

Abstract
Background
Transgenic mice with cardiac restricted overexpression of tumor necrosis factor (MHCsTNF mice) develop progressive myocardial fibrosis, diastolic dysfunction and adverse cardiac remodeling. Insofar as tumor necrosis factor (TNF) does not directly stimulate fibroblast collagen synthesis, we asked whether TNF-induced fibrosis was mediated indirectly through interactions between mast cells and cardiac fibroblasts.
Methods and Results
Cardiac mast cell number increased 2–3-fold (p < 0.001) in MHCsTNF mice compared to littermate (LM) controls. Outcrossing MHCsTNF mice with mast cell deficient (c-kit−/−) mice showed that the 11-fold increase (p < 0.001) in collagen volume fraction in MHCsTNF/c-kit+/− mice was abrogated in MHCsTNF/c-kit−/− mice, and that the leftward shifted LV pressure-volume curve in the MHCsTNF/c-kit+/− mice was normalized in the MHCsTNF/c-kit−/− hearts. Furthermore, the increase in TGF-β1 and type I TGF-β receptor (TβR I) mRNA levels was significantly (p = 0.03, p = 0.01 respectively) attenuated in MHCsTNF/c-kit−/− when compared to MHCsTNF/c-kit+/− mice. Co-culture of fibroblasts with mast cells resulted in enhanced α-smooth muscle actin expression, increased proliferation and collagen mRNA expression, and increased contraction of 3-D collagen gels in MHCsTNF fibroblasts compared to LM fibroblasts. The effects of mast cells were abrogated by TβR I antagonist NP-40208.
Conclusions
These results suggest that increased mast cell density with resultant mast cell-cardiac fibroblast cross-talk is required for the development of myocardial fibrosis in inflammatory cardiomyopathy. Cardiac fibroblasts exposed to sustained inflammatory signaling exhibit an increased repertoire of pro-fibrotic phenotypic responses in response to mast cell mediators.
Keywords: Tumor necrosis factor (TNF), Transforming growth factor-beta (TGF-β), cardiac fibroblasts, mast cells, myocardial fibrosis
INTRODUCTION
The response of the heart to injury shares many of the features of wound healing that are observed in the lung, liver, kidney, and skin. Indeed, for each of these tissues, fibrosis represents a final common pathway that leads to organ dysfunction and/or failure. Myocardial fibrosis can result in excessive muscle fiber entrapment, myocyte loss, myocyte atrophy, electrical anisotropy and reentrant arrhythmias and/or abnormal diastolic and systolic stiffness of the myocardium, each of which is sufficient to contribute to the development and progression of left ventricular dysfunction.1 Although it is clear that progressive myocardial fibrosis is deleterious to the heart, and although previous studies have identified a number of molecules that are sufficient to provoke increased collagen synthesis in isolated cardiac fibroblasts in vitro and in experimental models in vivo (e.g. platelet-derived growth factor, granulocyte colony-stimulating factor, angiotensin II, aldosterone, endothelin, connective tissue growth factor (CCN2/CTGF), and transforming growth factor-β (TGF-β)),2,3 it is not at all clear from existing studies exactly how or why wound healing becomes dysregulated in the adult mammalian heart. Indeed, whereas previous studies have suggested an important role for activation of the renin angiotensin system in the development of myocardial fibrosis (reviewed in 1), recent studies from this and other laboratories have suggested an important proximal role for proinflammatory cytokines in the initiation and progression of myocardial fibrosis.4,5,6
Germane to the present discussion is the observation from several laboratories that transgenic mice with cardiac restricted overexpression of the inflammatory mediator tumor necrosis factor (TNF) develop progressive myocardial fibrosis.4–6 Given that TNF inhibits collagen gene expression and/or collagen synthesis in cardiac fibroblasts,7,8 and therefore is not directly pro-fibrotic, the mechanism(s) for the progressive fibrosis in the setting of sustained inflammatory signaling remains unknown. Here we show that increased mast cell density within the heart is required for the development of myocardial fibrosis and diastolic dysfunction in mice with sustained TNF signaling (MHCsTNF mice). Moreover, we show for the first time that cardiac fibroblasts exposed to sustained inflammatory signaling develop increased sensitivity to TGF-β and exhibit an increased repertoire of pro-fibrotic phenotypic responses in response to mast cell mediators, suggesting that alterations in cardiac fibroblast phenotype may contribute to the development of dysregulated wound healing (fibrosis) during sustained inflammation in the heart.
METHODS
Generation of Mouse Lines
MHCsTNF transgenic mice
The hemizygous line of transgenic mice with cardiac restricted overexpression of cardiac restricted TNF (referred to as MHCsTNF mice) has been described elsewhere in detail (C57BL/6 background).6,9 Briefly, these mice develop a heart failure phenotype that is characterized by progressive LV dilation and progressive myocardial fibrosis from 8–12 weeks of age. Littermates (LM) lacking the TNF transgene were used as the appropriate controls.
Mast cell deficient mice (c-kit−/−)
Genetically mast cell deficient mice KitW-sh/HNihrJaeBsmJ (referred to as c-kit−/− mice)10 were purchased from Jackson Laboratory (Bar Harbor, Maine) and outcrossed with MHCsTNF mice. The resulting F2 generation harboring the TNF transgene (c-kit+/−) were then backcrossed with homozygous c-kit−/− mice to generate the lines of mice used in these studies (F3 generation): LM/c-kit+/−, MHCsTNF/c-kit+/− and MHCsTNF/c-kit−/−. All mice were housed under standard environmental conditions and were fed commercial chow and tap water ad libitum. All studies conformed to the principles of the National Institutes of Health “Guide for the Care and Use of Laboratory Animals,” and were approved by Washington University School of Medicine in St. Louis and Baylor College of Medicine Animal Care and Use Committee.
Cell Culture
Cardiac fibroblasts
Primary cultures of mouse cardiac fibroblasts were prepared from 12 week old MHCsTNF and LM, c-kit−/− mice using a modification of previously published methods (see data supplement).11
Mast cells
The murine MC/9 mast cell line was purchased from the American Type Culture Collection (Rockville, Maryland) and cultured in mast cell growth medium: DMEM (4.5 g/L glucose) with 6mM L-glutamine, 1.5 g/L sodium bicarbonate, 0.05mM 2-mercaptoethanol, 10% Rat T-STIM (Becton Dickenson, Catalog No. 354115) and 10% fetal bovine serum.
Cardiac fibroblasts were co-cultured with mast cells under two different experimental conditions: (1) a transwell co-culture system (Costar, Corning, New York)12 and (2) 3-dimensional rat tail collagen I gels (Invitrogen GIBCO, Grand Island, N.Y.).13 For the transwell co-culture studies, the fibroblasts were separated from the mast cells (final concentration 1×106 cells/ml) by a porous polycarbonate membrane insert (0.4 μm pore size). The mast cell/fibroblast co-cultures were treated with diluent, recombinant human TGF-β1 (2ng/ml, R&D Systems, Minneapolis, MN), basic fibroblast growth factor (bFGF 25ng/ml, Sigma-Aldrich, St. Louis, MO), NP-40208 (1 μM, a gift from Scios Inc., Freemont, CA) or losartan (100 μM, Sigma-Aldrich, St. Louis, MO). The mast cell/fibroblast co-cultures were incubated for 48 hours and the fibroblasts were harvested and analyzed for alpha-smooth muscle actin (α-SMA), BrdU incorporation and collagen gene expression (see below). Mast cell conditioned medium was also used to stimulate LM and MHCsTNF fibroblasts.
For the 3-D gel contraction assays,13 cardiac fibroblasts from LM or MHCsTNF hearts were suspended in DMEM. Rat tail type I collagen was then added to a final gel concentration of 1.5mg/ml. The number of fibroblasts in the final mixture was held constant at 1 × 105 cells per milliliter. Mast cells were added to the collagen gels alone, or along with the fibroblasts at a ratio (fibroblasts: mast cells) of 1: 0, 1:1 or 1:10 (see data supplement). The co-cultures were treated with diluent, NP-40208 (1 μM), losartan (100 μM), EGTA (5mM, Sigma-Aldrich, St. Louis, MO), verapamil (50 μM, Sigma-Aldrich, St. Louis, MO), and cromolyn (100 μM, Sigma-Aldrich, St. Louis, MO). The surface area of the collagen gel was determined as a measure of the degree of gel contraction and final results were expressed as a percent of gel area at baseline.
Characterization of Isolated Cardiac Fibroblasts
To determine the mechanism(s) for the increased fibrosis in the MHCsTNF mouse hearts, we characterized “fibroblast activation” in cardiac fibroblasts isolated from 12 week old MHCsTNF and LM mouse hearts. Fibroblast activation was measured as (1) the mean fluorescence intensity (MFI) of fibroblasts with α-SMA staining, (2) fibroblast proliferation (BrdU incorporation), and (3) fibroblast production of extracellular matrix components (collagen 1A1, 1A2 and 3A1 mRNA levels).14
a-SMA expression
Flow cytometry was used to determine the degree of α-SMA staining in cardiac fibroblasts isolated from LM and MHCsTNF mouse hearts (see data supplement). Cardiac fibroblasts were treated with diluent, recombinant human TNF (200U/ml, R&D Systems, Minneapolis, MN) or TGF-β1 (2ng/ml) for an additional 48 hours, and removed from the culture dishes. Flow cytometry (FACScalibur, Becton-Dickinson) was performed using a FITC conjugate anti-mouse α-SMA and an isotype-matched IgG2a mouse FITC conjugate, as the appropriate control. The extent α-SMA staining in the fibroblast cultures was expressed as MFI.
Fibroblast proliferation
Cardiac fibroblast proliferation was determined using the colorimetric Cell Proliferation ELISA, BrdU kit (Roche, Indianapolis, IN), as described.15 The cell cultures were stimulated with TNF (200U/ml) and bFGF (25ng/ml) for additional 48 hours in serum–free media, followed by the addition of BrdU for 24 hours. All experiments were performed in triplicate.
Collagen gene expression
Cardiac fibroblasts isolated from LM, MHCsTNF and c-kit−/− hearts were treated with diluent, TNF (200U/ml) or TGF-β1 (2ng/ml) for 48 hours and collagen gene expression collagen I (collagen 1A1 and 1A2) and collagen III (collagen 3A1) mRNA levels quantified by real-time PCR in cardiac fibroblasts, as described (see data supplement).16
Cardiac Histology, Morphometry and Function
Cardiac mast cells
Toluidine Blue staining was performed to identify the presence of mast cells in the LM, LM/c-kit+/−, LM/c-kit−/− and MHCsTNF, MHCsTNF/c-kit+/−, MHCsTNF/c-kit−/− mouse hearts at 4, 8 and 12 weeks of age (see data supplement).
Myocardial fibrosis
Myocardial collagen content was determined in the hearts from LM/c-kit+/−, MHCsTNF/c-kit+/−, and MHCsTNF/c-kit−/− mice at 4, 8 and 12 weeks of age using the picrosirius red technique, as described (see data supplement).6
Cardiac hypertrophy
Cardiac mass was assessed by determining the heart-weight to body-weight ratios of LM/c-kit+/−, MHCsTNF/c-kit+/−, and MHCsTNF/c-kit−/− mice at 4, 8 and 12 weeks of age.6
Left ventricular diastolic filling
Hearts from LM, LM/c-kit+/−, c-kit−/−, MHCsTNF/c-kit +/−, and MHCsTNF/c-kit−/− were isolated and perfused in the Langendorff mode, as described (see data supplement).17
Myocardial Gene Expression
Total RNA was extracted from the hearts of LM/c-kit+/−, MHCsTNF/c-kit+/− and MHCsTNF/c-kit−/− mice using TRIzol reagent (Invitrogen, Carlsbad CA), in order to assess TGF-β signaling and collagen gene expression (see data supplement).
Statistical Analysis
All results are expressed as mean values ± SEM. Data were assessed with the use of commercially available statistical software (PASW statistics 17, SPSS Inc.). Two way analysis of variance (ANOVA) was used to evaluate mean differences in mast cell number, fibrillar collagen content; MFI of α-SMA, proliferation, gene expression and % gel contraction; where appropriate, post-hoc ANOVA testing was performed. Two way repeated measures ANOVA was used to assess mean differences in % LVedp. A p value < 0.05 was considered statistically significant.
RESULTS
Characterization of Cardiac Fibroblast Phenotype in MHCsTNF Hearts
To determine the mechanism(s) for the increased fibrosis in the MHCsTNF mouse hearts, we characterized “fibroblast activation”14 in cardiac fibroblasts isolated from 12 week old MHCsTNF and LM mouse hearts. As shown by the representative flow cytometric analysis illustrated in Figure 1A, and the group data summarized in Figure 1B, the MFI for α-SMA staining was ~ 1.2 fold (p = 0.012) greater in fibroblasts from MHCsTNF mouse hearts compared to fibroblasts from LM control. To determine whether TNF signaling was directly responsible for the increased myofibroblast differentiation, we treated MHCsTNF and LM fibroblasts with exogenous TNF (200U/ml). Interestingly, exogenous TNF had no significant (p = 0.18) effect on myofibroblast differentiation, whereas treatment with TGF-β1 (2 ng/ml) resulted in a significant increase in α-SMA intensity in both MHCsTNF and LM fibroblasts when compared to diluent treated fibroblasts. Importantly, the degree of myofibroblast differentiation was significantly greater (p = 0.002) in the TGF-β1 treated MHCsTNF fibroblasts compared to the LM fibroblasts. Figure 1C shows that fibroblast proliferation was not significantly different in the LM and MHCsTNF fibroblasts. Moreover, treatment with TNF had no effect on fibroblast proliferation in LM and MHCsTNF fibroblasts. In contrast, treatment with bFGF, which was used as a positive control, resulted in a significant (p = 0.001) increase in LM and MHCsTNF fibroblast proliferation when compared to diluent treated cells; however, there was no significant difference in BrdU incorporation between the bFGF-treated LM and MHCsTNF fibroblasts. Figures 1D–1F show that there was no increase in collagen gene expression in the LM and MHCsTNF fibroblasts at baseline, nor after stimulation with TNF. In contrast TGF-β1 stimulation significantly increased in collagen 1A1 and 1A2 mRNA levels in LM and MHCsTNF fibroblasts, but had no effect on collagen 3A1 mRNA levels. Importantly, there was no significant difference in collagen 1A1 and 1A2 gene expression between the TGF-β1 treated LM and MHCsTNF fibroblasts. Viewed together these observations suggest that sustained TNF signaling had a small effect on fibroblast activation, and raised the interesting possibility that the myocardial fibrosis observed in the MHCsTNF mice was mediated indirectly.
Figure 1.
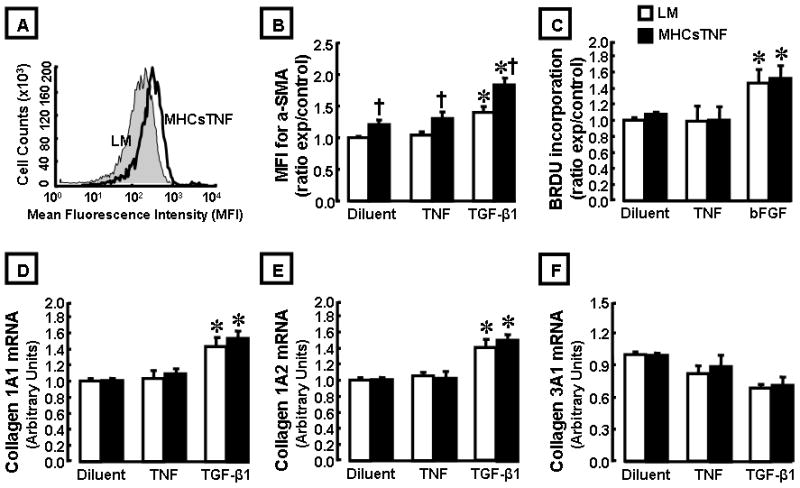
Characterization of cardiac fibroblast phenotype in MHCsTNF transgenic and littermate control (LM) mouse hearts. (A) Representative flow cytometry analysis for alpha-smooth muscle actin (α-SMA) staining in cardiac fibroblasts isolated from LM and MHCsTNF transgenic mouse hearts. (B) Group data for fold change of mean fluorescence intensity (MFI) of α-SMA staining in cardiac fibroblasts isolated from LM (n= 8) and MHCsTNF transgenic mouse hearts (n= 6) treated with diluent, TNF, and TGF-β1. (C) BrdU incorporation in fibroblasts isolated from LM (n= 8) and MHCsTNF transgenic mouse hearts (n= 6) treated with diluent, TNF, and bFGF. (D–F) Collagen 1A1, 1A2 and 3A1 gene expression in fibroblasts isolated from LM (n= 6) and MHCsTNF transgenic mouse hearts (n= 4) treated with diluent, TNF, and TGF-β1. All experiments were performed in triplicate. (* = p < 0.05 compared to diluent, † = p < 0.05 compared to LM).
Role of Mast Cells in TNF Induced Myocardial Fibrosis
Previous studies have shown that inflammation at the site of tissue injury leads to increased mast cell homing to the site of injury.18–20 To explore the possibility that mast cells accumulated in the hearts of the MHCsTNF transgenic mice, we determined mast cell number in the LM and MHCsTNF mouse hearts at 4, 8 and 12 weeks of age. Figure 2B shows there was a significant increase in mast cell number in the MHCsTNF mouse hearts at 4 (1.8-fold, p = 0.04), 8 (3.9-fold, p < 0.001), and 12 (2.4-fold, p < 0.001) weeks when compared to LM controls. As shown in Figure 2B, although mast cells were present in low numbers in the hearts of LM mice, there was no significant change in mast cell number in these hearts from 4 – 12 weeks of age. Noting previous studies which showed that mast cells accumulate in areas of ongoing tissue fibrosis,21–23 we next asked whether the increase in mast cell number observed in the MHCsTNF mouse hearts contributed to the myocardial fibrosis. To address this question we outcrossed MHCsTNF mice with mast cell deficient (c-kit−/−) mice. Crossing the MHCsTNF mice into a c-kit deficient background significantly attenuated the increase in mast cells observed in the MHCsTNF mice at 4, 8 and 12 weeks of age (Figure S-1, supplemental data). Figure 3A shows representative picrosirius red staining for myocardial collagen content in the LM/c-kit+/−, MHCsTNF/c-kit+/− and MHCsTNF/c-kit−/− hearts, whereas Figure 3B depicts the results of group data. As we reported previously, there was a progressive increase in myocardial collagen content from 4–12 weeks of age in the MHCsTNF mice compared to LM controls.6 However, the salient finding shown by Figure 3B is the myocardial collagen content was significantly (p = 0.031) attenuated by 8 weeks of age in the MHCsTNF/c-kit−/− hearts when compared to MHCsTNF/c-kit+/− hearts, and was completely abrogated by 12 weeks of age (p = 0.12) compared to LM/c-kit+/−. The difference in collagen content in the MHCsTNF/c-kit−/− mouse hearts was not secondary to a primary defect in c-kit−/− cardiac fibroblasts, insofar as the degree of α-SMA activation and BrdU incorporation were not significantly different in the c-kit−/− and LM fibroblasts at baseline, or in response to TGF-β1 and bFGF stimulation. Moreover, the c-kit−/− fibroblasts were more sensitive to TGF-β1 stimulated collagen gene expression when compared to LM controls (see data supplement Figure S-2).
Figure 2.
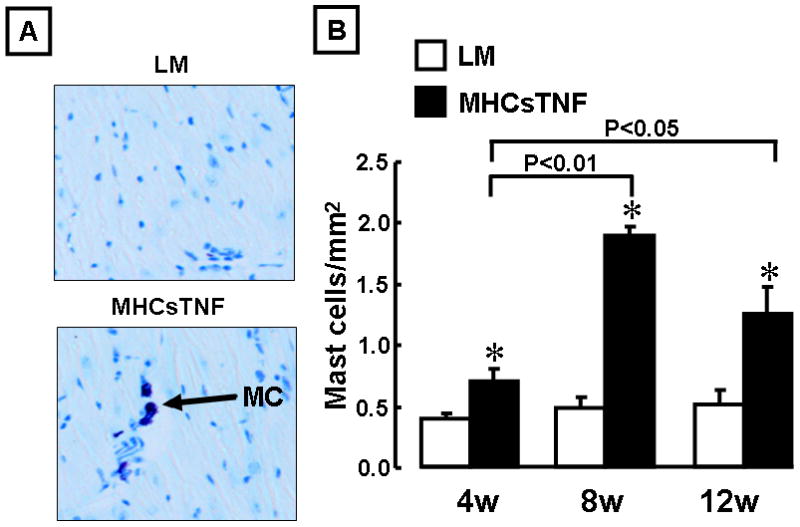
Mast cell density in littermate control (LM) and MHCsTNF transgenic mice. (A) Representative histological sections (200 × magnifications) of LM and MHCsTNF transgenic mouse hearts stained with Toluidine Blue at 12 weeks of age. Mast cells stained dark purple (indicated by arrows). (B) Results of group data for mast cell numbers in LM and MHCsTNF transgenic mouse hearts at 4, 8, and 12 weeks (n=7–9/group/time). (* = p < 0.01 compared to LM).
Figure 3.
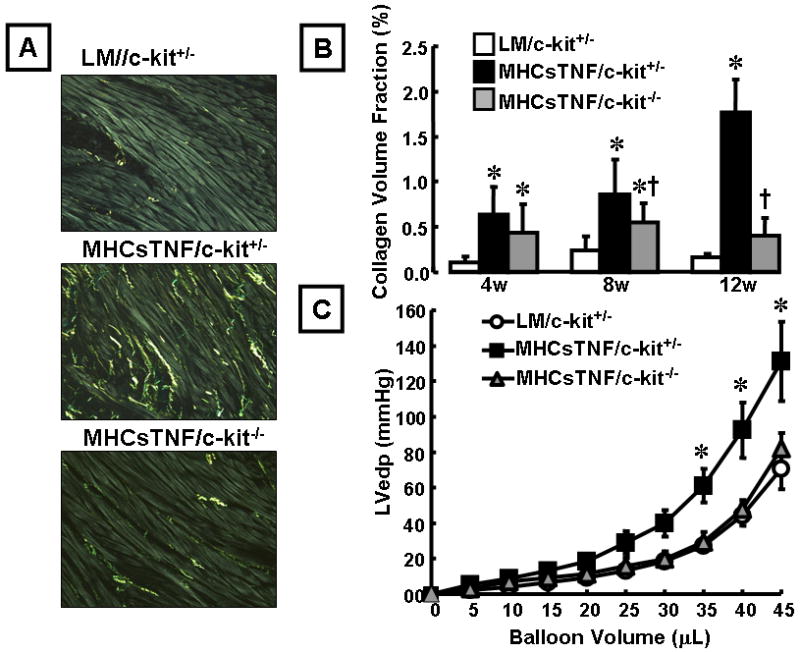
Effect of mast cells on myocardial fibrillar collagen content and LV pressure-volume relationship. (A) Representative picrosirius red staining of myocardial sections from LM/c-kit+/−, MHCsTNF/c-kit+/−, and MHCsTNF/c-kit−/− mice at 12 weeks of age. (B) Results of group data for fibrillar collagen content (expressed as percentage of myocardial area) at 4, 8 and 12 weeks of age (n = 4–7/group/time). Two way ANOVA testing revealed a significant (p < 0.001) time-dependent increase in overall myocardial overall collagen content from 4–12 weeks of age, as well as collagen content between groups (p < 0.001); There was a significant increase in fibrillar collagen content in MHCsTNF/c-kit+/− mice compared with LM/c-kit+/− mice at 4, 8, and 12 weeks of age, whereas there was a significant decrease in fibrillar collagen content in MHCsTNF/c-kit−/− mice compared with MHCsTNF/c-kit+/− mice at 8 and 12 weeks of age (Key: *= p < 0.05 compared to LM/c-kit+/− at the respective time points, †= p < 0.05 at 8 weeks of age and p < 0.001 at 12 weeks of age compared to MHCsTNF/c-kit+/− mice). (C) LV pressure-volume curves for LM/c-kit+/− (n = 8), MHCsTNF/c-kit+/− (n = 5), and MHCsTNF/c-kit−/− (n = 5) mice at 12 weeks of age. Two way repeated measures ANOVA revealed a significant difference (p = 0.004) in left ventricular end-diastolic pressure (LVedp) between MHCsTNF/c-kit+/− hearts and LM/c-kit+/− and MHCsTNF/c-kit−/− mouse hearts, whereas there was no significant difference (p = 0.58) in LVedp between LM/c-kit+/− and MHCsTNF/c-kit−/− mouse hearts. (Key: * = p < 0.05 compared to LM/c-kit+/−).
To determine the effect of mast cells on LV diastolic filling, we measured LV pressure-volume relationships in the LM/c-kit+/−, MHCsTNF/c-kit+/− and MHCsTNF/c-kit−/− mouse hearts by Langendorff buffer perfusion at 12 weeks of age. As shown in Figure 3C, there was a significant (p = 0.002) leftward shift in the LV pressure-volume curve in the MHCsTNF/c-kit+/− hearts compared to LM/c-kit+/− mouse hearts, consistent with our prior observations in this model.24 Importantly, the LV pressure-volume curve was normalized in MHCsTNF/c-kit−/− mice (p = 0.58) compared to LM/c-kit+/−, consistent with the normalization of collagen content in these mice. The rightward shift in the pressure volume curves from the MHCsTNF/c-kit−/− mouse hearts was not secondary to intrinsic differences in the LV pressure volume relationship in c-kit−/− mouse hearts, which were not different from LM control hearts (see supplemental data Figure S-3). The Table shows that the heart-weight to body-weight ratio was significantly greater (p = 0.002, p = 0.014, p = 0.002) in the MHCsTNF/c-kit+/− mice than in the LM/c-kit+/− mice at 4, 8 and 12 weeks of age. In contrast, the heart-weight to body-weight ratio in the MHCsTNF/c-kit−/− mice was not significantly different (p = 0.06) from LM/c-kit+/− mice at 12 weeks of age.
We have shown that TGF-β signaling through the type I TGF-β receptor (TβR I) contributes to the increase in fibrillar collagen content in MHCsTNF mice.6,24 Given that mast cells secrete preformed TGF-β, as well as components of the renin angiotensin system, which are capable of upregulating TGF-β signaling,25 we examined TGF-β1, TGF-β2, TβR I and type II TGF-β receptor (TβR II) mRNA expression in LM/c-kit+/−, MHCsTNF/c-kit+/− and MHCsTNF/c-kit−/− mouse hearts. Figure 4 shows that TGF-β1, TGF-β2 mRNA levels were increased significantly (p = 0.04 and p = 0.002 for TGF-β1, p = 0.02 and p < 0.001 for TGF-β2) at 8 and 12 weeks, and that TβR I and TβR II mRNA levels were increased (p < 0.001 for TβR I, p = 0.041 for TβR II) at 12 weeks of age in the MHCsTNF/c-kit+/− hearts when compared to LM/c-kit+/−. Importantly, both TGF-β1 and TβRI mRNA were attenuated significantly (p = 0.03, p = 0.01) at 12 weeks of age in the MHCsTNF/c-kit−/− hearts when compared to the MHCsTNF/c-kit+/− mice, suggesting a previously unanticipated role for mast cells in modulating TGF-β expression in this inflammatory cardiomyopathy model.6 Similarly, immunohistochemistry showed that TGF-β protein levels were decreased in the MHCsTNF/c-kit−/− mice when compared to MHCsTNF/c-kit+/− mice (see data supplement Figure S-4).
Figure 4.
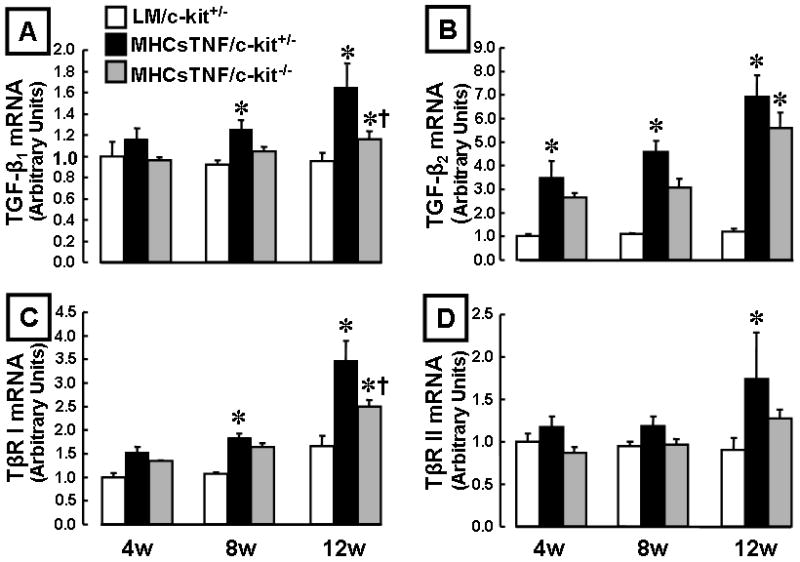
Myocardial TGF-β1, TGF-β2, TβR I, and TβR II mRNA expression in LM/c-kit+/−, MHCsTNF/c-kit+/−, and MHCsTNF/c-kit−/− mice at 4, 8 and 12 weeks (n=3–6/group/time) of age. (A) TGF-β1, (B) TGF-β2, (C) TβR I and (D) TβR II mRNA levels. Two-way ANOVA revealed a significant difference in TGF-β1 (p = 0.04), TGF-β2 (p = 0.01) and TβR I (p = 0.001) mRNA levels within groups from 4–12 weeks of age, as well as in LM/c-kit+/−, MHCsTNF/c-kit+/−, and MHCsTNF/c-kit−/− mice (p = 0.018, p < 0.001 and p < 0.001, respectively); there were significant differences in mRNA levels in LM/c-kit+/−, MHCsTNF/c-kit+/−, and MHCsTNF/c-kit−/− mice at 4, 8 and 12 weeks of age. (* = p < 0.05 compared to LM/c-kit+/−, †p < 0.05 compared to MHCsTNF/c-kit+/−).
Mast Cell – Fibroblast Interactions
The above studies suggested an important role for mast cells in the development and progression of myocardial fibrosis in the MHCsTNF mice. Accordingly, we next asked whether there were interactions between mast cells and cardiac fibroblasts, as well as whether there were qualitative and/or quantitative differences in the degree of interaction in cardiac fibroblasts exposed to sustained TNF signaling. Figure 5A shows two important findings. First, co-culture of mast cells with cardiac fibroblasts resulted in a significant (p < 0.001) increase in α-SMA staining in LM and MHCsTNF fibroblasts when compared to diluents treated fibroblasts; however, the degree was significantly greater (p < 0.001) in MHCsTNF fibroblasts. Moreover, these effects were mimicked by conditioned media from mast cells treated MHCsTNF fibroblasts. Second, treatment with the TβR I antagonist NP-40208 significantly (p = 0.02) attenuated the mast cell-induced the increase of α-SMA staining in fibroblasts isolated from MHCsTNF hearts, suggesting that the mast cell induced increase in myofibroblast activation was secondary to TGF-β signaling. Given that angiotensin II induces the expression of TGF-β1,26 and given that mast cells are capable of local regulation of the renin angiotensin system in the heart,27 we treated the mast cell/cardiac fibroblasts co-cultures with losartan, an angiotensin type 1 receptor (AT1) antagonist. Figure 5A shows that there was no significant (p = 0.13) difference in mast cell-induced α-SMA staining in MHCsTNF fibroblasts treated with losartan, whereas there was a significant decrease in α-SMA staining in MHCsTNF fibroblasts treated with losartan + NP-40208. Figure 5B shows that co-culturing mast cells with cardiac fibroblasts resulted in a significant (p < 0.001) increase in BrdU incorporation in LM and MHCsTNF fibroblasts when compared to diluent treated fibroblasts; however, the degree of BrdU incorporation was significantly greater (p < 0.001) in MHCsTNF fibroblasts. Similar findings were obtained using mast cell conditioned media. Treatment with NP-40208 significantly (p = 0.039) attenuated the mast cell-induced increase in BrdU incorporation in MHCsTNF fibroblasts, and completely abrogated the mast cell-induced increase in BrdU incorporation in LM fibroblasts. In contrast there was no significant difference (p = 0.11) in BrdU incorporation in the losartan + NP-40208 treated mast cell co-cultured fibroblasts from LM mouse hearts, whereas there was a significant (p = 0.048) difference in BrdU incorporation in the mast cell co-cultured fibroblasts from MHCsTNF mouse hearts treated with losartan + NP-40208, when compared to NP-40208 treated cells.
Figure 5.
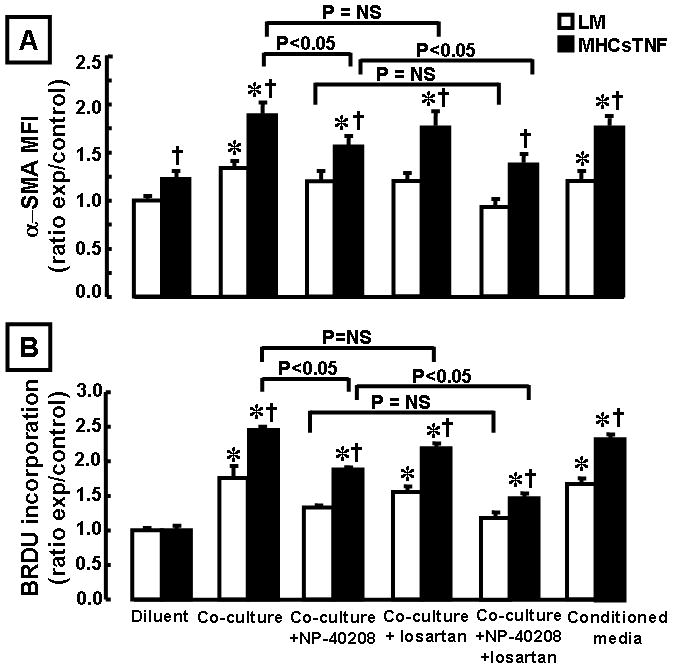
Littermate control (LM) and MHCsTNF fibroblast activation and proliferation in the presence and absence of mast cells, or mast cell conditioned media. LM and MHCsTNF cardiac fibroblasts (n = 4 dishes or wells per group/culture condition) were cultured in the presence of diluent or mast cells using a transwell co-culture system, and the mast cell/fibroblast co-cultures were treated with NP-40208 (1 μM), losartan (100 μM), NP-40208 + losartan. (A) fibroblast activation (α-SMA staining) and (B) fibroblast proliferation (BrdU incorporation) were determined (see methods for details). Two way ANOVA revealed a significant difference in MHCsTNF fibroblast activation (p < 0.001) and proliferation (p < 0.001) compared to LM fibroblasts; α-SMA staining and BrdU incorporation were significantly greater in MHCsTNF diluent treated and mast cell co-cultured fibroblasts when compared to the respective LM fibroblasts, and that these differences were sensitive to inhibition with NP-40208, but not to losartan. The increase in BrdU incorporation in mast cell co-cultured LM fibroblasts was also sensitive to inhibition with NP-40208, but not to losartan. α-SMA staining and BrdU incorporation were significantly decreased in MHCsTNF fibroblasts treated with NP-40208 + losartan when compared to NP-40208 treated alone. Similar findings were obtained using mast cell conditioned media. All experiments were performed in triplicate. (*= p < 0.05 compared to the respective diluent treated LM and MHCsTNF fibroblasts, †p < 0.05 compared to the LM mast cell co-cultured fibroblasts).
Insofar as our in vivo studies suggested that myocardial mast cells contributed to the increased levels of TGF-β and TβR mRNA (Figure 4) in the MHCsTNF mice, we asked whether co-culturing mast cells with cardiac fibroblasts was sufficient to increase TGF-β and TβR mRNA in vitro. As shown in the data supplement (Figure S-5) there was no significant difference in the mRNA expression levels for TGF-β1, TGF-β2, TβR I and TβR II mRNA in diluent treated LM and MHCsTNF fibroblasts. However, co-culturing mast cells with fibroblasts resulted in a significant increase in expression of TGF-β1 and TβR I mRNA in both LM and MHCsTNF fibroblasts; moreover, TGF-β1 and TβR I mRNA levels were significantly (p = 0.02, p =0. 045) greater in the MHCsTNF fibroblasts than in the LM fibroblasts. In contrast, co-culturing mast cells with cardiac fibroblasts had no effect on TGF-β2 and TβR II mRNA levels in either MHCsTNF or LM fibroblasts. Taken together, these results suggest that paracrine signaling between cardiac fibroblasts and mast cells provokes fibroblast activation in both LM and MHCsTNF fibroblasts, at least in part through a TGF-β mediated signaling pathway, but that MHCsTNF fibroblasts are qualitatively and quantitatively more sensitive to the paracrine effects of mast cells.
To determine whether mast cells directly influenced collagen gene expression in isolated cardiac fibroblasts, we examined collagen gene expression in the mast cell/cardiac fibroblast co-culture system. In control experiments we established that Ang II (1 μM) was sufficient to increase collagen 1A1, 1A2, and 3A1 mRNA expression in LM and MHCsTNF fibroblasts and that the concentration of losartan used was sufficient to block collagen gene expression (data not shown). Figure 6 shows that co-culturing fibroblasts with mast cells or mast cell conditioned media resulted in significant increases in collagen 1A1, 1A2, and 3A1 (p = 0.02, p = 0.03, p = 0.02) mRNA in LM fibroblasts (compared to diluent), and increases in collagen 1A1 (p < 0.001), 1A2 (p = 0.004), and collagen 3A1 (p = 0.001) mRNA levels in MHCsTNF fibroblasts. Moreover, the mast cell induced increase in collagen 1A1, 1A2, and 3A1 mRNA expression was significantly greater (p = 0.02, p = 0.03, p = 0.02) for MHCsTNF fibroblasts when compared to LM fibroblasts. Treatment with NP-40208 abrogated the mast cell induced increased collagen gene expression in both LM and MHCsTNF fibroblasts (p = 0.56 – 0.82 compared to diluent). Treatment with losartan also abrogated the mast cell induced increase in collagen 1A1 gene expression in both LM (p = 0.09) and MHCsTNF (p = 0.07) co-cultured fibroblasts when compared to diluent; however, the mast cell induced increase in collagen 1A2 and 3A1 gene expression was not sensitive to losartan in both LM and MHCsTNF fibroblasts. Treatment with losartan + NP-40208 resulted in a significant decrease in collagen 1A1 gene expression compared to NP-40208 alone in LM (p = 0.04) and MHCsTNF (p = 0.04) fibroblasts.
Figure 6.
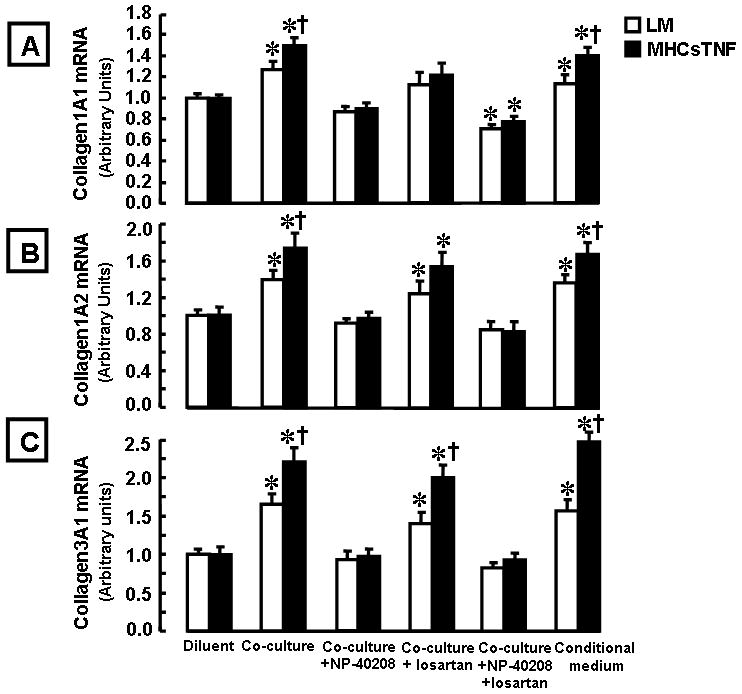
Collagen mRNA expression in littermate control (LM) and MHCsTNF cardiac fibroblasts in the presence and absence of mast cells, or mast cell conditioned media. LM and MHCsTNF cardiac fibroblasts were cultured in the presence of diluent or mast cells using a transwell co-culture system (n = 3 cultures per group/culture condition) and collagen 1A1, 1A2 and 3A1 mRNA levels were determined by real-time PCR (see methods for details). The mast cell/fibroblast co-cultures were treated with NP-40208 (1 μM), losartan (100 μM), NP-40208 + losartan. (A) mRNA expression for collagen 1A1, (B) mRNA expression for collagen 1A2, and (C) mRNA expression for collagen 3A1. All experiments were performed in triplicate. (*= p < 0.05 compared to the respective diluent treated LM and MHCsTNF fibroblasts, †p < 0.05 compared to the respective LM fibroblasts co-cultured with mast cells).
Effect of Mast Cells on 3-D Collagen Gel Contraction
To determine whether cross talk between mast cells and cardiac fibroblasts had an effect on collagen remodeling, we co-seeded mast cells and fibroblasts in a 3-D rat tail collagen I gel.13,28 Figure 7A depicts representative collagen gels seeded with mast cells alone, LM and MHCsTNF fibroblasts alone, collagen gels seeded with a 1:1 or 1:10 ratio of fibroblasts to mast cells. The fibroblast cell number was constant across all experiments. As shown by the group data depicted in Figure 7B, mast cells alone had no effect on contraction of the 3-D collagen gels. However, co-culturing mast cells with cardiac fibroblasts resulted in a mast cell dependent contraction of the 3-D gel (p < 0.001), with increasing gel contraction in relation to increasing concentration of mast cells in the gel.
Figure 7.
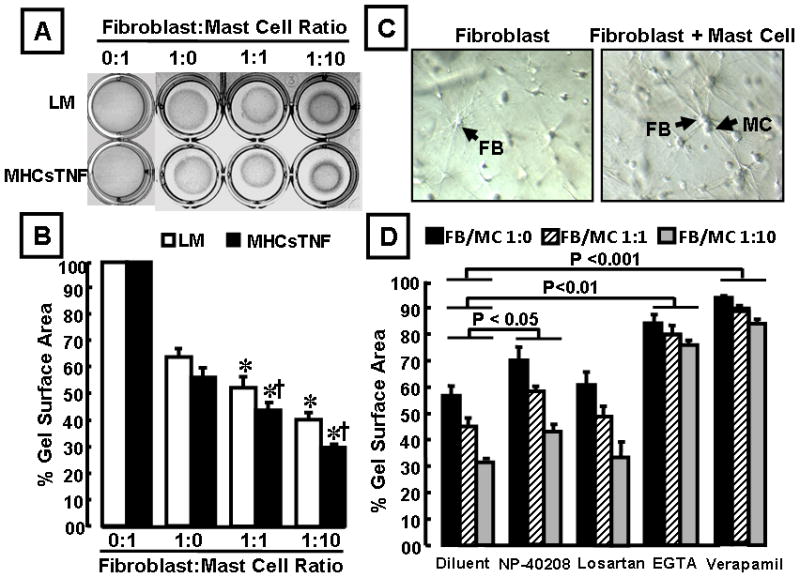
Effect of mast cells on 3-dimensional collagen gel contraction by cardiac fibroblasts. (A) representative collagen gels with mast cells alone, LM and MHCsTNF fibroblasts alone, a 1:1 and a 1:10 ratio of fibroblasts to mast cells (* = p < 0.05 compared to diluent treated LM and MHCsTNF fibroblasts, † p < 0.05 compared to respective LM fibroblasts). (B) Results of group data for 3-D collagen gel contraction in 0:1, 1:0, 1:1, and 1:10 fibroblast: mast cell ratio (n = 6 cultures per group/culture condition). Two-way ANOVA showed significant differences in gel contraction within and between groups (p < 0.001). (C) Representative morphology of MHCsTNF fibroblasts (indicated by arrows) in the absence and presence of mast cells (x200 magnification). (D) Effect of NP-40208 (1 μM), losartan (100 μM), EGTA (5mM) and verapamil (50 μM) on 3-D collagen gel contraction in MHCsTNF cardiac fibroblasts. (n = 6 cultures per culture condition). Two-way ANOVA testing revealed significant differences in gel contraction between groups for effect of NP-40208, EGTA and verapamil (p = 0.011, p = 0.002, and p < 0.001, respectively), but not to losartan. All experiments were performed in triplicate.
However, the important finding shown by Figure 7B is that mast cell induced gel-contraction was significantly (p < 0.001) greater for fibroblasts from the MHCsTNF transgenic mice than for fibroblasts from LM hearts. Interestingly, when LM and MHCsTNF fibroblasts were cultured in the 3-D collagen gels alone they demonstrated slender filopodia. However, when fibroblasts were co-cultured in the gel together with mast cells their filopodia became more prominent, consistent with the appearance of myofibroblasts, which may provide a mechanistic explanation for the increased gel contraction observed in the co-culture system (Figure 7C). As shown in Figure 7D, NP-40208 had a small but significant (p = 0.01) effect in terms of attenuating the degree of the gel contraction in the MHCsTNF fibroblasts when cultured alone or when seeded with mast cells. In contrast, losartan had no effect on the collagen gel contraction in the MHCsTNF fibroblasts when cultured alone or when seeded with mast cells. Interestingly, mast cell induced collagen gel contraction was significantly attenuated by EGTA (p = 0.002), and was almost completely abolished by verapamil (p < 0.001), suggesting that the observed increase in mast cell-induced gel contraction was calcium-dependent. Similar overall results were obtained in LM fibroblasts (Figure S-6 in the data supplement). The mast cell stabilizer cromolyn sodium had no effect on mast cell induced gel contraction (Table, data supplement), suggesting that the observed effects of mast cells on gel contraction were not histamine dependent.
DISCUSSION
The results of this study, in which we examined the mechanisms of myocardial fibrosis in a transgenic mouse model of sustained TNF signaling, suggest that increased mast cell density with resultant mast cell-cardiac fibroblast cross-talk is required for the development of myocardial fibrosis. Here we also show for the first time that cardiac fibroblasts exposed to sustained inflammatory signaling exhibit an increased repertoire of pro-fibrotic phenotypic responses in response to mast cell mediators. The following lines of evidence support these statements. First, we observed that the number of cardiac mast cells increased significantly in the hearts of MHCsTNF mice at 8–12 weeks of age (Figure 2A), which coincides with the development of myocardial fibrosis in this mouse model.6 Second, breeding the MHCsTNF mice onto a mast cell deficient background significantly attenuated the myocardial fibrillar collagen content and the progressive time-dependent upregulation of TGF-β1, β2, and TβR I, II mRNA expression in MHCsTNF mice (Figures 3 and 4).6 The changes in myocardial collagen content were functionally significant insofar as the leftward shift of the LV pressure-volume curve was abolished in MHCsTNF mice that were bred onto a mast cell deficient background (Figure 3B). Moreover, these results are internally consistent with our previous observation that the progressive myocardial fibrosis and leftward shift of the LV pressure-volume curve in this model was sensitive to inhibition with a TβR I antagonist.24 Third, the co-culture experiments suggest that mast cells provoked increased fibroblast activation in MHCsTNF fibroblasts, as well as increased type I (collagen 1A1) and type III (collagen 3A1) gene expression and fibroblast proliferation in LM and MHCsTNF fibroblasts (Figures 5 and 6). The mast cell-induced changes in LM and MHCsTNF fibroblasts were sensitive to inhibition with the TβR I antagonist NP-40208. Nonetheless, we cannot exclude a potentially important role other for profibrotic mast cell mediators (e.g. tryptase, chymase). Although not definitive, our results suggest that one mechanism for the sensitivity to TGF-β signaling in the LM and MHCsTNF fibroblasts may relate to increased TβR I expression (Figure S-4) in the fibroblasts that were co-cultured with mast cells. In contrast, the mast cell-induced changes in the fibroblasts were only partially sensitive to inhibition of the angiotensin type 1 receptor with losartan (Figure 5), suggesting that the pro-fibrotic effects of mast cells are predominately mediated through the effects of TGF-β on cardiac fibroblasts.
The mast cell-induced changes in LM and MHCsTNF fibroblasts in vitro were functionally significant, insofar as the degree of 3-D collagen gel contraction was directly correlated with an increased ratio of mast cells to fibroblasts in the gel (Figure 7B). Moreover, the degree of gel contraction in the co-cultures was greater for the MHCsTNF fibroblasts than for LM fibroblasts, consistent with the increase in α-SMA expression in the MHCsTNF fibroblasts. Further evidence that collagen gel contraction was mediated by α-SMA in the fibroblasts is supported by the observation that collagen gel contraction was completely abrogated by treating the cultures with EGTA or verapamil (Figure 7D and S-6), which would have decreased the amount of calcium available for α-actin induced fibroblast contraction. Consistent with the findings discussed above regarding the effects of mast cell released TGF-β, the mast cell-induced increase in 3-D collagen gel contraction was sensitive to inhibition with NP-40208, whereas losartan had no effect. One limitation of these studies is that we did not address the mechanism for the increased mast cell density in the MHCsTNF mice. Mast cells arise from hematopoietic progenitor cells, but do not circulate in the blood; instead they acquire their mature phenotype locally in the tissues where they reside.29 Insofar as intravenous transfer of bone marrow-derived cultured mast cells does not result in engraftment of mast cells into the heart,30 it was not possible to perform mast cell “knock-in” experiments in the MHCsTNF/c-kit−/− mice to determine whether increased mast cell homing or increased mast cell proliferation within the heart was responsible for the increased mast cell density in the MHCsTNF mice.
Role of Mast Cells in the Failing Heart
Although mast cells have been implicated in chronic inflammatory conditions that lead to fibrosis in the liver, lung and skin (reviewed in29), the role of mast cells in the failing heart, wherein sustained inflammation and myocardial fibrosis contribute to adverse cardiac remodeling, is comparatively less well understood. Although previous studies have demonstrated a direct relationship between mast cell density and collagen volume fraction in experimental31–33 and human heart failure,34–36 a direct cause and effect relationship between the mast cells and the development of myocardial fibrosis has not been established heretofore. Previous studies that have employed chymase inhibitors37 or mast cell stabilizing agents31,38 have demonstrated decreased myocardial fibrosis in experimental models, and thus provide indirect supportive evidence for a potential role for mast cell induced myocardial fibrosis in heart failure. However, it should be recognized that chymase inhibitors decrease the local production of angiotensin II, a profibrotic peptide, in the heart. Moreover, mast cell stabilizing agents (e.g. cromolyn, nedocromil and ketotifen) inhibit the release of neutrophil-derived proinflammatory mediators, and have a variety of other effects on nitric oxide production and phospholipase C activation and NADPH expression.39 Thus, mast cell stabilizing agents have mechanisms that are independent of their effects on stabilizing mast cell membranes. Here we show that mast cell density increases in the hearts of MHCsTNF mice in direct relationship to the development of myocardial fibrosis in a mast cell-sufficient background, and that breeding the MHCsTNF onto a mast cell-deficient background decreases mast cell density, abrogates myocardial fibrosis and improves diastolic filling, thereby establishing a direct causal relationship between mast cell density and progressive myocardial fibrosis. Our results are qualitatively similar, but quantitatively different from those of Hara and colleagues, who reported a significant decrease in perivascular fibrosis, with a only trend towards a decrease in interstitial fibrosis in mast cell deficient (WBB6F1-W/Wv) mice after pressure overload.40 With regard to the cellular mechanisms for mast cell-induced fibrosis, previous studies have shown that mast cells release a variety of preformed pro-fibrotic mediators, such as TGF-β and bFGF, which directly stimulate fibroblast activation and proliferation. Our mast cell co-culture experiments are consistent with these prior reports, and show that mast cell secretory products provoke a pro-fibrotic phenotype in both LM and MHCsTNF fibroblasts (Figures 5 and 6), and that fibroblasts isolated from a transgenic model of sustained inflammatory signaling exhibit an enhanced repertoire of pro-fibrotic phenotypic responses to mast cell mediators when compared to LM fibroblasts.
Conclusions
This study shows for the first time that sustained inflammatory signaling in the heart leads to progressive myocardial fibrosis and diastolic cardiac dysfunction through a mechanism that requires increased myocardial mast cell density, with a resultant increase in TGF-β mediated pro-fibrotic signaling between cardiac mast cells and resident cardiac fibroblasts. Apart from the novelty of these findings, this study further suggests that sustained TNF signaling results in increased sensitization of resident fibroblasts to the biological mediators released by mast cells, including enhanced responsiveness to TGF-β. From a teleological standpoint these inflammation-induced changes in the cardiac fibroblasts may be beneficial by allowing resident cardiac fibroblasts to be more efficient in terms of myocardial repair mechanisms at sites of injury and inflammation; however, in the long term these changes may be maladaptive and contribute to excessive myocardial fibrosis and pathological remodeling. Whether the observed pathological fibrosis observed in inflammatory cardiomyopathy represents increased fibroblast sensitivity to TGF-β signaling, or whether it represents a pro-fibrotic response secondary to increased mast cell density or both is not known, and whether these results will obtain in the human myocardium will require further study.
Supplementary Material
Table.
Heart-weight to Body-Weight Ratio
| LM/c-kit+/− | MHCsTNF/c-kit+/− | MHCsTNF/c-kit−/− | |
|---|---|---|---|
| 4 w | 5.07±0.18 (n=4) | 6.36±0.68 (n=3)* | 6.67±0.61 (n=3)* |
| 8 w | 5.08±0.37 (n=6) | 5.82±0.55 (n=6)* | 5.79±0.44 (n=6)* |
| 12 w | 4.77±0.23(n=5) | 5.98±0.56 (n=5)* | 5.39±0.69 (n=5) |
Heart-weight to body-weight ratios was determined in LM/c-kit+/−, MHCsTNF/c-kit+/−, and MHCsTNF/c-kit−/− mice at 4, 8 and 12 weeks of age.
p < 0.05 compared to LM/c-kit+/− mouse hearts at the respective time periods by two way ANOVA
Acknowledgments
This research was supported by research funds from the N.I.H. (RO1 HL58081, RO1 HL61543, RO1 HL-42250)
Sources of Funding
This research was supported by research funds from the National Institutes of Health (RO1 HL58081, RO1 HL61543, RO1 HL-42250 and T32 HL07816 NIH).
Footnotes
Disclosures
The authors have no disclosures relevant to this manuscript.
References
- 1.Weber KT, Brilla CG, Janicki JS. Myocardial Fibrosis - Functional Significance and Regulatory Factors. Cardiovasc Res. 1993;27:341–348. doi: 10.1093/cvr/27.3.341. [DOI] [PubMed] [Google Scholar]
- 2.Blobe GC, Schiemann WP, Lodish HF. Role of transforming growth factor beta in human disease. N Engl J Med. 2000;342:1350–1358. doi: 10.1056/NEJM200005043421807. [DOI] [PubMed] [Google Scholar]
- 3.Reisdorf P, Lawrence DA, Sivan V, Klising E, Martin MT. Alteration of transforming growth factor-beta1 response involves down-regulation of Smad3 signaling in myofibroblasts from skin fibrosis. Am J Pathol. 2001;159:263–272. doi: 10.1016/s0002-9440(10)61692-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kubota T, McTiernan CF, Frye CS, Slawson SE, Koretsky AP, Demetris AJ, Feldman AM. Dilated cardiomyopathy in transgenic mice with cardiac specific overexpression of tumor necrosis factor-alpha. Circ Res. 1997;81:627–635. doi: 10.1161/01.res.81.4.627. [DOI] [PubMed] [Google Scholar]
- 5.Bryant D, Becker L, Richardson J, Shelton J, Franco F, Pechock RM, Thompson M, Giroir BP. Cardiac Failure in transgenic mice with myocardial expression of tumor necrosis factor-α (TNF) Circulation. 1998;97:1375–1381. doi: 10.1161/01.cir.97.14.1375. [DOI] [PubMed] [Google Scholar]
- 6.Sivasubramanian N, Coker ML, Kurrelmeyer K, DeMayo F, Spinale FG, Mann DL. Left ventricular remodeling in transgenic mice with cardiac restricted overexpression of tumor necrosis factor. Circulation. 2001;2001:826–831. doi: 10.1161/hc3401.093154. [DOI] [PubMed] [Google Scholar]
- 7.Siwik DA, Chang DL, Colucci WS. Interleukin-1beta and tumor necrosis factor-alpha decrease collagen synthesis and increase matrix metalloproteinase activity in cardiac fibroblasts in vitro. Circ Res. 2000;86:1259–1265. doi: 10.1161/01.res.86.12.1259. [DOI] [PubMed] [Google Scholar]
- 8.Peng J, Gurantz D, Tran V, Cowling RT, Greenberg BH. Tumor necrosis factor-alpha-induced AT1 receptor upregulation enhances angiotensin II-mediated cardiac fibroblast responses that favor fibrosis. Circ Res. 2002;91:1119–1126. doi: 10.1161/01.res.0000047090.08299.d5. [DOI] [PubMed] [Google Scholar]
- 9.Li X, Moody MR, Engel D, Walker S, Clubb FJ, Jr, Sivasubramanian N, Mann DL, Reid MB. Cardiac-specific overexpression of tumor necrosis factor-alpha causes oxidative stress and contractile dysfunction in mouse diaphragm. Circulation. 2000;102:1690–1696. doi: 10.1161/01.cir.102.14.1690. [DOI] [PubMed] [Google Scholar]
- 10.Berrozpe G, Timokhina I, Yukl S, Tajima Y, Ono M, Zelenetz AD, Besmer P. The W(sh), W(57), and Ph Kit expression mutations define tissue-specific control elements located between -23 and -154 kb upstream of Kit. Blood. 1999;94:2658–2666. [PubMed] [Google Scholar]
- 11.Farivar R, Chobanian AV, Brecher P. Salicylate or aspirin inhibits the induction of the inducible nitric oxide synthase in rat cardiac fibroblasts. Circ Res. 1996;78:759–768. doi: 10.1161/01.res.78.5.759. [DOI] [PubMed] [Google Scholar]
- 12.Zhang Q, Oh CK, Messadi DV, Duong HS, Kelly AP, Soo C, Wang L, Le AD. Hypoxia-induced HIF-1 alpha accumulation is augmented in a co-culture of keloid fibroblasts and human mast cells: involvement of ERK1/2 and PI-3K/Akt. Exp Cell Res. 2006;312:145–155. doi: 10.1016/j.yexcr.2005.10.006. [DOI] [PubMed] [Google Scholar]
- 13.Rhee S. Fibroblasts in Three Dimensional Matrices: Cell Migration and Matrix Remodeling. Exp Mol Med. 2009;41:858–865. doi: 10.3858/emm.2009.41.12.096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bechtel W, McGoohan S, Zeisberg EM, Muller GA, Kalbacher H, Salant DJ, Muller CA, Kalluri R, Zeisberg M. Methylation determines fibroblast activation and fibrogenesis in the kidney. Nat Med. 2010;16:544–550. doi: 10.1038/nm.2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang F, Trial J, Diwan A, Gao F, Birdsall H, Entman M, Hornsby P, Sivasubramanian N, Mann D. Regulation of cardiac fibroblast cellular function by leukemia inhibitory factor. J Mol Cell Cardiol. 2002;34:1309. doi: 10.1006/jmcc.2002.2059. [DOI] [PubMed] [Google Scholar]
- 16.Divakaran V, Adrogue J, Ishiyama M, Entman ML, Haudek S, Sivasubramanian N, Mann DL. Adaptive and maladptive effects of SMAD3 signaling in the adult heart after hemodynamic pressure overloading. Circ Heart Fail. 2009;2:633–642. doi: 10.1161/CIRCHEARTFAILURE.108.823070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sakata Y, Dong JW, Vallejo JG, Huang CH, Baker JS, Tracey KJ, Tacheuchi O, Akira S, Mann DL. Toll-like receptor 2 modulates left ventricular function following ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol. 2007;292:H503–H509. doi: 10.1152/ajpheart.00642.2006. [DOI] [PubMed] [Google Scholar]
- 18.Powell DW, Mifflin RC, Valentich JD, Crowe SE, Saada JI, West AB. Myofibroblasts. I. Paracrine cells important in health and disease. Am J Physiol. 1999;277:C1–C9. doi: 10.1152/ajpcell.1999.277.1.C1. [DOI] [PubMed] [Google Scholar]
- 19.van den Borne SW, Diez J, Blankesteijn WM, Verjans J, Hofstra L, Narula J. Myocardial remodeling after infarction: the role of myofibroblasts. Nat Rev Cardiol. 2009;7:30–37. doi: 10.1038/nrcardio.2009.199. [DOI] [PubMed] [Google Scholar]
- 20.Wipff PJ, Hinz B. Myofibroblasts work best under stress. J Bodyw Mov Ther. 2009;13:121–127. doi: 10.1016/j.jbmt.2008.04.031. [DOI] [PubMed] [Google Scholar]
- 21.Rothe MJ, Kerdel FA. The mast cell in fibrosis. Int J Dermatol. 1991;30:13–16. doi: 10.1111/j.1365-4362.1991.tb05871.x. [DOI] [PubMed] [Google Scholar]
- 22.Mason GI, Hamburger J, Matthews JB. Mast cells, extracellular matrix components, TGFbeta isoforms and TGFbeta receptor expression in labial salivary glands in systemic sclerosis. Ann Rheum Dis. 2000;59:183–189. doi: 10.1136/ard.59.3.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gruber BL. Mast cells in the pathogenesis of fibrosis. Curr Rheumatol Rep. 2003;5:147–153. doi: 10.1007/s11926-003-0043-3. [DOI] [PubMed] [Google Scholar]
- 24.Sakata Y, Chancey AL, Divakaran VG, Sekiguchi K, Sivasubramanian N, Mann DL. Transforming growth factor-beta receptor antagonism attenuates myocardial fibrosis in mice with cardiac-restricted overexpression of tumor necrosis factor. Basic Res Cardiol. 2007;103:60–68. doi: 10.1007/s00395-007-0689-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hu ZQ, Yamazaki T, Cai Z, Yoshida T, Shimamura T. Mast cells display natural suppressor activity partially by releasing transforming growth factor-beta. Immunology. 1994;82:482–486. [PMC free article] [PubMed] [Google Scholar]
- 26.Wolf G, Zahner G, Schroeder R, Stahl RA. Transforming growth factor beta mediates the angiotensin-II-induced stimulation of collagen type IV synthesis in cultured murine proximal tubular cells. Nephrol Dial Transplant. 1996;11:263–269. doi: 10.1093/oxfordjournals.ndt.a027251. [DOI] [PubMed] [Google Scholar]
- 27.Silver RB, Reid AC, Mackins CJ, Askwith T, Schaefer U, Herzlinger D, Levi R. Mast cells: a unique source of renin. Proc Natl Acad Sci U S A. 2004;101:13607–13612. doi: 10.1073/pnas.0403208101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bachelet I, Munitz A, Levi-Schaffer F. Co-culture of mast cells with fibroblasts: a tool to study their crosstalk. Methods Mol Biol. 2006;315:295–317. doi: 10.1385/1-59259-967-2:295. [DOI] [PubMed] [Google Scholar]
- 29.Metz M, Grimbaldeston MA, Nakae S, Piliponsky AM, Tsai M, Galli SJ. Mast cells in the promotion and limitation of chronic inflammation. Immunol Rev. 2007;217:304–328. doi: 10.1111/j.1600-065X.2007.00520.x. [DOI] [PubMed] [Google Scholar]
- 30.Galli SJ, Tsai M. Mast cells: versatile regulators of inflammation, tissue remodeling, host defense and homeostasis. J Dermatol Sci. 2008;49:7–19. doi: 10.1016/j.jdermsci.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.De Jongste MJ, Haaksma J, Hautvast RW, Hillege HL, Meyler PW, Staal MJ, Sanderson JE, Lie KI. Effects of spinal cord stimulation on myocardial ischaemia during daily life in patients with severe coronary artery disease. A prospective ambulatory electrocardiographic study. Br Heart J. 1994;71:413–418. doi: 10.1136/hrt.71.5.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Patella V, Marino I, Arbustini E, Lamparter-Schummert B, Verga L, Adt M, Marone G. Stem cell factor in mast cells and increased mast cell density in idiopathic and ischemic cardiomyopathy. Circulation. 1998;97:971–978. doi: 10.1161/01.cir.97.10.971. [DOI] [PubMed] [Google Scholar]
- 33.Shiota N, Rysa J, Kovanen PT, Ruskoaho H, Kokkonen JO, Lindstedt KA. A role for cardiac mast cells in the pathogenesis of hypertensive heart disease. J Hypertens. 2003;21:1935–1944. doi: 10.1097/00004872-200310000-00022. [DOI] [PubMed] [Google Scholar]
- 34.Akgul A, Skrabal CA, Thompson LO, Loebe M, Lafuente JA, Noon GP, Youker KA. Role of mast cells and their mediators in failing myocardium under mechanical ventricular support. J Heart Lung Transplant. 2004;23:709–715. doi: 10.1016/j.healun.2003.06.006. [DOI] [PubMed] [Google Scholar]
- 35.Batlle M, Perez-Villa F, Lazaro A, Garcia-Pras E, Ramirez J, Ortiz J, Orus J, Roque M, Heras M, Roig E. Correlation between mast cell density and myocardial fibrosis in congestive heart failure patients. Transplant Proc. 2007;39:2347–2349. doi: 10.1016/j.transproceed.2007.06.047. [DOI] [PubMed] [Google Scholar]
- 36.Akgul A, Youker KA, Noon GP, Loebe M. Quantitative changes in mast cell populations after left ventricular assist device implantation. ASAIO J. 2005;51:275–280. doi: 10.1097/01.mat.0000150507.61120.00. [DOI] [PubMed] [Google Scholar]
- 37.Kanemitsu H, Takai S, Tsuneyoshi H, Yoshikawa E, Nishina T, Miyazaki M, Ikeda T, Komeda M. Chronic chymase inhibition preserves cardiac function after left ventricular repair in rats. Eur J Cardiothorac Surg. 2008;33:25–31. doi: 10.1016/j.ejcts.2007.09.040. [DOI] [PubMed] [Google Scholar]
- 38.Levick SP, McLarty JL, Murray DB, Freeman RM, Carver WE, Brower GL. Cardiac mast cells mediate left ventricular fibrosis in the hypertensive rat heart. Hypertension. 2009;53:1041–1047. doi: 10.1161/HYPERTENSIONAHA.108.123158. [DOI] [PubMed] [Google Scholar]
- 39.Vural KM, Liao H, Oz MC, Pinsky DJ. Effects of mast cell membrane stabilizing agents in a rat lung ischemia-reperfusion model. Ann Thorac Surg. 2000;69:228–232. doi: 10.1016/s0003-4975(99)01052-8. [DOI] [PubMed] [Google Scholar]
- 40.Hara M, Ono K, Hwang MW, Iwasaki A, Okada M, Nakatani K, Sasayama S, Matsumori A. Evidence for a role of mast cells in the evolution to congestive heart failure. J Exp Med. 2002;195:375–381. doi: 10.1084/jem.20002036. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.