

Abstract
The kisspeptin receptor (KISS1R) is a G protein-coupled receptor recognized as the trigger of puberty and a regulator of reproductive competence in adulthood 1,2,3. Inactivating mutations in KISS1R identified in patients have been associated with iodiopathic hypogonadotropic hypogonadism (IHH) 1,2 and precocious puberty 4. Functional studies of these mutants are crucial for our understanding of the mechanisms underlying the regulation of reproduction by this receptor as well as those shaping the disease outcomes, which result from abnormal KISS1R signaling and function. However, the highly GC-rich sequence of the KISS1R gene makes it rather difficult to introduce mutations or amplify the gene encoding this receptor by PCR.
Here we describe a method to introduce mutations of interest into this highly GC-rich sequence that has been used successfully to generate over a dozen KISS1R mutants in our laboratory. We have optimized the PCR conditions to facilitate the amplification of a range of KISS1R mutants that include substitutions, deletions or insertions in the KISS1R sequence. The addition of a PCR enhancer solution, as well as of a small percentage of DMSO were especially helpful to improve amplification. This optimized procedure may be useful for other GC-rich templates as well.
The expression vector encoding the KISS1R is been used to characterize signaling and function of this receptor in order to understand how mutations may change KISS1R function and lead to the associated reproductive phenotypes. Accordingly, potential applications of KISS1R mutants generated by site-directed mutagenesis can be illustrated by many studies 1,4,5,6,7,8. As an example, the gain-of-function mutation in the KISS1R (Arg386Pro), which is associated with precocious puberty, has been shown to prolong responsiveness of the receptor to ligand stimulation 4 as well as to alter the rate of degradation of KISS1R 9. Interestingly, our studies indicate that KISS1R is degraded by the proteasome, as opposed to the classic lysosomal degradation described for most G protein-coupled receptors 9. In the example presented here, degradation of the KISS1R is investigated in Human Embryonic Kidney Cells (HEK-293) transiently expressing Myc-tagged KISS1R (MycKISS1R) and treated with proteasome or lysosome inhibitors. Cell lysates are immunoprecipitated using an agarose-conjugated anti-myc antibody followed by western blot analysis. Detection and quantification of MycKISS1R on blots is performed using the LI-COR Odyssey Infrared System. This approach may be useful in the study of the degradation of other proteins of interest as well.
Protocol
1. Site-directed mutagenesis of highly GC-rich KISS1R gene sequence
Template: full cDNA sequence of the human KISS1R with a Myc-tag fused to its N-terminus. This sequence is cloned into the pCS2+ expression vector, which is compatible with the mammalian cell lines subsequently used for transfections. This expression vector is referred to herein as pCS2+MycKISS1R.
- Primer design: primers are designed to carry desired mutations, according to instructions of the Quikchange Site-Directed Mutagenesis kit (Stratagene). In summary:
- Both (forward and reverse) primers must contain the desired mutation and anneal to the same sequence on opposite strands of the plasmid (e.g. forward and reverse primers are complementary to each other)
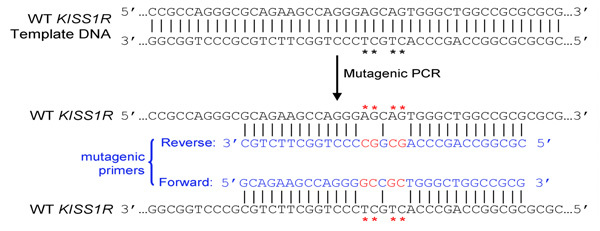
- Primers should be 25 to 45 bases long and end in one or more C or G bases
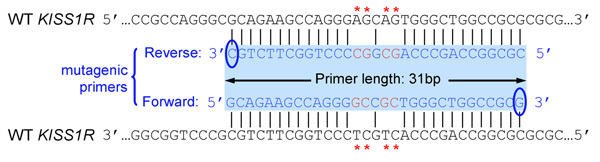
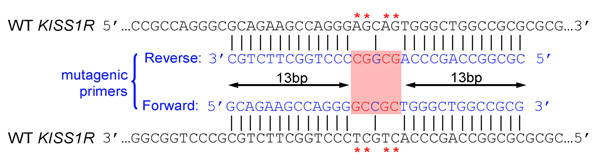
- Introduced mutation(s) should be in the middle of primer and flanked by ˜10–15 bases of correct sequence on both sides
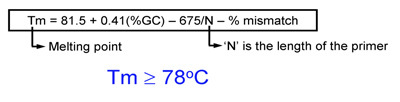
- Melting temperature (Tm) of primers should be equal or greater than 78°C. Use the following formulas to estimate Tm:
- When introducing mutations: Tm = 81.5 + 0.41(%GC) – 675/N – % mismatch (N is the primer length in bases)
- When introducing insertions or deletions: Tm = 81.5 + 0.41(%GC) – 675/N (N does not include the bases which are being inserted or deleted)
- Use desalted primers (no further purifications necessary).
The best amplification conditions include the addition of PCRx Enhancer solution (Invitrogen) and DMSO. At least 2 concentrations of DMSO, such as 4% and 8%, should be tested. PCR conditions are shown in Table I and in Figure 1; and results of a representative PCR amplification of pCS2+MycKISS1R is shown in Figure 2.
Eliminate parental DNA on amplification products by digestion of methylated DNA with DpnI for 1h 30min at 37°C: mix in a centrifuge tube 17.5μl of PCR product with 2μl of 10x NEBuffer 4 and 0.5μl of DpnI (10U; New England Biolabs).
Transform DpnI-treated PCR product: mix 2μl of DpnI-treated DNA with 45μl of XL10-Gold Ultracompetent E. coli (Stratagene) in pre-chilled 15 ml cell culture tube. Add 2μl of β-mercaptoethanol and follow Stratagene instructions. Plate 100-400μl of transformed bacteria in LB-agar plates containing 100μg/ml ampicillin and incubate at 37°C.
Miniprep 2 to 4 individual colonies to isolate plasmid DNA. Confirm the successful introduction of desired mutations by sequencing and analysis of isolated DNA.
2. Transient transfection of MycKISS1R into HEK-293 cells
The following experiments are performed in human embryonic kidney cells (HEK-293) cultured in a CO2 incubator (5% CO2) at 37°C in DMEM supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin.
Seed HEK-293 cells at 2.5x105 cells/ml in 6-well plates and let them grow overnight at 37°C before transfection. NOTE: (i) use triplicate wells for each experimental condition; (ii) ideal cell confluence at the time of transfection is 30%-50%.
Transfect HEK-293 cells using the GenePorter Transfection Reagent (Genlantis), according to the manufacturer's instructions: Mix half of the serum-free DMEM with 0.5μg pCS2+ MycKISS1R plus 0.5μg control (empty) vector to totalize 1μg of DNA/well. Mix the other half with 10μl transfection reagent per μg of DNA transfected. NOTE: Ideal plasmid DNA concentration may vary.
3. Cell treatment and lysis
24h after transfection, replace cell medium with 1ml DMEM containing 2.5% FBS (to decrease cell metabolism). NOTE: This decrease in serum may facilitate and/or amplify the detection of results.
Add lysosome inhibitor (100μg/ well of Leupeptin) directly into each well of one entire 6-well plate. Incubate at 37°C for 6 or 16 h (or other desired times)
Add freshly prepared proteasome inhibitor (10μM/ well of MG132) directly into all wells of two entire 6-well plates. Incubate at 37°C for 2, 4, 6 or 16h (or desired times). Add vehicle to all wells of the fourth 6-well plate (0 time-point) and incubate at 37°C for 16h
- When incubation is over, move plates to ice and perform this entire lysis procedure on ice to prevent protein degradation:
- To increase protein yield, combine the triplicates on 6-well plates in a single centrifuge tube
- Aspirate medium and wash cells once with 1ml of ice-cold phosphate buffered saline (PBS)
- Add 100μl of ice-cold lysis buffer (20mM HEPES, pH 7.4, 1% NP-40, 150mM NaCl, 1mM EDTA, 0.25% sodium deoxycholate) containing protease inhibitors (1x cocktail containing 100mM AEBSF-HCl, 80μM aprotinin, 5mM bestatin, 1.5mM E-64, 0.5M EDTA, 2mM leupeptin and 1mM pepstatin A, plus 2mM PMSF) to each well
- Remove cells with a cell scraper and transfer cell lysates to centrifuge tubes
- Pass cells ˜10 times through a 20-gauge needle. NOTE: Do not sonicate samples intended for western blot detection of membrane proteins. Sonication leads to aggregation of membrane proteins, which will not migrate properly during electrophoresis
- Incubate cell lysates for 1h at 4°C on a rocking platform
- Centrifuge cell lysates at 4°C for 10 min at 10,000 x rpm and transfer supernatants to new tubes. NOTE: Do not disturb pellets during this step
- Determine protein concentration in 10μl of supernatants using the BCA method (Pierce)
- Dilute lysates to 1mg/ml with lysis buffer containing protease inhibitors.
4. Immunoprecipitation and Western blot detection of MycKISS1R
- Perform the following immunoprecipitation steps on ice (or at 4°C):
- Wash the appropriate amount of agarose-conjugated anti-myc antibody (2.5μg/ sample) twice with ice-cold PBS and add this to 400μg of lysate protein.
- Immunoprecipitate the MycKISS1R on lysates overnight at 4°C in a rocking platform with gentle agitation.
- Spin down agarose beads by pulse centrifugation at 4°C (up to 10,000 x rpm)
- Aspirate and discard supernatants (without disturbing the pellets)
- Wash beads once with ice-cold lysis buffer and twice with ice-cold PBS. Invert tubes gently before spinning
- Ressuspend beads containing antibody-bound MycKISS1R in 2x sample loading buffer containing 10% β-mercaptoethanol.
- Western blot of MycKISS1R immunocomplexes:
- Heat samples for 30 min at 37°C. NOTE: Do not boil samples intended for western blot detection of membrane proteins. Like sonication, boiling also leads to aggregation of these proteins
- Move tubes immediately into ice for 5 min
- Separate proteins by SDS-PAGE in a 4-15% gradient gel.
- Transfer to Immobilon-FL PVDF membrane (for infra-red detection) at 25V for 30 min in Transfer buffer (48mM Tris base, 39mM glycine, 1.2mM SDS, 20% methanol, pH 9.2) using the Bio-Rad Semi-Dry Transfer Apparatus
- Wash membranes for 5 min at room temperature with Tris-Buffered Saline (TBS) and block for 1h at room temperature with Licor Odyssey blocking on a rocking platform (alternatively, 5% milk in TBS can be used to block non-specific binding).
- Incubate membranes overnight at 4°C with rabbit anti-myc antibody (1:500) in blocking solution containing 0.1% Tween-20
- Remove primary antibody and wash membranes 3 times of 5 min each with TBS containing 0.1% Tween-20 (TBST)
- Incubate membranes for 1h at room temperature with goat Infra-RedDye ® 800CW-labelled anti-rabbit IgG (1:10,000) in blocking buffer containing 0.1% Tween-20 and 0.01% SDS
- Remove secondary antibody, wash membranes 3 times of 5 min each with TBST and one last time with TBS only (to remove remaining Tween-20)
- Imaging and quantification of MycKISS1R using the LI-COR Odyssey Infra-Red Imager:
- The MycKISS1R on the membranes will be imaged using the LI-COR Odyssey Infra-Red Imager. To begin, place membrane on bottom left corner of Odyssey scanner, aligning it with the grid. Cover with the rubber mat, smooth out bubbles with roller and close the lid
- Create a new project file on the computer. Name the file, click "done", and then enter the scanner login on "scan". Size the scanner console box to fit your membrane, and then choose 169μm resolution and medium image quality
- Choose intensity settings for 700 (red) and 800 (green) channels according to expected strength of each signal. This is for signal visualization purposes only and will not influence quantification. Click "start scan"
- Name and save the scan, then click "OK" to open it on a new window for quantification. MycKISS1R monomers should be visible at approximately 43kD
- Using the "box" tool (on the left-hand sidebar), draw a box around the first band. Drag the box around to make sure all bands fit in, then "copy" and "paste" the box over all bands
- Select all boxes using "Ctrl+A", and then select the option to subtract median background. Click "report" on the top menu and a spreadsheet with the quantification values will appear. Representative results shown here are represented as fold-increase over untreated cells (time zero)
5. Representative Results:
Site-directed mutagenesis of highly GC-rich KISS1R gene sequence: Table 1. shows the combination of reagents to improve efficiency of KISS1R amplification. This combination is been successfully used to introduce several mutations directed against distinct regions of the KISS1R cDNA sequence, as well as for the amplification of this highly GC-rich gene. Figure 1 shows cycling and amplification conditions for the mutagenesis of KISS1R. These conditions have been modified from the QuikChange Site-Directed Mutagenesis kit (Stratagene). Figure 2. shows a representative result using this optimized protocol. The addition of 4% or 8% DMSO combined with the PCRx Enhancer significantly improves yield of amplification of the GC-rich KISS1R cDNA-containing plasmid. In this representative experiment, 4% DMSO provided slightly better amplification conditions compared to 8% DMSO. Transformation of DpnI-treated amplification products with ultracompetent E. coli typically yields 100 to over 1,000 colonies, and the rate of successful introduction of desired mutations is 80-90% as determined by DNA sequencing.
Use of MycKISS1R constructs to study receptor physiology: In this representative experiment, the wild type pCS2+MycKISS1R amplified according to the optimized protocol described here is used to study in vivo KISS1 receptor degradation in a relevant cell line (HEK-293) transiently expressing MycKISS1R. After treating the transfected HEK-293 cells with lysosome (leupeptin) or proteasome (MG132) inhibitors according to the protocol described in methods, the cells are lysed and processed for western blot. The top panel of Figure 3 shows the scan of MycKISS1R monomers whereas the bottom panel shows the quantification of the bands shown on top panel. Quantification of bands indicates that neither 6h nor 16h of leupeptin treatment affected MycKISS1R protein levels. Conversely, treatment with MG132 resulted in a time-dependent increase in MycKISS1R protein in these cells, which culminates with a 45-fold accumulation of the receptor after 16h of incubation with MG132. These observations indicate that, unlike most G protein-coupled receptors, KISS1R is degraded by the proteasome (rather than the lysosome).
| DMSO | |||
| Reagents | 0 | 4% | 8% |
| Water | 35 | 33 | 31 |
| 10x Pfu Ultra Taq buffer | 5 | 5 | 5 |
| dNTP mix (10mM) | 1 | 1 | 1 |
| Primer sense (25pmol/μl) | 1 | 1 | 1 |
| Primer antisense (25pmol/μl) | 1 | 1 | 1 |
| 10x PCRx Enhancer Solution | 5 | 5 | 5 |
| DMSO | 0 | 2 | 4 |
| Pfu Ultra Taq polymerase (2.5U/μl) | 1 | 1 | 1 |
| plasmid DNA (20ng/μl) | 1 | 1 | 1 |
Table 1. Combination of reagents successfully used to mutate and amplify the GC-rich KISS1R
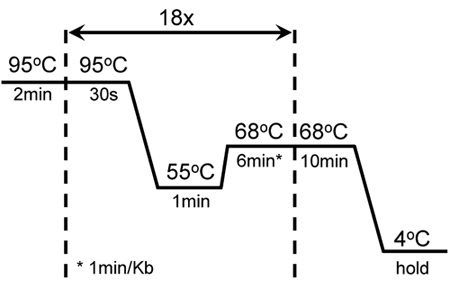 Figure 1. Cycling conditions of successful mutagenesis and amplification of GC-rich KISS1R: A 2min hot start was followed by 18 cycles of 30 sec melting at 95°C; 1 min annealing at 55°C and 6 min extension at 68°C. An additional 10 min extension at 68°C was added at the end of the last cycle. These settings were adjusted from the original mutagenesis protocol of the QuikChange II XL-Site-Directed Mutagenesis kit (Stratagene).
Figure 1. Cycling conditions of successful mutagenesis and amplification of GC-rich KISS1R: A 2min hot start was followed by 18 cycles of 30 sec melting at 95°C; 1 min annealing at 55°C and 6 min extension at 68°C. An additional 10 min extension at 68°C was added at the end of the last cycle. These settings were adjusted from the original mutagenesis protocol of the QuikChange II XL-Site-Directed Mutagenesis kit (Stratagene).
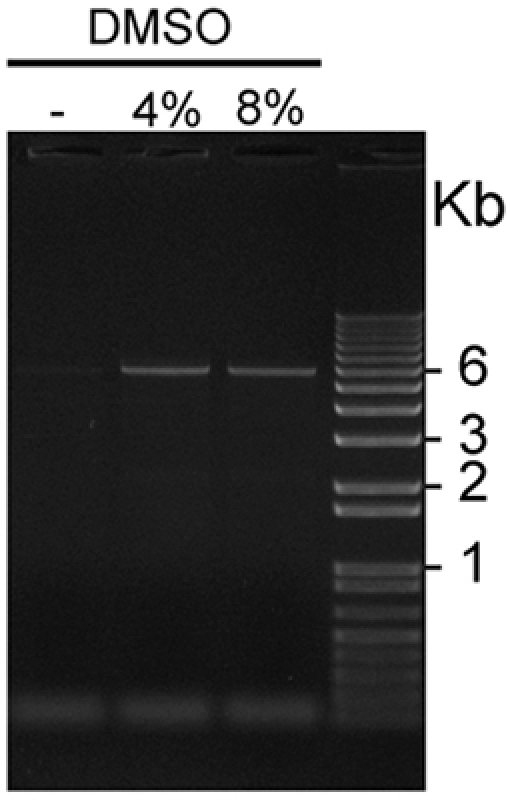 Figure 2. Visualization of GC-rich pCS2+MycKISS1R amplified in the presence or absence of DMSO: Five μl aliquots of pCS2+MycKISS1R amplified in the presence of 0, 4 or 8% DMSO were loaded in this representative 1% agarose gel stained with ethidium bromide and visualized using UV light. The plasmid bands of 6Kb are visible on both lanes loaded with PCR products amplified in the presence of 4% and 8% DMSO, but not on the first lane, which was loaded with a PCR product amplified in the absence of DMSO.
Figure 2. Visualization of GC-rich pCS2+MycKISS1R amplified in the presence or absence of DMSO: Five μl aliquots of pCS2+MycKISS1R amplified in the presence of 0, 4 or 8% DMSO were loaded in this representative 1% agarose gel stained with ethidium bromide and visualized using UV light. The plasmid bands of 6Kb are visible on both lanes loaded with PCR products amplified in the presence of 4% and 8% DMSO, but not on the first lane, which was loaded with a PCR product amplified in the absence of DMSO.
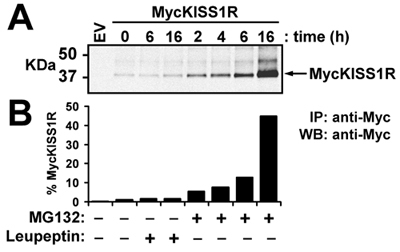 Figure 3. Effect of leupeptin or MG132 on levels of MycKISS1R protein in HEK-293 cells: HEK-293 cells expressing MycKISS1R were treated with 100μg/ml leupeptin or 10μM MG132 at 37°C for the designated times. MycKISS1R on 400μg of cell lysate was immunoprecipitated with 2.5μg of agarose-conjugated anti-myc antibody and analyzed by western blot. (A) LI-COR Odyssey detection of MycKISS1R after incubation of immunoblots with rabbit anti-Myc-tag antibody followed by incubation with IRDye 800CW-labeled anti-rabbit; (B) Quantification of MycKISS1R bands shown in (A) using the LI-COR Odyssey quantification software. Results are represented as fold-increase over untreated cells (time 0).
Figure 3. Effect of leupeptin or MG132 on levels of MycKISS1R protein in HEK-293 cells: HEK-293 cells expressing MycKISS1R were treated with 100μg/ml leupeptin or 10μM MG132 at 37°C for the designated times. MycKISS1R on 400μg of cell lysate was immunoprecipitated with 2.5μg of agarose-conjugated anti-myc antibody and analyzed by western blot. (A) LI-COR Odyssey detection of MycKISS1R after incubation of immunoblots with rabbit anti-Myc-tag antibody followed by incubation with IRDye 800CW-labeled anti-rabbit; (B) Quantification of MycKISS1R bands shown in (A) using the LI-COR Odyssey quantification software. Results are represented as fold-increase over untreated cells (time 0).
Discussion
Site-directed mutagenesis has been used to study protein function by introducing nucleotide changes in the coding sequence of targeted genes for over three decades. The original technique was described in 1978 by the British-Canadian Chemist and Nobel Prize winner Michael Smith 10. Michael Smith shared the 1993 Nobel Prize in Chemistry with Kary Mullis, the American Biochemist who invented the Polymerase Chain Reaction (PCR) 11. The original method described by Michael Smith has been improved over the years. The ability to introduce mutations, insertions or deletions into the desired gene sequence in a single PCR reaction has contributed to the high accuracy and success rates of this procedure, as well as to the production of mutants in a relatively short period of time.
Despite all technical improvements, mutations and amplification of highly GC-rich gene sequences such as KISS1R are particularly troublesome. The size of primers designed for mutagenesis of high GC content sequences must be adjusted to accommodate the GC content so that the melting temperature is within the recommended range. Accordingly, primers to introduce mutations in KISS1R are sized to have a melting point between 78°-86°C. Some of the KISS1R gene regions have a GC content of over 85%. The size of primers designed to introduce mutations in these regions must be adjusted to meet the melting point range mentioned above. Primers with melting points above that limit may not separate at 95°C, impairing amplification. On the other hand, primers with a Tm lower than 78°C are more likely to bind to and amplify non-specific sequences.
Likewise, amplification conditions and reagents are optimized for the high GC content sequence of KISS1R. A combination of 4% DMSO and a PCR Enhancer Solution from Invitrogen (Table 1 and Figure 2) is shown to increase amplification efficiency of KISS1R. Alternatively, other reagents such as betaine have been successfully used to improve amplification of other GC rich sequences 12,13. However, there was no noticeable positive effect of betaine on KISS1R amplification in our hands. Also, PCR enhancer solutions available from other suppliers may be equally capable of improving amplification of GC rich sequences.
Using the optimized settings described here, we have successfully introduced several mutations in distinct regions of the KISS1R cDNA, which included up to 6 nucleotide changes at once (carried by same primers) and the deletion of up to 27 nucleotides. Transformation of these mutants generated hundreds to thousands of colonies with a high (80-90%) rate of colonies containing the correct mutant sequence confirmed by DNA sequencing.
Site-directed mutagenesis can also be used to add antigenic tags such as Myc or Flag to the amino or carboxyl terminus of proteins. These tags allow for selective immunoprecipitation and/or detection of proteins, and are especially helpful when reliable antibodies against the protein of interest are not available, as for KISS1R. In the example presented here, the expression vector encoding for the KISS1R, which is fused to an eleven amino acid Myc-tag at its amino-terminus (MycKISS1R), is used to study the degradation pathway of KISS1R in HEK-293 cells. The efficiency of transfection of HEK-293 cells with MycKISS1R using the method described here is 25-30%. Thus, immunoprecipitation is used to concentrate MycKISS1R from cell lysates thus improving detection and visualization of the receptor. This approach is often used to facilitate visualization of proteins expressed endogenously at low levels as well 14.
Importantly, G protein-coupled receptors such as KISS1R, as well as other membrane proteins, should not be homogenized by sonication or boiled before gel loading. While these procedures are typically used to process samples for immunoprecipitation or western blot, sonication and boiling lead to the aggregation of membrane proteins, impairing their proper migration during electrophoresis 15. In fact, replacement of these procedures with milder homogenization, such as needle-passage, and lower sample heating, such as 37°C for 30 min (rather than boiling) is required for detection of KISS1R monomers.
Immunoprecipitated MycKISS1R is detected using Odyssey Imager (LI-COR Biosystems) after incubation of blots with infrared dye-conjugated secondary antibody. The advantages of this system include a sustaining strong signal that can be detected with high accuracy for months, as opposed to the short-lived signal emitted by chemiluminescence. Moreover, the quantification software of Odyssey Imager offers greater sensitivity and linearity of detection when compared to traditional quantification of scanned x-ray films following chemiluminescence. However, detection of horseradish peroxidase (HRP)-conjugated antibodies by chemiluminescence may be used as well.
The absence of classical lysosomal degradation and the detection of proteasomal degradation of KISS1R described here in HEK-293 cells have also been observed in COS-7 cells transfected with the same MycKISS1R expression vector 9. Although unusual, proteasomal degradation of G protein-coupled receptors has been previously reported in the literature 16,17.
In summary, this manuscript demonstrates how to investigate the effect of genetic mutations in the KISS1R gene, which is a highly GC-rich sequence, in order to understand how mutations in this receptor can lead to reproductive disorder phenotypes such as hypogonadotropic hypogonadism or precocious puberty.
Disclosures
No conflicts of interest declared.
Acknowledgments
This work was partially funded by the Reproductive Branch of the National Institute of Child Health and Human Development (NICHD – R21 HD059015) and by the Charles H. Hood Foundation Young Investigator Child Health Research Award (Boston, MA).
References
- Seminara SB, Messager S, Chatzidaki EE. The GPR54 Gene as a Regulator of Puberty. N Engl J Med. 2003;349:1614–1627. doi: 10.1056/NEJMoa035322. [DOI] [PubMed] [Google Scholar]
- de Roux N, Genin E, Carel JC. Hypogonadotropic Hypogonadism Due to Loss of Function of the KiSS1-Derived Peptide Receptor GPR54. Proc Natl Acad Sci U S A. 2003;100:10972–10976. doi: 10.1073/pnas.1834399100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messager S, Chatzidaki EE, Ma D. Kisspeptin Directly Stimulates Gonadotropin-Releasing Hormone Release via G Protein-Coupled Receptor 54. Proc Natl Acad Sci U S A. 2005;102:1761–1766. doi: 10.1073/pnas.0409330102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teles MG, Bianco SD, Brito VN. A GPR54-Activating Mutation in a Patient with Central Precocious Puberty. N Engl J Med. 2008;358:709–715. doi: 10.1056/NEJMoa073443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenenbaum-Rakover Y, Commenges-Ducos M, Iovane A. Neuroendocrine Phenotype Analysis in Five Patients with Isolated Hypogonadotropic Hypogonadism due to a L102P Inactivating Mutation of GPR54. J Clin Endocrinol Metab. 2007;92:1137–1144. doi: 10.1210/jc.2006-2147. [DOI] [PubMed] [Google Scholar]
- Semple RK, Achermann JC, Ellery J. Two Novel Missense Mutations in G Protein-Coupled Receptor 54 in a Patient with Hypogonadotropic Hypogonadism. J Clin Endocrinol Metab. 2005;90:1849–1855. doi: 10.1210/jc.2004-1418. [DOI] [PubMed] [Google Scholar]
- Wacker JL, Feller DB, Tang XB. Disease-Causing Mutation in GPR54 Reveals the Importance of the Second Intracellular Loop for Class A G-Protein-Coupled Receptor Function. J Biol Chem. 2008;283:31068–31078. doi: 10.1074/jbc.M805251200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szereszewski JM, Pampillo M, Ahow MR. GPR54 regulates ERK1/2 activity and hypothalamic gene expression in a Galpha(q/11) and beta-arrestin-dependent manner. PLoS One. 2010;5:e12964–e12964. doi: 10.1371/journal.pone.0012964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianco SDC, Vandepas L, Correa-Medina M, Gereben B, Mukherjee A, Kuohung W, Carroll R, Teles MG, Latronico AC, Kaiser UB. KISS1R Intracellular Trafficking and Degradation: Effect of the Arg386Pro Disease-Associated Mutation. Endocrinology. 2011 doi: 10.1210/en.2010-0903. Forthcoming. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchison CA, Phillips S, Edgell MH. Mutagenesis at a Specific Position in a DNA Sequence. J Biol Chem. 1978;253:6551–6560. [PubMed] [Google Scholar]
- Mullis KB. The Unusual Origin of the Polymerase Chain Reaction. Sci Am. 1990;262:56–61. doi: 10.1038/scientificamerican0490-56. [DOI] [PubMed] [Google Scholar]
- Henke W, Herdel K, Jung K, Schnorr D, Loening SA. Betaine improves the PCR amplification of GC-rich DNA sequences. Nucleic Acids Res. 1997;25:3957–3958. doi: 10.1093/nar/25.19.3957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahdev S, Saini S, Tiwari P, Saxena S, Singh Saini K. Amplification of GC-rich genes by following a combination strategy of primer design, enhancers and modified PCR cycle conditions. Mol Cell Probes. 2007;21:303–307. doi: 10.1016/j.mcp.2007.03.004. [DOI] [PubMed] [Google Scholar]
- Kong KC. Chapter 10. In: Poyner DR, Wheatly M, editors. MSTA. in G Protein-Coupled Receptors: Essential Methods. Wiley-Blackwell; 2010. pp. 197–204. [Google Scholar]
- Hall RA. Chapter 9. In: George SR, O'Dowd BF, editors. G protein-Coupled Receptor-Protein Interactions. John Wiley & Sons, Inc; 2005. pp. 170–171. [Google Scholar]
- Chaturvedi K, Bandari P, Chinen N, Howells RD. Proteasome Involvement in Agonist-Induced Down-Regulation of Mu and Delta Opioid Receptors. J Biol Chem. 2001;276:12345–12355. doi: 10.1074/jbc.M008054200. [DOI] [PubMed] [Google Scholar]
- Shenoy SK, McDonald PH, Kohout TA, Lefkowitz RJ. Regulation of Receptor Fate by Ubiquitination of Activated Beta 2-Adrenergic Receptor and Beta-Arrestin. Science. 2001;294:1307–1313. doi: 10.1126/science.1063866. [DOI] [PubMed] [Google Scholar]