Scheme 1B) (10–16), proceeds by intramolecular hydride transfer. By comparison, the general-base-catalyzed isomerization of triosephosphates in water proceeds by a proton transfer reaction mechanism (17, 18), while competing proton and hydride transfer reaction pathways are observed for the nonenzymatic isomerization of glyceraldehyde to give dihydroxyacetone (DHA) in alkaline aqueous solution (19, 20).
Scheme 1.

TIM satisfies at least two criteria for perfection in enzymatic catalysis (3), the most important being that the second-order rate constant kcat/Km = 2 x 108 M−1 s−1 for isomerization of the free carbonyl form of the substrate is close to the maximum possible for a diffusion-limited reaction (21).2 By comparison, kcat/Km ≈ 103 M−1 s−1 for the XI-catalyzed isomerization of D-xylose (22–24) falls far short of the limiting rate constant of ca. 108 M−1 s−1 for a diffusion-limited reaction catalyzed by a perfect enzyme (3). We are interested in understanding how this difference in catalytic efficiency is related to the difference in the structure of the substrates of the TIM- and XI-catalyzed isomerization reactions (15, 25).
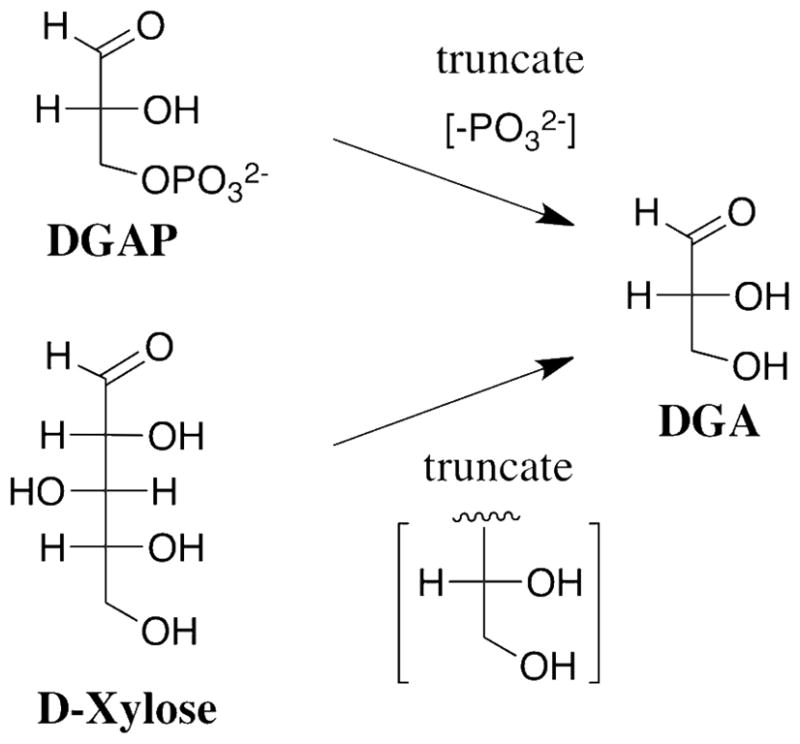
Truncation of the phosphoryl dianion from DGAP or the terminal -CH(OH)CH2OH fragment from D-xylose gives D-glyceraldehyde (DGA), the fragment common to the substrates for the two enzymes (Scheme 2). TIM catalyzes the slow isomerization of the free carbonyl form of DGA to give DHA in D2O with kcat/Km = 0.34 M−1 s−1 that is much smaller than kcat/Km = 2 x 108 M−1 s−1 for isomerization of DGAP, and only marginally larger than kB = 6.5 x 10−3 M−1 s−1 for deprotonation of DGA by the small Brønsted base 3-quinuclidinone (26).2 We report here that the observed kinetic parameter kcat/Km for the XI-catalyzed isomerization of DGA to give DHA in D2O is similar to that for the TIM-catalyzed isomerization under the same reaction conditions (26). The greater efficiency of TIM compared to XI for catalysis of the reaction of their whole substrates is therefore related to a difference in the activation of the respective enzymes by interactions with the nonreacting phosphodianion of DGAP and the terminal ethylene glycol fragment of D-xylose, respectively.
Scheme 2.
We have complemented these kinetic studies with the report of an ultra-high resolution (0.97 Å) X-ray crystal structure of the XI•triose complex obtained by soaking crystals of XI with DGA. This X-ray crystal structure shows that the more abundant (95% of total DGA) hydrate of DGA (DGAH) was bound to XI. By contrast, TIM shows selectivity for binding and catalysis of the reactive free carbonyl form of the substrate DGAP (27). The formation of the complex of XI with the unreactive hydrated triose induces movement of the metal cofactor and associated shifts in the position of active site residues that are remarkably similar to the conformational changes observed for complexes of XI with pentose and hexose sugar substrates.
MATERIALS AND METHODS
Materials
Reagent grade organic chemicals and inorganic salts from commercial sources were used without further purification. Water was distilled and then passed through a Milli-Q water purification system. Deuterium oxide (99.9% atom D), deuterium chloride (35% w/w, 99.5% atom D) and CD3COOD (99.5% atom D) were purchased from Cambridge Isotope Laboratories. D-Glyceraldehyde (90 wt % in water) was purchased from Aldrich. NADH (disodium salt), D-xylose, D-fructose, and sorbitol dehydrogenase from sheep liver (lyophilized powder) were purchased from Sigma. A crystalline suspension of D-xylose isomerase from Streptomyces rubiginosus was purchased from Hampton Research. D-Threonohydroxamic acid was prepared by a published procedure (28).
General methods
Imidazole and NaCl were dried in a vacuum oven overnight before preparation of buffered solutions in D2O. Values of pD were determined by adding 0.40 to the observed reading of the pH meter at 25 °C (29). Stock solutions (2 M) of D-xylose and D-fructose in D2O at I = 0.1 (NaCl) were kept at room temperature for one day to ensure formation of an equilibrium mixture of sugar anomers. Commercial D-glyceraldehyde (90 wt % in water) exists mainly as the dimer. This was dissolved in D2O, the D2O was removed in vacuo, and the compound was then dissolved in 0.1 M NaCl in D2O to give a ca. 50 mM solution at I = 0.1 (NaCl). This solution was kept at room temperature for ≥ 1 week, until ≥ 95% of monomeric glyceraldehyde was observed by 1H NMR analysis. 1H NMR showed the presence of only ca. 0.04% of DHA. Solutions of authentic DHA (50 mM at I = 0.1, NaCl) were prepared in D2O and were used on the day that they were prepared.
Enzymes were dialyzed against the appropriate buffer at 5 °C. Crystalline XI used in kinetic studies was freed of ammonium sulfate by extensive dialysis against 30 mM imidazole buffer in H2O (pH 7.0 or 8.0) that contained 12.5 mM MgCl2 (I = 0.1, NaCl). The protein was then dialyzed exhaustively against 30 mM imidazole buffer in D2O (pD 7.0 or 8.0) that contained 12.5 mM MgCl2 at I = 0.1 (NaCl). Typically, a 3 – 4-fold volume excess of dialysis buffer over sample was used, and the buffer was changed 7 times. The concentration of XI in stock solutions used for kinetic and product studies was determined from the absorbance at 280 nm using ε = 0.96 mg−1 ml cm−1 (30) and a molecular weight of 43,000 Dalton/monomer (23). The concentration of XI in stock solutions used for crystallization studies was determined using the Bio-Rad protein assay (Bio-Rad, Hercules, CA). Sorbitol dehydrogenase was dialyzed against 30 mM imidazole buffer in D2O (pD 7.0 or 8.0) that contained 12.5 mM MgCl2 (I = 0.1, NaCl). The enzyme was stable (< 5% loss of activity) at 5 °C for more than 6 weeks. One unit of enzyme activity is defined as the amount of enzyme that will catalyze the transformation of 1 μmole of substrate per minute under our assay conditions at 25 °C.
Enzyme assays
Enzyme assays were performed at 25 °C. The activity of sorbitol dehydrogenase in D2O was determined by monitoring the enzyme-catalyzed oxidation of NADH by D-fructose at 340 nm. The standard assay solution (1.0 mL) contained 100 mM D-fructose, 0.15 mM NADH, 10 mM MgCl2 and 0.06 units of sorbitol dehydrogenase in 24 mM imidazole buffer at pD 7.0 or 8.0 (I = 0.1, NaCl). XI was assayed in D2O by coupling the isomerization of D-xylose to the reduction of the product D-xylulose by NADH using sorbitol dehydrogenase. The standard assay solution (1.0 mL) contained 50 mM D-xylose, 0.23 mM NADH, 10 mM MgCl2, 0.012 units of XI and 0.6 units of sorbitol dehydrogenase in 24 mM imidazole buffer at pD 7.0 or pD 8.0 (I = 0.1, NaCl).
X-Ray crystallographic analysis
XI from S. rubiginosus was prepared for crystallization by overnight dialysis of the commercial crystalline suspension (Hampton Research, Inc.) against 4 L of distilled H2O followed by concentration to 20 mg/mL using an Amicon centrifugal concentrator (Millipore, Inc.) with a 10,000 MWCO membrane. XI was crystallized by the vapor diffusion method with hanging drop geometry over wells containing a solution of 0.2 – 0.3 M Mg•formate at pH 7.0. Large crystals (ca. 0.4 x 0.4 x 0.2 mm) were obtained after two days of growth at room temperature (ca. 22 °C). The structure of the complex of XI with D-glyceraldehyde was obtained by soaking crystals of unliganded XI for 9 h in a solution containing 0.4 M Mg•formate and 50 mM DGA. Crystals were transferred sequentially to solutions containing 0.4 M Mg•formate, 50 mM DGA, and 5 %, 15 %, or 30 % 2-methyl-2,4-pentanediol (MPD) and flash-cooled in a gaseous N2 stream at 100K. Diffraction data were collected at the National Synchrotron Light Source (NSLS), beamline X12B, using an ADSC Quantum 4 CCD detector. The data were integrated and scaled using DENZO and SCALEPACK (31).
Model refinement
The initial phases were derived from the 1.5 Å resolution structure of the unliganded S. rubiginosus enzyme (PDB entry 1OAD) (32) in space group P212121. The coordinates of the unliganded enzyme were used as the search model in determining the orientation of the molecule in the I222 unit cell with EPMR (33). Once the initial phases were obtained, the structure was refined to 1.5 Å resolution using CNS (34). Solvent molecules were added that showed reasonable hydrogen bond distances and geometry to donor and acceptor atoms. Care was taken to avoid modeling the density for the bound ligands as solvent. After the water structure was deemed to be satisfactory and the Rcryst reached approximately 19% (~21% Rfree), further refinement was done using SHELX-97 (35). Following isotropic refinement at full resolution, the model was refined anisotropically. Refinement of the individual anisotropic displacement parameters resulted in a 5.3% decrease in Rfree. Comparable drops in Rcryst accompanied the changes in Rfree. Iterative cycles of fitting in COOT (36) followed by refinement in SHELXL continued until the R factors converged. At this point, all protein hydrogen atoms except for amino and hydroxyl hydrogens were added using the riding model in SHELXL. This led to a ~1% decrease in the Rfree value.
1H NMR analyses
1H NMR spectra at 500 MHz were acquired at 25 °C using a Varian Unity Inova spectrometer with a 90 degree pulse angle, a 6000 Hz sweep width, a 4 – 6 s acquisition time, and a relaxation delay between pulses of 120 s, as described in our earlier work (20, 26, 37, 38). Normalized areas of the peaks corresponding to a single proton of DGA (ADGA), h-DHA (Ah-DHA) and d-DHA (Ad-DHA) in mixtures of these compounds were determined from the integrated areas of the peaks due to the C-1 proton of DGA hydrate (fhyd = 0.95) and the protons due to the free keto forms of h-DHA and of d-DHA (fhyd = 0.16), respectively, as described previously (20). The concentration of DGA was determined relative to the known concentration of the imidazole buffer from the ratio of the peak areas for the C-4 and C-5 protons of imidazole and the C-1 proton of DGA hydrate using fhyd = 0.95 for the fraction of DGA present as the hydrate in D2O (20, 26). A similar method was used to determine the concentration of D-threonohydroxamic acid using the peak area for its C-2 proton.
Reactions of DGA and DHA monitored by 1H NMR spectroscopy
The XI-catalyzed reactions of DGA and DHA in D2O at pD 7.0 or 8.0 in the presence of 24 mM imidazole buffer and 10 mM MgCl2 at 25 °C and I = 0.1 (NaCl) were monitored by 1H NMR spectroscopy. The reactions (10 mL volume) were initiated by making a 5-fold dilution of a ca. 50 mM stock solution of DGA or DHA (I = 0.1, NaCl) into 30 mM imidazole buffer (I = 0.1, NaCl) containing 12.5 mM MgCl2 to give final concentrations of 11 mM substrate, 24 mM imidazole, 10 mM MgCl2 and 0.30 mM XI for the reaction at pD 7.0 and 0.20 mM XI for the reaction at pD 8.0. At timed intervals an aliquot (1 mL) was withdrawn and the solution was adjusted to pD ≈ 5 with 10 μL or 25 μL of 1 M CD3COOD for reactions at pD 7.0 or 8.0, respectively. It was shown in control experiments that this drop in pD effectively quenches the slow XI-catalyzed reaction. The enzyme was removed by ultrafiltration at 5 °C using a Microcon YM-10 centrifugal filtration device. The filtrate was then flash frozen over solid CO2 and stored at −15 °C. 1H NMR analyses of the mixture were performed within one week of freezing. It has been shown that DGA is stable to nonenzymatic isomerization and deuterium exchange reactions over this period of time (20). The same procedure was used to monitor the XI-catalyzed isomerization of DGA in the presence of 0.5 mM of the competitive inhibitor D-threonohydroxamic acid (28).
The fraction of DGA converted to DHA, (fDHA)tot, during reaction of ≤10% of the starting DGA was calculated from the normalized peak areas due to a single proton of the reactant, ADGA, and the two products using eq 1, where Ah-DHA and Ad-DHA are the normalized peak areas for a single proton of the two products h-DHA and d-DHA, respectively, determined as described previously (see above) (20). The apparent first-order rate constants for the XI-catalyzed conversion of DGA to h-DHA and d-DHA, kobsd (s−1), were then calculated as the slopes of plots of (fDHA)tot against time according to eq 2, which were linear over the initial 10% of the reaction (see Figure 1). The ratio of the yields of h-DHA and d-DHA were obtained using eq 3, where ACH2 is the observed area of the singlet at 4.300 ppm due to the α-CH2OD groups of the free carbonyl form of both h-DHA and d-DHA, and ACHD is the area of the 0.024 ppm upfield-shifted triplet due to the single α-CHDOD group of d-DHA (see Figure 2) (20). The fraction of the total DHA product labeled with deuterium, fd-DHA/(fDHA)tot, was then calculated from this ratio using eq 4.
Figure 1.

Data for the XI-catalyzed isomerization reaction of 11 mM DGA to give DHA in D2O in the presence of 24 mM imidazole buffer and 10 mM MgCl2 at 25 °C and I = 0.1 (NaCl) monitored by 1H NMR spectroscopy. A. The time course for the fractional formation of total DHA where (fDHA)tot was determined from the area of the signals for the products h-DHA and d-DHA and the remaining substrate using eq 1: (●) Reaction catalyzed by 0.30 mM XI at pD 7.0; (■) Reaction catalyzed by 0.20 mM XI at pD 8.0. B. The change with time in the observed fractional yield of d-DHA (see eq 4): (●) Reaction catalyzed by 0.30 mM XI at pD 7.0; (▲) Reaction catalyzed by 0.27 mM XI at pD 7.0; (□) Reaction catalyzed by 0.20 mM XI at pD 8.0.
Figure 2.

Representative partial 1H NMR spectra at 500 MHz obtained during the XI-catalyzed isomerization reaction of 11 mM DGA in D2O in the presence of 24 mM imidazole buffer and 10 mM MgCl2 at 25 °C and I = 0.1 (NaCl). The large singlet at 4.300 ppm is the signal for the two α-CH2OD groups of h-DHA and the single α-CH2OD group of d-DHA. The upfield-shifted triplet at 4.276 ppm is the signal for the single α-CHDOD group of d-DHA. A. Reaction catalyzed by 0.30 mM XI at pD 7.0. The area of the triplet is 2.5 % that of the singlet. B. Reaction catalyzed by 0.20 mM XI at pD 8.0. The area of the triplet is 1.2 % that of the singlet.
| (1) |
| (2) |
| (3) |
| (4) |
The slow uncatalyzed reaction of DGA in the absence of XI in D2O at pD 7.0 in the presence of 24 mM imidazole buffer and 10 mM MgCl2 at 25 °C and I = 0.1 (NaCl) was followed directly by 1H NMR spectroscopy (26) by monitoring the decrease in the area of the doublet due to the C-1 proton of DGA hydrate, using the signals for the C-4 and C-5 hydrogens of imidazole as an internal standard (26). The observed first-order rate constant for this nonenzymatic reaction of DGA, (kN)app, was determined as the slope of a semilogarithmic plot of the fraction of remaining substrate against time.
Steady state kinetic studies
Values of Vmax and Km for the XI-catalyzed isomerization of D-xylose to give D-xylulose in D2O at pD 7.0 or 8.0 in the presence of 24 mM imidazole and 10 mM MgCl2 at 25 °C and I = 0.10 (NaCl) were determined from the nonlinear least squares fit of the initial velocity data to the Michaelis-Menten equation. The value of Ki for competitive inhibition of XI by DGA was determined from the nonlinear least squares fit of the initial velocities v to eq. 5. The quoted errors are standard deviations. The initial velocities determined in these experiments were reproducible to better than ± 5%.
| (5) |
RESULTS
Kinetic and product studies
D-xylose isomerase from Streptomyces rubiginosus was assayed in D2O by coupling the enzyme-catalyzed isomerization of D-xylose to form D-xylulose to the reduction of D-xylulose by NADH using sorbitol dehydrogenase. Table 1 reports the values of kcat and Km determined for the XI-catalyzed isomerization of D-xylose in D2O at pD 7.0 in the presence of 24 mM imidazole buffer and 10 mM MgCl2 at 25 °C and I = 0.1 (NaCl). Values of kcat = 3.3 s−1 and Km = 5.0 mM at pD 8.0 were determined for this isomerization reaction under otherwise similar conditions. By comparison, the XI-catalyzed (Streptomyces violaceoruber) isomerization of D-xylose in H2O at 35 °C exhibits a broad pH optimum at neutral pH, with kcat = 10 s−1 and Km = 2.8 mM (23).
Table 1.
Kinetic Parameters for Isomerization of D-Glyceraldehyde (DGA) and D-Xylose Catalyzed by D-Xylose Isomerase from Streptomyces rubiginosus in D2O at 25 °C.a
| Substrate | Pathway | kcat (s−1) | Km (M) | kcat/Km (M−1 s−1) |
|---|---|---|---|---|
| DGA | Total | 1.0 x 10−4 | 3 x 10−3 | 0.034 (0.035)b |
| DGA c | Hydride Transfer | 9 x 10−5 | 3 x 10−3 | 0.031 |
| DGA d | Proton Transfer | 1 x 10−5 | 3 x 10−3 | 3 x 10−3 |
| D-Xylose | Hydride Transfer | 2.4 | 4.9 x 10−3 | 490 |
At pD 7.0 in the presence of 24 mM imidazole buffer and 10 mM MgCl2 at I = 0.1 (NaCl), unless noted otherwise. Rate constants are reported for the reaction of total DGA or D-xylose.
Data for reaction at pD 8.0.
Kinetic parameters for the reaction with intramolecular transfer of hydrogen to form h-DHA, calculated from the observed kinetic parameters and the 90% fractional yield of h-DHA.
Kinetic parameters reaction with transfer of deuterium from solvent to product to form d-DHA, calculated from the observed kinetic parameters and the 10% fractional yield of d-DHA.
The inhibition of the XI-catalyzed isomerization of D-xylose by DGA at pD 7.0 was examined using six concentrations of D-xylose between 2.5 mM and 30 mM and fixed concentrations of 3.2 and 6.3 mM DGA (data not shown). The presence of DGA does not change the maximum velocity for the reaction in the presence of saturating D-xylose. This shows that inhibition by DGA is competitive. An inhibition constant for DGA of Ki = 3.0 ± 0.1 mM was determined from the nonlinear least-squares fit of the initial velocity data to eq 5. The initial velocities determined for the XI-catalyzed isomerization of 21 mM D-xylose in the presence of 5.3 mM and 10.3 mM DGA at pD 8.0 are also consistent with Ki = 3 mM for DGA which shows that there is no large change in Ki for the enzyme-catalyzed reaction on moving to the higher pD.
The nonenzymatic reaction of DGA in D2O at pD 7.0 in the presence of 24 mM imidazole buffer and 10 mM MgCl2 at 25 °C and I = 0.1 (NaCl) was monitored by 1H NMR for three weeks, during which time there was ca. 25% disappearance of the starting DGA. The major reaction products were formate and glycolaldehyde from the oxidative cleavage of DGA. There was no detectable formation of DHA from a nonenzymatic aldose-ketose isomerization reaction. These data gave (kN)app ≈ 1.4 x 10−7 s −1 as the net observed rate constant for all of the nonenzymatic reactions of DGA (Scheme 3).
Scheme 3.

The XI-catalyzed isomerization of DGA in D2O at pD 7.0 or 8.0 in the presence of 24 mM imidazole buffer and 10 mM MgCl2 at 25 °C and I = 0.1 (NaCl) was monitored by 1H NMR spectroscopy for ca. 9 h during which time there was disappearance of ca. 10% of the starting DGA. Figure 1A shows the linear initial rate time courses, with slope kobsd, for the fractional conversion of 11 mM DGA to give h-DHA and d-DHA, (fDHA)tot (eq 1), catalyzed by XI at pD 7.0 ([E] = 0.30 mM) or pD 8.0 ([E] = 0.20 mM). Values of the apparent first-order rate constants kobsd = (2.2 ± 0.1) x 10−6 s−1 and (1.5 ± 0.1) x 10−6 s−1 at pD 7.0 and 8.0, respectively, were determined from the slopes of the linear correlations in Figure 1A (eq 2), where the quoted error is the standard deviation in this slope. The values of kcat and kcat/Km (Table 1) for the XI-catalyzed isomerization of DGA in D2O (Scheme 3) were obtained from the apparent first-order rate constants using eq. 6, with the assumption that Ki = 3 mM determined for competitive inhibition by DGA of the XI-catalyzed reaction of D-xylose is equal to Km for the XI-catalyzed reaction of DGA.
| (6) |
Figure 2 shows representative partial 1H NMR spectra of in the region of the free keto form of DHA, obtained during the XI-catalyzed isomerization of DGA in D2O. The singlet at 4.300 ppm with an area ACH2 is due to the α-CH2OD groups of both h-DHA and d-DHA, and the 0.024 ppm upfield-shifted triplet with an area ACHD is due to the α-CHDOD group of d-DHA (Scheme 3). In these spectra the area ACHD is ca. 2.5% and 1.2%, respectively, that of the area ACH2 for reactions at pD 7.0 (Figure 2A) and pD 8.0 (Figure 2B). Figure 1B shows the change with time in the fraction of the product DHA that is labeled with deuterium at pD 7.0 and 8.0, determined from the peak areas ACH2 and ACHD using eqs 3 and 4. The increase in fd-DHA/(fDHA)tot with time results from the slow XI-catalyzed deuterium exchange of the methylene protons of the product h-DHA to give additional d-DHA. The initial fractions of the product DHA that contains deuterium, 0.095 and 0.05 at pD 7.0 and 8.0, respectively, were determined by making a short linear extrapolation of the data in Figure 1B to zero reaction time. Table 1 reports the values of kcat/Km for the XI-catalyzed isomerization of DGA with intramolecular hydride transfer to give h-DHA and with proton transfer with incorporation of deuterium from solvent D2O to give d-DHA, calculated from the kinetic parameters for the overall XI-catalyzed isomerization of DGA and these fractional product yields.
No products of XI-catalyzed isomerization were detected during the reaction of 11 mM DGA for 9 h in D2O at pD 7.0 in the presence of 24 mM imidazole buffer, 10 mM MgCl2, 0.27 mM XI and 0.5 mM (5,000 Ki) of the competitive inhibitor D-threonohydroxamic acid (28) at 25 °C and I = 0.1 (NaCl).
The XI-catalyzed isomerization of DHA (11 mM) in D2O at pD 7.0 in the presence of 24 mM imidazole buffer and 10 mM MgCl2 and 0.30 mM XI at 25 °C and I = 0.1 (NaCl) was monitored by 1H NMR spectroscopy for ca. 25 h during which time ca. 6% of the starting h-DHA had undergone deuterium exchange with solvent to form d-DHA. Small amounts of h-DGA and d-DGA (ca. 3% of total DHA) from thermodynamically unfavorable isomerization were also detected. No products of the enzyme-catalyzed deuterium exchange or isomerization reactions of DHA were detected by 1H NMR after a 25 h reaction time under the same conditions, but in the presence of 0.5 mM D-threonohydroxamic acid.
X-Ray crystallographic analyses
XI crystallized in space group I222 with unit cell dimensions a = 92.6, b = 98.1, and c = 102.6. XI from S. olivochromogenes has been shown to crystallize in space group P21212 with pseudo I222 symmetry (15). However, these data were not collected from crystals cooled to 100 K. The first XI structure determined from cryogenic data displayed true I222 symmetry (16). Given that attempts at refinement of the present structures in several orthorhombic space groups failed, we conclude that I222 is also the true space group for these crystals. Data collection and refinement statistics for the structures of XI crystals obtained after soaking for nine h in a solution that contained 50 mM DGA are presented in Table 2. The structure at 0.97 Å resolution (Figure 3) showed electron density that was modeled as two ligands bound at the enzyme active site: the hydrate of DGA (DGAH, 49% occupancy) and formate ion (51% occupancy). The C3 hydroxyl of DGAH interacts with the protein through a hydrogen bond to Nε of the imidazole side chain of His54 (2.6 Å) and to a bridging water molecule, with the γ-oxygen of Thr90 (2.8 Å). XI shows good catalytic activity with either Mg2+ or Mn2+ bound at the two metal sites (22). We have worked with the Mg2+ enzyme. The C2 hydroxyl is coordinated to M1 (2.1 Å). The two C1 hydroxyls of DGAH, Oa and Ob, interact with M1 (2.2 Å) and with an ordered solvent molecule. One oxygen atom of formate ion is found to bridge M1 and M2a (2.2 Å and 2.5 Å, respectively). The second oxygen is coordinated to M2a (2.3 Å) and interacts by hydrogen bonding with the ζ-ammonium ion of Lys183 (3.1Å). The water/hydroxide ion ligand of M2a is 2.0 Å from the Mg2+ atom. In this arrangement the binding of DGAH and formate appears to be mutually exclusive. A second structure (not shown) of XI soaked with DGA for a period of 15 minutes is nearly identical to the structure DGAH despite the much shorter soaking time.
Table 2.
Statistics from X-Ray Data Collection and Refinement.
| Data Collection Statistics | DGAH |
|---|---|
| Resolution (Å) (last shell)a | 20.00-0.97 (1.00-0.97) |
| Wavelength (Å) | 0.9399 |
| No. of reflections | |
| Observed | 7,151,163 |
| Unique | 272,713 |
| Completeness (%)a | 99.7 (96.7) |
| Rmerge (%)a,b | 5.5 (39.4) |
| Redundancy | 8.5 (5.4) |
| I/s(I)a | 39.5(3.4) |
|
| |
| Refinement Statistics
| |
| No. of reflections in working set | 262,140 |
| No. of reflections in test set | 13,001 |
| Rcryst (Rfree) | 0.116 (0.125) |
| No. of non-hydrogen atoms | 3,206 |
| No. of solvent atoms | 523 |
| Average B-factor (Å2) | |
| Protein atoms | 11.0 |
| DHA/Formate | 8.7/8.7 |
| Mg/Mn(II) ions | 12.6 |
| Solvent | 25.1 |
| RMS deviations | |
| Bond lengths (Å) | 0.007 |
| Bond angles (º) | 1.329 |
Data for the highest resolution shell are given in parenthesis.
Rmerge = Σhkl Σi| Ihkl, i-<Ihkl> I |/ΣhklΣi | Ihkl, i |, where <Ihkl> is the mean intensity of the multiple Ihkl, i observations for symmetry-related reflections.
Figure 3.

The structure of the XI active site complexed with DGAH. A. Shown as a stereo-view with the |Fo|-|Fc| simulated annealing omit electron density map shown as grey wireframe, the metal ions as silver spheres, and the water molecules as blue spheres. These figures were generated with the POVSCRIPT+ modification of the MOLSCRIPT program (60) and POV-Ray (61) (http://www.povray.org/). B) A diagram of the structure which shows the waters as blue spheres and the hydrogen bonds shown as dashed lines labeled with distances in Å. The atoms and corresponding distances in red indicate those with partial occupancy.
DISCUSSION
There are two pathways for the slow TIM-catalyzed reactions of the neutral truncated substrates DGA and glycolaldehyde in D2O: (1) a reaction at the enzyme active site that is shut down by binding of the competitive inhibitor 2-phosphoglycolate (PGA); and, (2) a reaction that is insensitive to PGA and/or to mutations that cripple the reaction of the natural substrate DGAP at the enzyme active site (8). This second nonspecific reaction pathway has also been documented for the reaction of glycolaldehyde catalyzed by bovine serum albumin (39). By contrast, the observed XI-catalyzed isomerization of DGA to give DHA is completely abolished by the tight-binding competitive inhibitor D-threonohydroxamic acid (28), which shows that this isomerization occurs exclusively at the enzyme active site. Similarly, the very slow XI-catalyzed deuterium exchange reaction of DHA is also completely inhibited by the binding of D-threonohydroxamic acid.
XI-catalyzed isomerization of DGA
The XI-catalyzed isomerization of [2-3H]-D-glucose in water at pH 7 proceeds with transfer of tritium from substrate to solvent about once every 109 substrate turnovers to form product (13). This is consistent with intramolecular hydride transfer in conversion of the α-hydroxy aldehyde to the α-hydroxy ketone, and a very rare competing enzyme-catalyzed proton transfer reaction. The improbable alternative proton transfer mechanism would require an extraordinary level of shielding of a putative catalytic side-chain that shuttles the proton between the two reacting carbon atoms (13). By contrast, the XI-catalyzed isomerization of DGA to give DHA in D2O reported here proceeds with incorporation of 0.10 and 0.05 mole fraction of deuterium for the reactions at pD 7.0 and 8.0, respectively (Figure 1B), so that at least 5 – 10% of the reaction of the truncated substrate proceeds by a proton transfer mechanism (Scheme 4). We also find that XI catalyzes the slow transfer of deuterium from solvent D2O into DHA to give d-DHA (kex, Scheme 4). The XI-catalyzed reactions of both DGA and DHA are abolished by the binding of the competitive inhibitor D-threonohydroxamic acid (28).
Scheme 4.
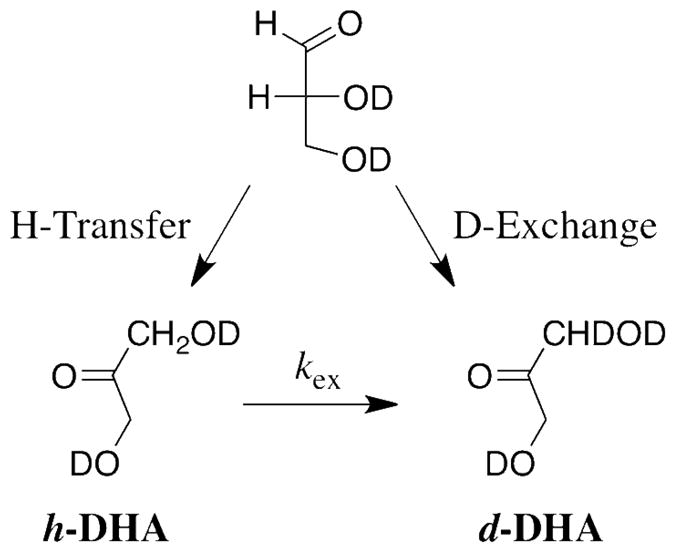
We conclude that turnover of the minimal substrate DGA by XI proceeds by the competing hydride and proton transfer pathways (Scheme 4) previously observed for the nonenzymatic isomerization of DGA in aqueous solution (19, 20). Therefore, the interaction between the terminal CH(OH)CH2OH fragment of D-xylose and XI appears to provide a specific stabilization of the transition state for enzyme-catalyzed isomerization with intramolecular hydride transfer, so that the truncation of this fragment results in a large increase in the relative rate of the reaction with proton transfer. We suggest that the very rare tritium exchange reaction of [2-3H]-D-glucose and the isomerization of DGA with transfer of deuterium from solvent to product DHA catalyzed by XI proceed by adventitious deprotonation of bound substrate by a basic residue at the enzyme active site. There is ample precedent for nonspecific protein-catalyzed deprotonation of α-carbonyl carbon with second-order rate constants similar to that reported here for XI. The value of kcat/Km = 3 x 10−3 M−1 s−1 for the XI-catalyzed isomerization of DGA by proton transfer to give d-DHA (Table 1) is 10-fold larger than kN = 3.2 x 10−4 M−1 s−1 for nonenzymatic deprotonation of DGA by the tertiary amine 3-quinuclidinone (Table 3) (26), but it is similar to kcat/Km = 0.013 and 3 x 10−3 M−1 s−1 determined for the deuterium exchange reactions of glycolaldehyde in D2O catalyzed by bovine serum albumin and the crippled K12G mutant of yeast TIM, respectively (8, 39).
Table 3.
Enzymatic Rate Accelerations for Turnover of the Whole Substrates and the Common Substrate Fragment D-Glyceraldehyde by D-Xylose Isomerase and Triosephosphate Isomerase.
| Catalyst | Substrate | Pathway | kNa (M−1 s−1) | kcat/Km (M−1 s−1) | Rate Effect b | Fragment Binding Energy |
|---|---|---|---|---|---|---|
| Zn2+ | DGA | Hydride Transfer | 1.6 x 10−5 c | |||
| Zn2+ | D-Xylose | 3.2 x 10−8 d | ||||
| XI | DGA | 0.034 e | 2100 | 9.3 kcal/mol f | ||
| XI | D-Xylose | 490 e | 1.5 x 1010 | |||
|
| ||||||
| Buffer g | DGA | Proton Transfer | 3.2 x 10−4 h | |||
| TIM | DGA | 0.017 h | 50 | 11.9 kcal/mol j | ||
| TIM | DGAP | 9.6 x 106 i | 3 x 1010 | |||
Second-order rate constants for the reference nonenzymatic isomerization reactions of total DGA or D-xylose in D2O at pD 7.0 and 25 °C.
The enzymatic rate acceleration calculated as the ratio of the second-order rate constants for the enzyme-catalyzed reaction and the reference nonenzymatic reaction catalyzed by Zn2+ or the Brønsted base 3-quinuclidinone in aqueous solution.
Calculated as kZn = kT[DO−] where kT = 950 M−2 s−1 is the third-order rate constant for the lyoxide-ion promoted Zn2+-catalyzed isomerization of total DGA and [DO−] = 1.7 x 10−8 M at pD 7.0 (Data from Ref. 20).
Calculated from kZn for isomerization of DGA with a 500-fold correction for the smaller fraction of D-xylose than of DGA present in the reactive carbonyl form. We make the assumption that the free carbonyl forms of DGA and D-xylose have similar reactivities in Zn2+-catalyzed isomerization by hydride transfer (see text).
Data at pD 7.0 from Table 1.
The intrinsic binding energy of the terminal CH(OH)CH2OH fragment of D-xylose, calculated from the ratio of the enzymatic rate accelerations for the XI-catalyzed reactions of D-xylose and DGA.
Data for nonenzymatic deprotonation of DGA by the tertiary amine base 3-quinuclidinone. We make the assumption that DGA and DGAP have similar reactivities in nonenzymatic proton transfer.
Data from Ref. 26.
Calculated from kcat = 4300 s−1 and Km = 0.45 mM for turnover of total DGAP by rabbit muscle TIM in H2O at pH 7.5 and 25 °C (Ref. 26).
The intrinsic binding energy of the phosphodianion group of DGA, calculated from the ratio of the enzymatic rate accelerations for the TIM-catalyzed reactions of DGAP and DGA.
There is a decrease in the fractional yield of d-DHA from the XI-catalyzed isomerization of DGA in D2O from 9.5% to 5% on moving from pD 7.0 to 8.0 (Figure 1B), but this increase in pD results in no significant change in the value of kcat/Km for the overall reaction that gives h-DHA as the major product (Table 1). Therefore, the rate-determining step for the overall isomerization reaction is pD-independent in this range, but the dependence on pD of the steps that determine the relative yields of h-DHA and d-DHA are different. We are unable to provide a simple explanation for these data.
DGA is a competitive inhibitor of the XI-catalyzed isomerization of D-xylose in D2O with Ki = 3.0 mM that is similar to Km = 4.9 mM for isomerization of D-xylose under the same conditions (Table 1). Therefore, the addition of an ethylene glycol fragment to DGA to give D-xylose does not change the apparent stability of the complex with XI. The observation that the addition of this fragment to DGA results in a large increase in kcat/Km for XI-catalyzed isomerization by hydride transfer from 0.034 to 490 M−1 s−1 (Table 1) shows that its interactions with XI are expressed only in the transition state and are therefore utilized to stabilize the linear sugar in the transition state relative to the cyclic sugar in the Michaelis complex.
The data in Table 3 show that at pD 7.0 TIM provides only a 50-fold rate acceleration of the reference nonenzymatic deprotonation of DGA by the small Brønsted base catalyst 3-quinuclidinone in D2O (26), while XI provides a somewhat larger 2100-fold rate acceleration of the reference nonenzymatic Zn2+-catalyzed isomerization reaction of DGA that is promoted by lyoxide ion shown in Scheme 5 (20). The similar observed catalytic activities of XI (kcat/Km = 0.034 M−1 s1) and TIM (kcat/Km = 0.017 M−1 s1) towards isomerization of the common minimal substrate DGA by hydride transfer and proton transfer, respectively (Table 3), therefore requires that the difference in the kinetic parameters for catalysis of the isomerization of the whole substrates D-xylose and DGAP, respectively, by these enzymes results from different contributions of the intrinsic binding energy of the nonreacting portions of the substrate to stabilization of the transition state (40).
Scheme 5.
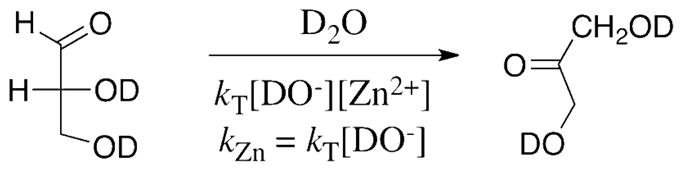
Insights from Crystallography
TIM shows a large selectivity for the binding and reaction of the reactive carbonyl form of DGAP (27). This probably reflects the snug fit of ligands at the active loop-closed enzyme (41, 42) and the exclusion of the sterically demanding hydrated substrate. By contrast, we report here that the X-ray crystal structure of the complex obtained by soaking XI in a solution that contains 50 mM total DGA shows that the ligand is bound as DGA hydrate (DGAH, Figure 3). Since XI forms an initial Michaelis complex with the cyclic form of glucose/xylose (14, 43, 44), there is no obvious requirement that the enzyme show selectivity towards binding the carbonyl form of the truncated substrate DGA. It would appear that this selectivity for the reactive carbonyl form of substrate is provided by the subsequent development of strong interactions of the enzyme with the C-4 and C-5 hydroxyls of the linear form of the full substrate D-xylose in the transition state (see above). While the binding mode observed herein for the DGAH/formate complex is strikingly similar to that of the cyclic form of glucose (PDB ID 1XIF; Figure 4a), it is a closer match to that of the linear xylulose (PDB ID 1XII; Figure 4b). Since these small fragments can adopt multiple binding orientations in the active site, this finding highlights a preference of XI for binding the linear forms of pentose/hexose sugars.
Figure 4.

Overlays of the structure of XI complexed with DGAH/formate/Mg2+ (DGHA and FMT, yellow backbone, Mg2+ as light grey spheres, H2O as cyan spheres) with those of: (A) the cyclic form of D-glucose (GLC, grey backbone, Mg2+ as dark grey spheres, H2O as yellow sphere, PDB ID 1XIF); and, (B) the linear form of D-xylulose (XUL, grey backbone, Mg2+ as dark grey spheres, H2O as yellow sphere, PDB ID 1XII). The hydrogen and coordination bonds are shown for the D-glucose and D-xylose ligands as grey dashed lines. Models were superimposed with the SSM algorithm as implemented in COOT. The Figures prepared using MOLSCRIPT (60).
The structure of XI with bound hydrated triose does not represent a productive complex due to both the presence of the hydrate form and the orientation of C1/C2 with respect to the catalytic metals (C2 hydroxyl group does not bridge M1 and M2 as expected for the hydride transfer mechanism). However, the DGAH structure does show that binding of the triose substrate can induce conformational changes in active site residues that are remarkably similar to those observed in structures with bound pentose and hexose substrates (Figure 4). Specifically, the X-ray structures demonstrate that in addition to mobility of the M2 metal center, the catalytic residues Glu181, Glu186, Asp255, and Asp257 adopt alternate conformations upon substrate binding compared to those observed in unliganded XI. This finding is also consistent with EPR spectroscopy studies on XI which show that binding of the cyclic form of substrates or substrate analogue inhibitors is sufficient to induce the shift of the metal ion in this site (45). A recent joint X-ray and neutron diffraction study shows that the catalytic metal, M2, occupies two sites after ring-opening and before isomerization (46). Thus, the metal ion movement may well be independent and not necessarily coupled to the isomerization reaction hypothesized to take place from the linear form of the sugar.
Rate acceleration and intrinsic binding energies for XI-catalyzed isomerization
The linear open form of D-xylose and the free carbonyl form of DGA are expected to show a similar intrinsic chemical reactivity in nonenzymatic Zn2+-catalyzed isomerization by hydride transfer in aqueous solution. However, the apparent rate constant for Zn2+-catalyzed isomerization of D-xylose should be ca. 500-fold smaller than the observed rate constant for isomerization of DGA at the same pD (Scheme 5). This is because the fraction of D-xylose present as the reactive linear sugar (0.01%, Scheme 6, (47)) is 500-fold smaller than the fraction of DGA present as the reactive free aldehyde (5% (26)). A 500-fold correction of the apparent second-order rate constant for nonenzymatic Zn2+-catalyzed isomerization of DGA by hydride transfer at pD 7.0, kZn = kT[DO−] = 1.6 x 10−5 M−1 s−1 (Scheme 5), gives an estimated rate constant of kZn = 3.2 x 10−8 M−1 s−1 for the Zn2+-catalyzed isomerization of D-xylose under the same conditions (Table 3). This can then be combined with kcat/Km = 490 M−1 s−1 for the XI-catalyzed isomerization of D-xylose to give an estimated enzymatic rate acceleration for isomerization of D-xylose of 1.5 x 1010-fold (Table 3).
Scheme 6.

The data for the XI-catalyzed reaction of DGA allow us to portion the 1.5 x 1010-fold rate acceleration for the XI-catalyzed isomerization of D-xylose into a smaller 2100-fold rate acceleration for reaction of the truncated substrate DGA (Table 3) and a much larger 7 x 106-fold rate acceleration that is due to specific stabilization of the transition state for isomerization by interactions between XI and the terminal ethylene glycol fragment of D-xylose. This corresponds to an apparent 9.3 kcal/mol intrinsic binding energy for this substrate fragment. By comparison, a larger ca. 12 kcal/mol intrinsic binding energy is estimated for the nonreacting substrate phosphodianion group in the TIM-catalyzed isomerization of DGAP (26, 48). The larger phosphodianion binding energy underscores the greater strength of electrostatic interactions between fully charged species, such as between the phosphodianion of DGAP and the alkylammonium side chain of Lys-12 at TIM (8, 49), compared with interactions with neutral hydrogen bond donors/acceptors.
The 9.3 kcal/mol intrinsic binding energy of the terminal ethylene glycol fragment of D-xylose is similar to that observed for other enzymatic reactions of sugars. For example, intrinsic transition state binding energies of 4 – 6 kcal/mol per sugar hydroxyl group have been reported for enzyme-catalyzed glycosyl transfer reactions (50). An unusually large 10 kcal/mol intrinsic binding energy was reported for the galactosyl C-2 hydroxyl in the breakdown of the covalent β-D-galactopyranosyl intermediate of β-galactosidase-catalyzed glycosyl transfer reactions (51).
The structure of XI determined by the time-of-flight neutron Laue method has been used to identify the location of the hydrogen atoms of enzyme-bound D-xylulose (52, 53). The neutron diffraction and X-ray crystal structures (15, 16, 25, 54) of XI show that the C-4 hydroxyl of substrate interacts with the metal ion designated as M1, and that the C-5 hydroxyl is hydrogen bonded to Nε2 of His54. The neutron diffraction structure requires a surprising perturbation in the relative pKas for the C-5 sugar hydroxyl and Nε 2 of His54 that results in formal proton transfer from the substrate oxygen to the protein imidazole. This suggests that the complex of the O-ionized sugar is stabilized by a strong, single potential minimum hydrogen bond (55, 56) to the cationic Nε 2 of His54 (53).
Ground state and transition state effects
The value of Km = 4.9 mM for the XI-catalyzed isomerization of D-xylose at pD 7.0 is not significantly different from Km ≈ Ki = 3.0 mM for DGA (Table 1). This shows that the binding interactions between XI and the terminal ethylene glycol fragment of D-xylose are not expressed in the Michaelis complex with D-xylose. Rather, these interactions develop on proceeding from the Michaelis complex between XI and the cyclic sugar to the transition state for sugar isomerization, and are expressed in the much larger value of kcat = 2.4 s−1 for D-xylose compared with kcat = 9.0 x 10−5 s−1 for XI-catalyzed isomerization of DGA by hydride transfer (Table 1).
There is a normal primary kinetic deuterium isotope effect on the XI-catalyzed isomerization of [2-2H]-D-glucose which shows that the ring-opening of glucose is a fast and reversible step that precedes rate-determining intramolecular hydride transfer (57, 58). A simple explanation for the observed substituent effects on kcat and Km is that the interactions between XI and the C-4 hydroxyl and the C-5 ring oxygen of D-xylose are minimal in the ground state Michaelis complex with the cyclic sugar, and develop only after ring opening of D-xylose to form the linear sugar. This must be the case for the hydrogen bond between the C-5 hydroxyl of the linear sugar and the cationic Nε2 of His54 (53). We propose that the interaction between the C-4 hydroxyl of D-xylose and the metal ion designated as M1 is also weak or absent at the productive complex with the cyclic sugar. If the interactions between XI and the C-4 and C-5 hydroxyl develop only upon conversion of cyclic D-xylose to the linear form, then the affinity of the linear sugar for XI will be much higher than that of the cyclic form of D-xylose. In fact, linear D-xylitol typically shows a higher affinity for XI than does D-xylose (22, 59). For example, the value of Ki = 0.026 mM for inhibition of XI-catalyzed [Streptomyces sp. (A.T.C.C. 21132)] isomerization by xylitol at pH 7.1 is more than 100-fold smaller than Km = 3.1 mM for the isomerization of D-xylose (22).
XI and TIM use different strategies to obtain an increase in kcat through the utilization of enzyme-ligand binding interactions to obtain specific transition state stabilization relative to the Michaelis complex. In the case of XI, the substrate ring-opening is a step on the pathway from the Michaelis complex to the transition state, and the new binding interactions that develop with the open linear sugar are expressed as an increase in kcat. Similarly, there is good evidence that a tightening of the electrostatic interactions between the cationic side chain of Lys-12 and the bound substrate DGAP on proceeding from the Michaelis complex (substrate dianion) to the transition state (enediolate-like trianion) result in a ca. 105-fold increase in kcat for TIM-catalyzed isomerization (8).
SUMMARY AND CONCLUSIONS
XI achieves its catalytic power through the summation of several stabilizing interactions between the protein catalyst and the substrate pieces. The enzyme provides a 2100-fold increase in the second-order rate constant for metal-ion-catalyzed isomerization by hydride transfer in D2O. The Km of 3 mM for the XI-catalyzed isomerization of DGA is consistent with a modest stabilization of the Michaelis complex. However, the X-ray crystal structure of this complex shows the triose bound as the unreactive hydrate (DGAH). If the dominant XI•DGAH complex is nonproductive, then the binding of DGA as the productive Michaelis complex must be relatively weak, and the weak binding compensated by effective protein activation of catalysis by the metal cation. The transition state for aldose-ketose isomerization of D-xylose is stabilized by 9.3 kcal/mol by interactions of XI with the C-4 and C-5 hydroxyls of substrate. This significant, but not extraordinarily large, interaction can be rationalized by an examination of the neutron diffraction (52, 53) and X-ray crystal structures (15, 16, 25, 54) of XI•sugar complexes. The larger kinetic parameters for catalysis by TIM compared with XI are due to: (i) The smaller catalytic burden borne by TIM, which must overcome only a 1.7 kcal/mol barrier to dehydration of DGAP to give the free carbonyl form, compared with XI, which must overcome a 5.4 kcal/mol barrier for ring-opening of D-xylose. (ii) The larger ca. 12 kcal/mol intrinsic binding energy for the phosphodianion fragment of DGAP, compared with the 9.3 kcal/mol binding energy of the terminal ethylene glycol fragment of D-xylose (Table 3).
The major impediment to perfection in enzymatic catalysis of isomerization of D-xylose is the evolution of an enzyme active site capable of accommodating the very different structures of the cyclic (99.99%) and linear (0.01%) forms of D-xylose (47). The higher affinity of XI for linear xylitol compared with the cyclic sugars suggests that this catalyst has evolved to optimize binding interactions with linear sugars, while the large Km of 4.9 mM for D-xylose in D2O (Table 1) shows that cyclic sugars bind relatively weakly. It is interesting that the apparent kcat/Km = 490 M−1 s−1 for XI-catalyzed isomerization of D-xylose in D2O (Table 1) and the fraction of sugar present in the linear form (10−4) are consistent with a value of kcat/Km = 4.9 x 106 M−1 s−1 for direct catalysis of the isomerization of linear D-xylose. This rate constant is not far removed from that for a diffusion-controlled reaction, such as that carried out by TIM.
Footnotes
This work was supported by Grant GM 39754 from the National Institutes of Health.
The coordinates of the refined structure have been deposited with the Protein Data Bank as entry #3U3H.
Abbreviations: DGA, D-glyceraldehyde; h-DGA, [1(R)-1H]-glyceraldehyde; d-DGA, [2(R)-2H]-glyceraldehyde; DGAH, hydrate of D-glyceraldehyde; DGAP, (R)-glyceraldehyde 3-phosphate; DHA, dihydroxyacetone; h-DHA, [1-1H]-dihydroxyacetone; d-DHA, [1-2H]-dihydroxyacetone; NADH, nicotinamide adenine dinucleotide, reduced form; NMR, nuclear magnetic resonance; PGA, 2-phosphoglycolate; TIM, triosephosphate isomerase; XI, D-xylose isomerase.
References
- 1.Knowles JR. Enzyme catalysis: not different, just better. Nature. 1991;350:121–124. doi: 10.1038/350121a0. [DOI] [PubMed] [Google Scholar]
- 2.Knowles JR. To build an enzyme. Philos Trans R Soc London, Ser B. 1991;332:115–121. doi: 10.1098/rstb.1991.0039. [DOI] [PubMed] [Google Scholar]
- 3.Knowles JR, Albery WJ. Perfection in enzyme catalysis: the energetics of triosephosphate isomerase. Acc Chem Res. 1977;10:105–111. [Google Scholar]
- 4.Malabanan MM, Amyes TL, Richard JP. A role for flexible loops in enzyme catalysis. Curr Opin Struct Biol. 2010;20:702–710. doi: 10.1016/j.sbi.2010.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rieder SV, Rose IA. Mechanism of the triose phosphate isomerase reaction. J Biol Chem. 1959;234:1007–1010. [PubMed] [Google Scholar]
- 6.Amyes TL, Richard JP. Enzymatic catalysis of proton transfer at carbon: Activation of triosephosphate isomerase by phosphite dianion. Biochemistry. 2007;46:5841–5854. doi: 10.1021/bi700409b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Go MK, Amyes TL, Richard JP. Hydron Transfer Catalyzed by Triosephosphate Isomerase. Products of the Direct and Phosphite-Activated Isomerization of [1-13C]-Glycolaldehyde in D2O. Biochemistry. 2009;48:5769–5778. doi: 10.1021/bi900636c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Go MK, Koudelka A, Amyes TL, Richard JP. Role of Lys-12 in Catalysis by Triosephosphate Isomerase: A Two-Part Substrate Approach. Biochemistry. 2010;49:5377–5389. doi: 10.1021/bi100538b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lodi PJ, Knowles JR. Neutral imidazole is the electrophile in the reaction catalyzed by triosephosphate isomerase: structural origins and catalytic implications. Biochemistry. 1991;30:6948–6956. doi: 10.1021/bi00242a020. [DOI] [PubMed] [Google Scholar]
- 10.Farber GK, Petsko GA, Ringe D. The 3.0 Å crystal structure of xylose isomerase from Streptomyces olivochromogenes. Prot Eng. 1987;1:459–466. doi: 10.1093/protein/1.6.459. [DOI] [PubMed] [Google Scholar]
- 11.Farber GK, Machin P, Almo SC, Petsko GA, Hajdu J. X-ray Laue diffraction from crystals of xylose isomerase. Proc Natl Acad Sci U S A. 1988;85:112–115. doi: 10.1073/pnas.85.1.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Glasfeld A, Farber GK, Ringe D, Marcel T, Drocourt D, Tiraby G, Petsko GA. Characterization of crystals of xylose isomerase from Streptomyces violaceoniger. J Biol Chem. 1988;263:14612–14613. [PubMed] [Google Scholar]
- 13.Allen KN, Lavie A, Farber GK, Glasfeld A, Petsko GA, Ringe D. Isotopic Exchange plus Substrate and Inhibition Kinetics of D-Xylose Isomerase Do Not Support a Proton-Transfer Mechanism. Biochemistry. 1994;33:1481–1487. doi: 10.1021/bi00172a026. [DOI] [PubMed] [Google Scholar]
- 14.Allen KN, Lavie A, Glasfeld A, Tanada TN, Gerrity DP, Carlson SC, Farber GK, Petsko GA, Ringe D. Role of the Divalent Metal Ion in Sugar Binding, Ring Opening, and Isomerization by D-Xylose Isomerase: Replacement of a Catalytic Metal by an Amino Acid. Biochemistry. 1994;33:1488–1494. doi: 10.1021/bi00172a027. [DOI] [PubMed] [Google Scholar]
- 15.Lavie A, Allen KN, Petsko GA, Ringe D. X-ray Crystallographic Structures of D-Xylose Isomerase-Substrate Complexes Position the Substrate and Provide Evidence for Metal Movement during Catalysis. Biochemistry. 1994;33:5469–5480. doi: 10.1021/bi00184a016. [DOI] [PubMed] [Google Scholar]
- 16.Fenn TD, Ringe D, Petsko GA. Xylose Isomerase in Substrate and Inhibitor Michaelis States: Atomic Resolution Studies of a Metal-Mediated Hydride Shift. Biochemistry. 2004;43:6464–6474. doi: 10.1021/bi049812o. [DOI] [PubMed] [Google Scholar]
- 17.Richard JP, Amyes TL. Proton transfer at carbon. Curr Opin Chem Biol. 2001;5:626–633. doi: 10.1016/s1367-5931(01)00258-7. [DOI] [PubMed] [Google Scholar]
- 18.Richard JP. Acid-base catalysis of the elimination and isomerization reactions of triose phosphates. J Am Chem Soc. 1984;106:4926–4936. [Google Scholar]
- 19.Nagorski RW, Richard JP. Mechanistic Imperatives for Enzymatic Catalysis of Aldose-Ketose Isomerization: Isomerization of Glyceraldehyde in Weakly Alkaline Solution Occurs with Intramolecular Transfer of a Hydride Ion. J Am Chem Soc. 1996;118:7432–7433. [Google Scholar]
- 20.Nagorski RW, Richard JP. Mechanistic Imperatives for Aldose-Ketose Isomerization in Water: Specific, General Base- and Metal Ion-Catalyzed Isomerization of Glyceraldehyde with Proton and Hydride Transfer. J Am Chem Soc. 2001;123:794–802. doi: 10.1021/ja003433a. [DOI] [PubMed] [Google Scholar]
- 21.Blacklow SC, Raines RT, Lim WA, Zamore PD, Knowles JR. Triosephosphate isomerase catalysis is diffusion controlled. Biochemistry. 1988;27:1158–1165. doi: 10.1021/bi00404a013. [DOI] [PubMed] [Google Scholar]
- 22.Van Bastelaere P, Vangrysperre W, Kersters-Hilderson H. Kinetic studies of magnesium2+ -, cobalt2+ -, and manganese2+-activated D-xylose isomerases. Biochem J. 1991;278:285–292. [PMC free article] [PubMed] [Google Scholar]
- 23.Kersters-Hilderson H, Callens M, Van Opstal O, Vangrysperre W, De Bruyne CK. Kinetic characterization of D-xylose isomerases by enzymic assays using D-sorbitol dehydrogenase. Enzyme Microb Technol. 1987;9:145–148. [Google Scholar]
- 24.Farber GK, Glasfeld A, Tiraby G, Ringe D, Petsko GA. Crystallographic studies of the mechanism of xylose isomerase. Biochemistry. 1989;28:7289–7297. doi: 10.1021/bi00444a022. [DOI] [PubMed] [Google Scholar]
- 25.Whitaker RD, Cho Y, Cha J, Carrell HL, Glusker JP, Karplus PA, Batt CA. Probing the roles of active site residues in D-xylose isomerase. J Biol Chem. 1995;270:22895–22906. doi: 10.1074/jbc.270.39.22895. [DOI] [PubMed] [Google Scholar]
- 26.Amyes TL, O’Donoghue AC, Richard JP. Contribution of phosphate intrinsic binding energy to the enzymatic rate acceleration for triosephosphate isomerase. J Am Chem Soc. 2001;123:11325–11326. doi: 10.1021/ja016754a. [DOI] [PubMed] [Google Scholar]
- 27.Trentham DR, McMurray CH, Pogson CI. The active chemical state of D-glyceraldehyde 3-phosphate in its reactions with D-glyceraldehyde 3-phosphate dehydrogenase, aldolase and triose phosphate isomerase. Biochem J. 1969;114:19–24. doi: 10.1042/bj1140019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Allen KN, Lavie A, Petsko GA, Ringe D. Design, Synthesis, and Characterization of a Potent Xylose Isomerase Inhibitor, D-Threonohydroxamic Acid, and High-Resolution X-ray Crystallographic Structure of the Enzyme-Inhibitor Complex. Biochemistry. 1995;34:3742–3749. doi: 10.1021/bi00011a032. [DOI] [PubMed] [Google Scholar]
- 29.Glasoe PK, Long FA. Use of glass electrodes to measure acidities in deuterium oxide. J Phys Chem. 1960;64:188–190. [Google Scholar]
- 30.Callens M, Kersters-Hilderson H, Vandekerckhove J, Van Opstal O, De Bruyne CK. Purification and some physicochemical properties of D-xylose isomerase from Streptomyces violaceus-ruber. Biochem Int. 1985;11:467–475. [Google Scholar]
- 31.Otwinowski Z, Minor W. Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 32.Ramagopal UA, Dauter M, Dauter Z. SAD manganese in two crystal forms of glucose isomerase. Acta Crystallogr, Sect D. 2003;D59:868–875. doi: 10.1107/s0907444903005663. [DOI] [PubMed] [Google Scholar]
- 33.Kissinger CR, Gehlhaar DK, Fogel DB. Rapid automated molecular replacement by evolutionary search. Acta Crystallogr, Sect D. 1999;D55:484–491. doi: 10.1107/s0907444998012517. [DOI] [PubMed] [Google Scholar]
- 34.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr, Sect D. 1998;D54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 35.Sheldrick GM. A short history of SHELX. Acta Crystallogr, Sect A. 2008;A64:112–122. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- 36.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr, Sect D. 2004;D60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 37.Amyes TL, Richard JP. Determination of the pKa of ethyl acetate: Brønsted correlation for deprotonation of a simple oxygen ester in aqueous solution. J Am Chem Soc. 1996;118:3129–3141. [Google Scholar]
- 38.O’Donoghue AC, Amyes TL, Richard JP. Hydron Transfer Catalyzed by Triosephosphate Isomerase. Products of Isomerization of (R)-Glyceraldehyde 3- Phosphate in D2O. Biochemistry. 2005;44:2610–2621. doi: 10.1021/bi047954c. [DOI] [PubMed] [Google Scholar]
- 39.Go MK, Malabanan MM, Amyes TL, Richard JP. Bovine Serum Albumin-Catalyzed Deprotonation of [1-13C]Glycolaldehyde: Protein Reactivity toward Deprotonation of the α-Hydroxy α-Carbonyl Carbon. Biochemistry. 2010;49:7704–7708. doi: 10.1021/bi101118g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jencks WP. Binding energy, specificity and enzymic catalysis: The Circe effect. Adv Enzymol Relat Areas Mol Biol. 1975;43:219–410. doi: 10.1002/9780470122884.ch4. [DOI] [PubMed] [Google Scholar]
- 41.Alahuhta M, Wierenga RK. Atomic Resolution crystallography of a complex of triosephosphate isomerase with a reaction intermediate analog: new insight in the proton transfer reaction mechanism. Prot: Struc, Funct & Bioinform. 2010;78:1878–1888. doi: 10.1002/prot.22701. [DOI] [PubMed] [Google Scholar]
- 42.Zhang Z, Sugio S, Komives EA, Liu KD, Knowles JR, Petsko GA, Ringe D. Crystal Structure of Recombinant Chicken Triosephosphate Isomerase-Phosphoglycolohydroxamate Complex at 1.8-Å Resolution. Biochemistry. 1994;33:2830–2837. doi: 10.1021/bi00176a012. [DOI] [PubMed] [Google Scholar]
- 43.Collyer CA, Henrick K, Blow DM. Mechanism for aldose-ketose interconversion by D-xylose isomerase involving ring opening followed by a 1,2-hydride shift. J Mol Biol. 1990;212:211–235. doi: 10.1016/0022-2836(90)90316-E. [DOI] [PubMed] [Google Scholar]
- 44.Schray KJ, Rose IA. Anomeric specificity and mechanism of two pentose isomerases. Biochemistry. 1971;10:1058–1062. doi: 10.1021/bi00782a019. [DOI] [PubMed] [Google Scholar]
- 45.Bogumil R, Kappl R, Huettermann J, Witzel H. Electron Paramagnetic Resonance of D-Xylose Isomerase: Evidence for Metal Ion Movement Induced by Binding of Cyclic Substrates and Inhibitors. Biochemistry. 1997;36:2345–2352. doi: 10.1021/bi962406n. [DOI] [PubMed] [Google Scholar]
- 46.Kovalevsky AY, Hanson L, Fisher SZ, Mustyakimov M, Mason SA, Trevor Forsyth V, Blakeley MP, Keen DA, Wagner T, Carrell HL, Katz AK, Glusker JP, Langan P. Metal Ion Roles and the Movement of Hydrogen during Reaction Catalyzed by D-Xylose Isomerase: A Joint X-Ray and Neutron Diffraction Study. Structure (Cambridge, MA, U S) 2010;18:688–699. doi: 10.1016/j.str.2010.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Drew KN, Zajicek J, Bondo G, Bose B, Serianni AS. 13C-labeled aldopentoses: detection and quantitation of cyclic and acyclic forms by heteronuclear 1D and 2D NMR spectroscopy. Carb Res. 1998;307:199–209. [Google Scholar]
- 48.Tsang WY, Amyes TL, Richard JP. A substrate in pieces: Allosteric activation of glycerol 3-phosphate dehydrogenase (NAD+) by phosphite dianion. Biochemistry. 2008;47:4575–4582. doi: 10.1021/bi8001743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Go MK, Amyes TL, Richard JP. Rescue of K12G Triosephosphate Isomerase by Ammonium Cations: The Reaction of an Enzyme in Pieces. J Am Chem Soc. 132:13525–13532. doi: 10.1021/ja106104h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Street IP, Rupitz K, Withers SG. Fluorinated and Deoxygenated Substrates as Probes of Transition-State Structure in Glycogen Phosphorylase. J Am Chem Soc. 1989;28:1581–1587. doi: 10.1021/bi00430a024. [DOI] [PubMed] [Google Scholar]
- 51.Richard JP, McCall DA, Heo CK, Toteva MM. Ground-State, Transition-State, and Metal-Cation Effects of the 2-Hydroxyl Group on β-D-Galactopyranosyl Transfer Catalyzed by β-Galactosidase (Escherichia coli, lac Z) Biochemistry. 2005;44:11872–11881. doi: 10.1021/bi050936q. [DOI] [PubMed] [Google Scholar]
- 52.Katz AK, Li X, Carrell HL, Hanson BL, Langan P, Coates L, Schoenborn BP, Glusker JP, Bunick GJ. Locating active-site hydrogen atoms in D-xylose isomerase: time-of-flight neutron diffraction. Proc Natl Acad Sci U S A. 2006;103:8342–8347. doi: 10.1073/pnas.0602598103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kovalevsky AY, Katz AK, Carrell HL, Hanson L, Mustyakimov M, Fisher SZ, Coates L, Schoenborn BP, Bunick GJ, Glusker JP, Langan P. Hydrogen Location in Stages of an Enzyme-Catalyzed Reaction: Time-of-Flight Neutron Structure of D-Xylose Isomerase with Bound D-Xylulose. Biochemistry. 2008;47:7595–7597. doi: 10.1021/bi8005434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Carrell HL, Glusker JP, Burger V, Manfre F, Tritsch D, Biellmann JF. X-ray analysis of D-xylose isomerase at 1.9 Å: native enzyme in complex with substrate and with a mechanism-designed inactivator. Proc Natl Acad Sci U S A. 1989;86:4440–4444. doi: 10.1073/pnas.86.12.4440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cleland WW, Frey PA, Gerlt JA. The Low Barrier Hydrogen Bond in Enzymatic Catalysis. J Biol Chem. 1998;273:25529–25532. doi: 10.1074/jbc.273.40.25529. [DOI] [PubMed] [Google Scholar]
- 56.Gerlt JA, MKM, Cleland WW, Frey PA. Understanding enzymic catalysis: The importance of short, strong hydrogen bonds. Chem & Biol. 1997;4:259–267. doi: 10.1016/s1074-5521(97)90069-7. [DOI] [PubMed] [Google Scholar]
- 57.Van Bastelaere PBM, Kersters-Hilderson HLM, Lambeir A-M. Wild-type and mutant D-xylose isomerase from Actinoplanes missouriensis: metal-ion dissociation constants, kinetic parameters of deuterated and non-deuterated substrates and solvent-isotope effects. Biochem J. 1995;307:135–142. doi: 10.1042/bj3070135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lee C, Bagdasarian M, Meng M, Zeikus JG. Catalytic mechanism of xylose (glucose) isomerase from Clostridium thermosulfurogenes. Characterization of the structural gene and function of active site histidine. J Biol Chem. 1990;265:19082–19090. [PubMed] [Google Scholar]
- 59.Rangarajan M, Hartley BS. Mechanism of D-fructose isomerization by Arthrobacter D-xylose isomerase. Biochem J. 1992;283:223–233. doi: 10.1042/bj2830223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kraulis PJ. MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J Appl Cryst. 1991;24:946–950. [Google Scholar]
- 61.Fenn TD, Ringe D, Petsko GA. POVScript+: a program for model and data visualization using persistence of vision ray-tracing. J Appl Cryst. 2003;36:944–947. [Google Scholar]