

Abstract
Synaptotagmin 1 (Syt1) is a Ca2+ sensor for SNARE-mediated, Ca2+-triggered synaptic vesicle fusion in neurons. It is composed of luminal, transmembrane, linker, and two Ca2+-binding (C2) domains. Here we describe expression and purification of full-length mammalian Syt1 in insect cells along with an extensive biochemical characterization of the purified protein. The expressed and purified protein is properly folded and has increased α-helical content compared to the C2AB fragment alone. Post-translational modifications of Syt1 were analyzed by mass spectrometry, revealing the same modifications of Syt1 that were previously described for Syt1 purified from brain extract or mammalian cell lines, along with a novel modification of Syt1, tyrosine nitration. A lipid binding screen with both full-length Syt1 and the C2AB fragments of Syt1 and Syt3 isoforms revealed new Syt1–lipid interactions. These results suggest a conserved lipid binding mechanism in which Ca2+-independent interactions are mediated via a lysine rich region of the C2B domain while Ca2+-dependent interactions are mediated via the Ca2+-binding loops.
The Ca2+-triggered fusion of docked synaptic and secretory vesicles with the plasma membrane causes temporal release of neurotransmitter and hormone release, respectively. Members of the SNARE family (soluble N-ethylmaleimide sensitive factor attachment protein receptor) and accessory proteins juxtapose vesicles and target membranes and, along with Ca2+ sensor synaptotagmin and other factors, catalyze fusion. Synaptotagmins, a family of ∼16 isoforms, consist of a short N-terminal lumenal (or extracellular) domain, a single transmembrane α-helix, an unstructured linker, and two C2 Ca2+-binding domains, termed C2A and C2B. Depending on the isoform, they are localized to synaptic/secretory vesicles or the plasma membrane. Synaptotagmin 1 (Syt1) acts as a primary Ca2+ sensor for fast synchronous synaptic vesicle release and insulin secretion. Synaptotagmins constitute a highly evolutionarily conserved protein family, especially for their Ca2+-binding domains. Although some biochemical studies of Syt1 have been conducted on brain extract or mammalian cell line immunoprecipitated protein, the bulk of biochemical characterization, which requires larger amounts of Syt1, has been conducted on the Escherichia coli-expressed C2AB fragment of Syt1 consisting of only C2A and C2B domains. Clearly, Syt1 expressed in E. coli lacks post-translational modifications, which have been reported in the mammalian-derived Syt1 studies.
Syt1 is N-glycosylated(1) at Asn24(2) and O-glycosylated at Thr15 and Thr16(3) in the luminal domain. In the COS-7 mammalian cell line, O-glycosylation has been reported to occur only in the presence of synaptobrevin.(3) In PC12 cells, both N- and O-glycosylations contribute to the proper targeting of Syt1 to secretory vesicles and Syt1 endocytosis.2,4 In addition to glycosylation, all five cysteines (Cys74, Cys75, Cys77, Cys79, and Cys82) in the Syt1 transmembrane proximal region are stably palmitoylated in PC12 and CV1 cells.5,6 Palmitoylation appears to be required for complete N-glycosylation of Syt1.(6) Apart from Syt12, all Syt isoforms have at least one transmembrane proximal cysteine residue.
Syt1 can be phosphorylated at Thr125 and Thr128 by casein kinase II7,8 (Thr128 of Syt1 in Rattus norvegicus corresponds to Thr129 of Syt1 in Bos taurus identified in ref (8)) and at Thr112 by Ca2+/calmodulin-dependent protein kinase II and protein kinase C(7) as shown in vitro. In vivo, K+-evoked depolarization or treatment with phorbol ester in rat brain synaptosomes and PC12 cells induces an increase in the level of phosphorylation at Thr112.(7)
Here we describe the expression, purification, and biochemical properties of insect cell-expressed full-length mammalian Syt1. In addition, we compare the biochemical properties of insect cell-expressed full-length mammalian Syt1 to those of the still widely used, prokaryotically expressed, Syt1-C2AB fragment. Using mass spectrometry, we have performed the first comprehensive analysis of several post-translational modifications of Syt1. Our results show that insect cell-expressed Syt1 carries some of the previously described post-translational modifications (glycosylation and phosphorylation) along with several new phosphorylation and O-glycosylation sites. Moreover, we have identified a novel post-translational modification of Syt1, tyrosine nitration. Furthermore, we have screened for lipid binding partners of Syt1 and Syt3 using a new large set of lipids. In addition to previously reported interactions with anionic lipids, phosphatidylserine (PS), phosphatidylinositol 4,5-bisphosphate [PtdIns(4,5)P2],9−14 and phosphatidylinositol 3,4,5-trisphosphate [PtdIns(3,4,5)P3],(11) Syt1 also interacts with additional phosphorylated phosphatidylinositides and other anionic lipids. Furthermore, Syt1 interacts with different phosphorylated phosphatidylinositides in a similar fashion, where the lysine rich region on the C2B domain (with minor contributions from Arg398 and Arg399) mediates Ca2+-independent interactions and the C2A, C2B Ca2+-binding loops mediate Ca2+-dependent interactions.
Experimental Procedures
Protein Expression
Full-length rat synaptotagmin 1 (1–421) and full-length Syt1 Gln154Cys, Cys74Ser, Cys75Ser, Cys77Ser, Cys79Ser, Cys82Ser, Cys277Ser coding sequences were cloned into the pvl1393 vector (Pharmingen). For affinity purification, a 3C protease cleavable His10 tag and a 1D4 tag were added at the C-terminus. Spodoptera frugiperda (Sf9) cells (Invitrogen) were transfected with pvl1393 carrying the Syt1 gene and linearized AcNPV DNA (Sapphire Baculovirus DNA, Orbigen) using Cellfectin (Invitrogen). Baculovirus was amplified in Sf9 cells in 10% fetal bovine serum containing SF900-II (Gibco) medium.
Large-scale protein expression was performed by infection of Trichoplusia ni (Hi5) cells in Insect-Xpress medium (Biowhitaker) at a cell density of 2.0 × 106 cells/mL with an infection course of 48 h. Cells were harvested by centrifugation and lysed with an M-110EH microfluidizer processor at 18000 psi (Microfluidics).
Protein Purification
Detergent Screen for Extraction of Syt1 from Insect Cell Membranes
The efficiency of different detergents in extracting Syt1 from insect membranes was screened by adding 1 wt % (g)/v (mL) of a detergent to 1 mL of membranes and solubilizing them for 4 h at 4 °C. Samples were spun at 100000 rpm for 30 min. All detergents were from Anatrace. Sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) (4 to 15%) was performed on the supernatant, containing detergent-solubilized Syt1, and Syt1 was detected by Western blotting with an anti-His antibody. Syt1 was observed in samples solubilized with n-decyl β-d-maltopyranoside (DDM), FOS-10, FOS-14, n-ocytl β-d-glucopyranoside (OG), and HEGA-10. Syt1 was not observed in samples solubilized with CHAPS, CHAPSO, CYGLU-3, and LDAO. For further purification, after membrane purification, Syt1 was extracted from membranes with either FOS-14 or DDM.
Purification
After membrane purification and detergent extraction, Syt1 was subsequently “batch” purified with Ni-NTA resin (Qiagen) at 4 °C, followed by 1D4 antibody affinity purification, in 20 mM Tris (pH 8.0), 0.5 M NaCl, 10 mM CaCl2, 5% glycerol, 1% FOS-14 (or DDM), and protease inhibitors. In general, although the purity after 1D4 affinity purification was higher, the yield was lower, so we frequently skipped this step. From the NiNTA resin, the protein was eluted with 300 mM imidazole (pH 7.5), 500 mM NaCl, and 0.02% DDM, and from the 1D4 antibody column using the 1D4 peptide. Syt1 was further purified by ion exchange chromatography [MonoS resin (GE Healthcare)] and/or size exclusion chromatography [KW-803.5 (Shodex)] to remove the Syt1 fragment of residues 111–421 from the full-length protein. For ion exchange, Syt1 was loaded in 0.1 M NaCl, 25 mM MES (pH 6.0), 0.02% DDM, and 2 mM β-mercaptoethanol. Size exclusion chromatography was conducted in 50 mM sodium phosphate (pH 8.0), 150 mM NaCl, and 0.02% DDM with or without 2 mM β-mercaptoethanol. For experiments other than lipid-overlay assay tags were removed using PreScission Protease (GE Healthcare) in size exclusion chromatography buffer.
Dye Labeling
The mutant full-length Syt1 (1–421, Cys74Ser, Cys75Ser, Cys77Ser, Cys79Ser, Cys82Ser, Cys277Ser, Gln154Cys) fragment was purified in the same way as the wild-type protein, and the purified sample was mixed with Cy5-maleimide (GE Healthcare) in 50 mM sodium phosphate (pH 8.0), 150 mM NaCl, 0.5 mM TCEP, and 0.02% DDM and incubated overnight at 4 °C. Free dye was separated from labeled protein using ion exchange (MonoS). The presence of Cy5 dye on Gln154Cys was confirmed by mass spectrometry. The labeling efficiency, estimated by ultraviolet–visible (UV–vis) dye and protein absorption at respective wavelengths, was ∼55–80%.
Syt1-C2AB and Syt3-C2AB Expression and Purification
R. norvegicus Syt1(96–421), Syt3(292–587), Syt1(96–421) Arg398Glu/Arg399Glu (termed Syt1-C2AB RR), and Syt1-C2AB(96–421) Lys326Glu/Lys327Ala (termed Syt1-C2AB KK) fragments were tagged with biotin, expressed in E. coli as GST fusion proteins, and purified as previously described.(15) Syt1(96–421) Arg398Glu/Arg399Glu and Syt1(96–421) Lys326Glu/Lys327Ala fragments with an N-terminal Avitag were generated by gene synthesis (Geneart, AG) and subcloned into the pGEX6P-1 vector (GE Healthcare). The GST tag was cleaved with PreScission Protease (GE Healthcare) for all experiments. The absence of DNA contamination(16) of purified C2AB fragments was confirmed by the lack of UV–vis absorption at 260 nm.
Circular Dichroism (CD) Analysis
CD data were collected on an Aviv202-01 spectrometer equipped with a thermoelectric unit using a 1 mm path-length cell. Protein samples were at a concentration of 0.2 mg/mL in buffer containing 20 mM potassium phosphate (pH 7.0), 150 mM KCl, and 0.02% dodecyl maltoside (DDM). The protein concentration was determined by absorption at 280 nm. Wavelength scans were measured from 190 to 270 nm every 1 nm with an averaging time of 1 s.
Lipids
Egg phosphatidylcholine (eggPC), dioleoylphosphatidylethanolamine (DOPE), 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (POPE), brain phosphatidylserine (PS), brain sphingomyelin (SM), 1,2-dioleoyl-sn-glycero-3-phospho(1′-myoinositol) (PtdIns), 1,2-dioleoyl-sn-glycero-3-phospho(1′-myoinositol-3′-phosphate) (PtdIns3P), brain phosphatidylinositol 4-phosphate (PtdIns4P), 1,2-dioleoyl-sn-glycero-3-phospho(1′-myoinositol-5′-phosphate) (PtdIns5P), brain phosphatidylinositol 4,5-bisphosphate [PtdIns(4,5)P2], 1,2-dioleoyl-sn-glycero-3-phospho(1′-myoinositol-3′,5′-bisphosphate) [PtdIns(3,5)P2], 1,2-dioleoyl-sn-glycero-3-phospho(1′-myoinositol-3′,4′,5′-trisphosphate) [PtdIns(3,4,5)P3], 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl) (Lissamine Rhodamine PE), 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), and 1-stearoyl-2-oleoyl-sn-glycero-3-phospho-l-serine (SOPS) were purchased from Avanti Polar Lipids as chloroform or chloroform/methanol/water solutions. Dihydrocholesterol was from Sigma-Aldrich, and it was used instead of cholesterol to prevent cholesterol oxidation.
Reconstitution of Full-Length Syt1 and Binding to Supported Lipid Bilayers
Liposomes for Syt1 reconstitution were made from either 100% POPC or an 80:20 POPC/SOPS mixture. Liposomes used to make supported lipid bilayers were made from an 80:20 POPC/SOPS mixture. Liposomes were made by a dry-film method, in which we dried the chloroform solution of lipids under argon while continuously manually rotating a round glass tube to make a thin lipid film. The thin lipid film was further dried overnight under vacuum. Lipid films were rehydrated in 50 mM Hepes (pH 7.0) and 150 mM NaCl at room temperature with intermittent vortexing. An aqueous content dye, 250 mM calcein, was added during rehydration to lipids used for Syt1 reconstitution. Both calcein and calcein-free liposomes were frozen and thawed in liquid nitrogen five times and extruded through a 50 nm pore filter (Avanti Polar Lipids). For reconstitution of full-length Syt1 into calcein-POPC/SOPS liposomes, Cy5-labeled Gln154Cys, Cys74Ser, Cys75Ser, Cys77Ser, Cys79Ser, Cys82Ser, Cys277Ser Syt1 was mixed with liposomes at protein:lipid ratio of 1:4000 and decyl maltoside (Anatrace, Inc.) to a lipid ratio of 1:2. The mixture was incubated for 30 min at 4 °C, followed by 15 min at room temperature, and passed over a 12 cm × 0.5 cm CL4B resin (GE Healthcare) column in detergent-free buffer. Fractions containing proteoliposomes were collected on the basis of the calcein absorption wavelength (495 nm). Protein-free POPC/SOPS supported lipid bilayers were formed on quartz slides by incubation of quartz/glass sample chambers with 7 mg/mL liposomes for 20–30 min at room temperature. Sample chambers were constructed from quartz slides and borosilicate glass coverslips that were cleaned as previously described.(15) The single-molecule/particle images were collected with a prism-based total internal reflection (TIR) fluorescence microscope as previously described.(17) An alternating 488 nm/635 nm laser illumination sequence was used to determine the location of calcein-liposome and Cy5-Syt1 spots. Collected images were corrected for background fluorescence and fluorescent spots localized as previously described.(15) In Figure 2, calcein and Cy5 spots were colocalized and the number of colocalized spots was counted; 75% of supported lipid bilayer-bound calcein-liposomes contained Cy5-Syt1.
Figure 2.

Full-length Syt1 binds PS bilayers in a Ca2+-dependent manner. (A) Binding of full-length Syt1 reconstituted into 100% PC liposomes, fluorescently labeled with the aqueous content dye, calcein, to 80:20 PC/PS supported lipid bilayers is dependent on Ca2+ and Syt1. The left panel shows a cartoon of the experiment. The middle panels show representative images of supported lipid bilayers in the presence and absence of Syt1 where bright spots are calcein-labeled liposomes. The blue lines on top of the images represent the wavelength of the excitation laser (488 nm). The right panel shows the mean number of calcein-bound liposomes bound to the bilayer after a 5 min incubation and after unbound liposomes were washed off with buffer containing the respective amount of Ca2+. Error bars indicate the standard deviation. (B) Binding of fluorescently labeled full-length Syt1 reconstituted into 80:20 PC/PS liposomes to supported lipid bilayers is dependent on Ca2+. Fluorescently labeled Syt1 was reconstituted in 80:20 PC/PS aqueous content labeled liposomes and incubated over 80:20 PC/PS bilayers for 20 min at different Ca2+ concentrations. The left panel shows a cartoon of the experiment. The three middle panels show representative images of supported lipid bilayers in the absence of Ca2+ (left image) and presence of Ca2+ (middle and right images). The lines on top of the images represent the wavelengths of the excitation laser (blue for 488 nm and red for 635 nm). The two images in the presence of Ca2+ show the same field of view that is optically split into two channels to monitor labeled liposomes and labeled Syt1. An example of a colocalized Syt1 and liposome spot is marked in red and blue, respectively. The right panel shows the average number of Syt1-liposomes bound by counting the number of spots colocalized to the supported lipid bilayer imaged after unbound liposomes were washed off with buffer containing the respective amount of Ca2+. Error bars indicate the standard deviation. Note that the concentration of Syt1-liposomes was different from that for the experiments shown in panel A. For both experiments, the number of spots was counted in four different 80 μm × 80 μm areas of the bilayer for two different bilayers.
Lipid Overlay Assay
Membrane Lipid Strips, PIP Strips, and SphingoStrips (Echelon Biosciences) contained 100 pmol of a single lipid species spotted on a hydrophobic membrane. Full-length Syt1(1–421), Syt1-C2AB(96–421), and Syt3-C2AB(292–587) constructs were incubated with Membrane Lipid Strips, PIP Strips, and SphingoStrips (Echelon Biosciences) following the manufacturer’s instructions in an identical buffer containg 50 mM Tris (pH 8.0), 150 mM NaCl, 0.02% dodecyl maltoside, 1% nonfat milk, and either 0.1 mM EGTA or 0.1 mM EGTA and 1.1 mM CaCl2. Binding of Syt to lipids was detected in the following manner. Full-length Syt1 carrying a 1D4 tag was detected with an anti-1D4 antibody followed by an alkaline phosphatase secondary antibody. Alkaline phosphatase was detected with the BCIP/NBT substrate system (Thermo Scientific). Syt1-C2AB(96–421) and Syt3-C2AB(292–587) carrying a biotin tag were detected with streptavidin conjugated to fluorophore AlexaFluor647 (Life Technologies). Streptavidin-AlexaFluor647 fluorescence was imaged using a fluorescence scanner (Typhoon, Amersham). To quantify binding of full-length Syt1, Syt1-C2AB(96–421), and Syt3-C2AB(292–587) constructs to lipids, the chromogenic or fluorescence intensity of lipid spots on Membrane Lipid Strips, PIP Strips, and SphingoStrips was integrated and background subtracted using UN-SCAN-IT (Silk Scientific) gel and blot analysis software.
Biosensor Assay
Site specifically biotinylated Syt1-C2AB and Syt3-C2AB were immobilized on a streptavidin tip, and binding of various liposomes to Syt was assessed using biolayer interferometry. Syt3 immobilized in this configuration was previously found to be functional as determined by its ability to bind to the SNARE complex.(15) Because of the high avidity of the interaction (many Syt1 on the tip with many lipids on the vesicle), the binding is nearly irreversible, and thus, equilibrium has not been reached in these measurements. However, because all interactions are evaluated under identical conditions, such measurements are informative, and similar caveats apply to commonly used pull-down experiments. Liposomes used in the biosensor assay were composed of various phospholipids, cholesterol, and a single anionic lipid species used as a target lipid for assaying its interaction with Syt. Compositions of liposomes in molar ratios are listed in Table 1. The headgroup composition of phospholipids mimicked the ratio reported for total brain extracts,18,19 the exception being that anionic lipids (PS, PtdIns, and phosphorylated phosphatidylinositols) were added at twice the concentration reported for total brain extract and synaptic vesicles because 90–100% of those lipids are located in the inner leaflet of the plasma membrane and the outer leaflet of synaptic vesicles. Liposomes were made by a dry-film method in the following way. All chloroform solutions of lipids were mixed at the molar ratios mentioned above and dried under argon as a thin lipid film on the wall of a round glass tube while the glass tube was continuously manually rotated. The thin lipid film was further dried overnight under vacuum. Thin lipid film was then rehydrated in 20 mM Hepes (pH 7.2), 150 mM KCl, and 0.1 mM EGTA at 40 °C with occasional vortexing. The liposome solution was then frozen and thawed five times in liquid nitrogen and extruded through a 50 nm pore filter (Avestin, Inc.). Lissamine rhodamine absorption was used to determine the concentration of extruded liposomes. The size of the extruded liposomes was checked by dynamic light scattering; all mixtures had radii of approximately 50 nm (data not shown).
Table 1. Lipid Compositions (mole percent) of Liposomes in Figure 5a.
| 0% PS, 0% PtdIns | 14% PS, 0% PtdIns | 2% PS, 0% PtdIns | 0% PS, 2% PtdIns | 0% PS, 2% PtdIns3P | 0% PS, 2% PtdIns4P | 0% PS, 2% PtdIns5P | 0% PS, 2% PtdIns(4,5)P2 | 0% PS, 2% PtdIns(3,5)P2 | 0% PS, 2% PtdIns(3,4,5)P3 | |
|---|---|---|---|---|---|---|---|---|---|---|
| cholesterol | 40 | 40 | 40 | 40 | 40 | 40 | 40 | 40 | 40 | 40 |
| eggPC | 29 | 22 | 28 | 28 | 28 | 28 | 28 | 28 | 28 | 28 |
| DOPE | 13 | 9 | 12 | 12 | 12 | 12 | 12 | 12 | 12 | 12 |
| POPE | 13 | 9 | 12 | 12 | 12 | 12 | 12 | 12 | 12 | 12 |
| SM | 5 | 5 | 5 | 5 | 5 | 5 | 5 | 5 | 5 | 5 |
| lissamine rhodamine PE | 0.06 | 0.06 | 0.06 | 0.06 | 0.06 | 0.06 | 0.06 | 0.06 | 0.06 | 0.06 |
| PS | 0 | 14 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| PtdIns | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 0 |
| PtdIns3P | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 0 |
| PtdIns4P | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 0 |
| PtdIns5P | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 0 |
| PtdIns(4,5)P2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 |
| PtdIns(3,5)P2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 |
| PtdIns(3,4,5)P3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 |
Lipid abbreviations are defined in Experimental Procedures.
An Octet system (ForteBio, Inc.) equipped with streptavidin SA biosensors (ForteBio, Inc.) was used to study the Syt1-C2AB– and Syt3-C2AB–lipid interactions in the absence and presence of Ca2+. N-Terminally biotinylated Syt1-C2AB, Syt1-C2AB KK, Syt1-C2AB RR, and Syt3-C2AB constructs at 2.5 μg/mL in 20 mM Hepes (pH 7.2), 150 mM KCl, 100 μM EGTA, and 0.05% Tween 20 were agitated at 1000 rpm and bound to streptavidin SA biosensors (ForteBio, Inc.) with typical capture levels of 2.0–2.9 nm between different proteins. Syt-loaded biosensors were equilibrated in Tween 20-free buffer. Syt-loaded biosensors were incubated with 5 μM liposomes in the case of Syt3C2AB and 20 μM liposomes in the case of Syt1-C2AB, Syt1-C2AB RR, and Syt1-C2AB KK in 20 mM Hepes (pH 7.2), 150 mM KCl, 100 μM EGTA or 100 μM EGTA, and 1.1 mM Ca2+ at 25 °C. To ensure that binding of liposomes is synaptotagmin specific, 5 μM liposomes were incubated with streptavidin biosensors to which synaptotagmin was not attached, and no binding was observed (Figure 5A). As a control to show that binding of Syt to streptavidin biosensors is specific and mediated by the N-terminal biotin tag, we incubated streptavidin biosensors with 5 mM biotin for 2 h at room temperature prior to the addition of 2.5 μg/mL N-terminally biotinylated Syt1-C2AB under the conditions described above. When the streptavidin biosensors are preblocked with biotin, we observe no binding of N-teminally biotinylated Syt to biosenors (data not shown).
Figure 5.

Anionic lipid binding assay using liposomes. (A) Schematic of the experiment and sensograms of liposome binding to bare streptavidin biosensors in the presence of Ca2+ (left sensogram) and streptavidin biosensors coated with N-terminally biotin-tagged Syt1 in the absence (middle sensogram) and presence (right sensogram) of Ca2+. We measured interactions between C2AB fragments of Syt3, Syt1, Syt1 Arg398Glu/Arg399Glu (Syt1-C2AB RR), and Syt1 Lys326Glu/Lys327Ala (Syt1-C2AB KK) with liposomes containing PC, PE, SM, cholesterol, and a single anionic lipid species {either PS, PtdIns, or various phosphorylated phosphatidylinositols [PtdIns3P, PtdIns4P, PtdIns5P, PtdIns(3,5)P2, PtdIns(4,5)P2, and PtdIns(3,4,5)P3] as indicated in the legend} at ratios similar to those observed in synaptic vesicles18,19 (for the ratios of different lipids in liposomes, see Experimental Procedures and Table 1). The binding levels for the sensograms are in units of nanometers. Similar sensograms were observed for the Syt3-C2AB, Syt1-C2AB KK, and Syt1-C2AB RR fragments (not shown). The solid arrow indicates the time of the addition of liposomes. The dotted arrow indicates the time of the switch to liposome-free buffer. (B–E) Bars represent the mean amount of bound liposomes detected at the same time point for all lipid mixtures and normalized by the level of N-terminally biotin-tagged Syt1-C2AB, Syt1-C2AB KK, Syt1-C2AB RR, and Syt3-C2AB fragments, respectively, captured by streptavidin biosensors. The error bars represent standard deviations obtained from two or three experiments. Comparison of interaction of Syt1-C2AB (B) and Syt3-C2AB (C) with a panel of anionic lipids in the presence and absence of Ca2+. Comparison of the interaction of Syt1-C2AB and Syt1-C2AB KK and Syt1-C2AB RR mutants with the lipids in the absence (D) and presence (E) of Ca2+.
Detection of Nitrated Tyrosines in Synaptic Vesicle Proteins
Frozen brains from 3–4-week-old female Sprague-Dawley rats were obtained from Charles River Laboratories International. Synaptic vesicles were purified as previously described,(20) with an exception that the controlled-pore-glass chromatography step was omitted and the peripheral protein removal step was followed. The P3 fraction described by Hell et al.(20) was used for our experiments. The presence of synaptic vesicles in the P3 fraction was confirmed by enrichment of α-synaptophysin, and the presence of Syt1 and synaptobrevin as detected by Western blotting with their respective antibodies (Synaptic Systems) (data not shown). The diameter of particles in the P3 fraction was ∼43 ± 9 nm as determined by dynamic light scattering (Malvern Instruments) (data not shown), consistent with reported synaptic vesicle size.(20) For detection of nitrated tyrosines of synaptic vesicle proteins, purified synaptic vesicles (P3 fraction) were mixed with 10% (w/v) sodium dodecyl sulfate in 20 mM Hepes (pH 7.2), 100 mM KCl, and 0.1 mM EGTA, incubated at 40 °C for 30 min, boiled, loaded onto a 4 to 15% SDS–PAGE gel, and blotted on PVDF membranes. Proteins with nitrated tyrosines were detected with primary monoclonal anti-nitrotyrosine rabbit IgG (Life Technologies) and horseradish peroxidase-linked secondary (Jackson ImmunoResearch Laboratories) antibodies and detected on the film using enhanced chemiluminescence (GE Healthcare). For the detection of Syt1, anti-nitrotyrosine primary and secondary antibodies were stripped using a Western blot stripping buffer (Thermo Scientific) and blots were restained with primary monoclonal anti-Syt1 (BD Transduction Laboratories) and horseradish peroxidase-linked secondary (Jackson ImmunoResearch Laboratories) antibodies and detected on the film using enhanced chemiluminescence (GE Healthcare). Nitrotyrosine bovine serum albumin (Cayman Chemicals) was used as a positive control for anti-nitrotyrosine antibody and antibody stripping, and as a negative control for the anti-Syt1 antibody. The location of the SDS–PAGE markers (Rainbow full range, GE Healthcare) on the film relative to the developed bands was manually marked with a pen.
Mass Spectrometry
Sample Preparation
The SDS–PAGE gel band of insect cell-expressed and -purified full-length Syt1 was Coomassie-stained (Bio-Rad) and destained in 200 μL of 25 mM ammonium bicarbonate in 50% (v/v) acetonitrile (ACN) for 15 min, and then 200 μL of 100% ACN was applied for 15 min at room temperature. Dithiothreitol (DTT) was added to a final concentration of 10 mM and incubated at 65 °C for 30 min to reduce the disulfide bonds. The reduced cysteines were then alkylated with the addition of iodoacetamide (IAA) at a final concentration of 20 mM and incubated at room temperature in the dark for 30 min. The iodoacetamide was then removed, and washes were performed with 200 μL of distilled water followed by addition of 100 μL of ACN. Then ACN was removed, and 50 μL of the 0.01 μg/μL trypsin solution was added to the diced gel band, allowed to rehydrate at 4 °C for 30 min, and then incubated at 37 °C overnight. The tryptic mixtures were acidified with formic acid (FA) to a final concentration of 1%. Peptides were extracted three times from the gel plugs using 50% ACN and 1% FA, concentrated with a SpeedVac to a desired volume (∼18 μL), and subjected to liquid chromatography–tandem mass spectrometry analysis.
Liquid Chromatography–Tandem Mass Spectrometry (LC–MS/MS)
Nanoflow reverse phase LC–MS/MS was performed using a capillary HPLC system (Agilent 1200, Agilent Technologies) coupled with a linear ion trap mass spectrometer LTQ-FT Ultra Hybrid ion cyclotron resonance mass spectrometer (ThermoFisher) through a nanoelectrospray ionization source built in house. Eight microliters of the tryptic peptides was preconcentrated and desalted onto a ZORBAX 300SB-C18 trap column [5 μm (inside diameter) × 5 mm, Agilent Technologies] with 5% ACN and 0.1% FA at a flow rate of 15 μL/min for 5 min. The separation of the tryptic peptides was performed on a C18 reverse phase column [75 μm (inside diameter) × 360 μm (outside diameter) × 100 mm (length)] packed in house with a 4 μm 100 Å pore size C18 reversed phase stationary phase (Synergy, Phenomenex) kept at a constant 40 °C using a column heater built in house at a flow rate of 380 nL/min. The mobile phases consisted of 5% acetonitrile with 0.1% formic acid (A) and 95% acetonitrile with 0.1% formic acid (B). A 90 min linear gradient from 18 to 35% B was used. Data were acquired using the Xcalibur (version 2.0.6) software supplied with the instrument. The LC runs were monitored in positive ion mode by sequentially recording survey MS scans (m/z 400–2000), in the ICR cell, while three MS2 were obtained in the ion trap via CID for the most intense ions.
Database Searching and Protein Identification
MS/MS spectra were extracted from raw data files and converted into mgf files using PAVA (University of California, San Francisco, CA). Mascot version 2.2 (Matrix Science Inc., London, U.K.) and X!Tandem(21) were used to perform database searches against the human subset SwissProt database of the extracted MS/MS data. The peptide tolerance was set at ±10 ppm with the MS/MS tolerance set at ±0.6 Da. Trypsin specificity was used allowing for one missed cleavage. The modifications of Met oxidation, protein N-terminal acetylation, nitrotyrosine, and peptide N-terminal pyroglutamic acid formation were allowed for, and Cys carbamidomethylation was set as a fixed modification. Search results can be viewed in the gpmDB (http://rat.thegpm.org) using accession code GPM64500003242.
Results
Insect Cell-Expressed Full-Length Synaptotagmin 1
Full-length Syt1 has been expressed in various mammalian cell lines for cell-based assays, but not in quantities amenable for purification and biochemical studies. To obtain the post-translational modifications found in natively expressed Syt1, we expressed Syt1 in a eukaryotic expression system using insect cells. After purification with affinity chromatography (utilizing His and 1D4 tags), a SDS–PAGE gel showed that insect cell-expressed Syt1 contains three bands: a full-length construct with an apparent molecular mass of ∼65 kDa (the sequence-predicted molecular mass is 47.4 kDa) that constituted the majority of the protein, a minor degradation product of ∼40 kDa, and a small amount of SDS-resistant dimer (running slightly above the 105 kDa molecular mass marker) (Figure 1A). N-Terminal sequencing of the 65 and ∼40 kDa SDS–PAGE bands showed that the former band starts with residue Val2 while the latter band starts with residue Lys111. Western blots against the C-terminal tag confirmed that both species end at residue Lys421 (data not shown). Thus, the degradation product corresponds to the C2AB fragment. The observation of these three SDS–PAGE bands (the main product at ∼65 kDa, a small amount of SDS-resistant dimer, and the C2AB fragment) is consistent with the SDS–PAGE profile of Syt1 purified from Rattus synaptic vesicles.(1) The C2AB degradation fragment can be separated from the full-length species using either ion exchange or size exclusion chromatography (Figure 3D).
Figure 1.

Purity and circular dichroism spectra of insect cell-expressed and -purified Syt1. (A) Coomassie-stained 4 to 15% SDS–PAGE gel of affinity-purified, Hi5-expressed, full-length Syt1. (B) Circular dichroism spectra of full-length Syt1 in the presence and absence of Ca2+. (C) Circular dichroism spectra of the E. coli-expressed Syt1-C2AB fragment in the presence and absence of Ca2+.
Figure 3.

Post-translational modifications of Syt1. (A) Shown is the sequence of Syt1 from R. norvegicus along with Syt1 post-translational modifications. The transmembrane domain is colored cyan (residues 58–79), the C2A domain yellow (residues 143–264), and the C2B domain gray (residues 274–377). Cysteine residues are colored orange (Cys74, Cys75, Cys77, Cys79, Cys82, and Cys277). Cysteines predicted to be palmitoylated are marked with wavy lines (Cys74, Cys75, Cys77, Cys79, and Cys82). Residues reported in the literature as glycosylated are colored brown (Thr15, Thr16, and Asn24). Residues reported in the literature as phoshorylated (Thr112, Thr127, and Thr128) are colored magenta. Post-translational modifications identified here by mass spectrometry are marked by black squares: Thr26 (O-glycosylated, colored brown), Ser30 and Thr128 (phoshorylated, colored magenta), and Tyr151, Tyr216, Tyr229, Tyr311, Tyr364, and Tyr380 (nitrated, colored green). (B) Local sequence alignments of R. norvegicus synaptotagmin isoforms for residues carrying post-translational modifications identified here by mass spectrometry: tyrosine nitration, phosphorylation, and O-glycosylation, as indicated on top of the sequences. The post-translationally modified residue numbers correspond to those of Syt1. (C) Detection of proteins with nitrated tyrosines in synaptic vesicles. Shown is a representative Western blot of rat synaptic vesicles (SV). A 4 to 15% SDS–PAGE gel was blotted first with primary anti-nitrotyrosine antibody (left) and then with primary anti-synaptotagmin 1 antibody (right). SDS–PAGE molecular mass markers (shown as ticks in three lanes in each gel) have been marked on both films with a pen on the basis of the locations of the SDS–PAGE molecular mass markers on the Western blot membrane. Arrows point to the location of Syt1. Nitrotyrosine BSA (nBSA) was used as a positive and negative control. (D) SDS–PAGE gel (4 to 15%) stained with Commassie dye of insect cell-expressed and -purified Syt1 treated with PNGaseF or not treated, as indicated. Right and left lanes contained molecular mass markers.
Full-Length Syt1 Is Folded
To determine whether insect cell-expressed and -purified Syt1 is folded, we measured its circular dichroism spectrum and compared it to that of the prokaryotically expressed Syt1-C2AB fragment (Figure 1B,C). The spectrum of the C2AB fragment has a minimum at 218 nm indicative of a mainly β-sheet rich fold (Figure 1C), as previously reported.(22) The circular dichroism spectrum of full-length Syt1 (Figure 1B) is indicative of a folded protein with an increased α-helical contribution (with minima at 208 and 222 nm) compared to that of the C2AB fragment. This increase in α-helical content is expected as full-length Syt1 contains a transmembrane α-helix. In addition, the luminal domain of Syt1, which is highly homologous to that of Syt2, probably encompasses an additional α-helical segment, as suggested by the crystal structure of the Syt2–botulinum neurotoxin B complex.(23) While it is unknown if the luminal segment remains α-helical in the absence of botulinum neurotoxin B, it is nevertheless predicted to be α-helical by secondary structure prediction algorithms.
The E. coli-expressed Syt1-C2AB fragment binds anionic lipids, phosphatidylserine (PS) and phosphatidylinositol 4,5-bisphosphate [PtdIns(4,5)P2], by simultaneously coordinating Ca2+ to Ca2+-binding loops of the C2A and C2B domains and to PS and PtdIns(4,5)P2.10,12,14 Thus, to further confirm that insect cell-expressed Syt1 is functionally folded, we tested its ability to bind PS-containing lipid membranes in a Ca2+-dependent manner. We reconstituted full-length Syt1 into liposomes (here termed Syt1-liposomes) with or without PS and assayed the Ca2+ dependence of binding of Syt1-liposomes to PS-containing supported lipid bilayers by single-molecule fluorescence microscopy (Figure 2). Liposomes were fluorescently labeled by entrapment of the water-soluble dye, calcein, inside the liposomes, and the number of fluorescent liposomes bound to the supported lipid bilayer was counted. To confirm that binding of Syt1-liposomes to the supported lipid bilayers is mediated by Syt1, we removed the cytoplasmic domain of Syt1 from the Syt1-liposomes via digestion with the chymotrypsin protease (Figure 2A, right bar chart). As a further conformation that binding of Syt1-liposomes to Syt1-free supported lipid bilayers is mediated by Syt1, we labeled Syt1 with a fluorescent dye at a specific site and counted the number of optically colocalized Syt1 and liposome particles bound to supported lipid bilayers (Figure 2B). Thus, binding of liposomes with and without PS to PS-containing supported lipid bilayers is dependent on the presence of both Syt1 and Ca2+.
Identification of Post-Translational Modifications of Insect Cell-Expressed Mammalian Syt1
Syt1 expressed in mammalian cell lines or immunoprecipitated from synaptic vesicles has several post-translational modifications.1−3,5,7,8,11,20 We have analyzed post-translational modifications of Hi5 insect cell-expressed Syt1 to evaluate how closely the pattern of post-translational modifications match the ones reported for mammalian cells. Although we obtained good coverage of peptides (70–86% of the Syt1 sequence), we were unable to observe the transmembrane domain (residues 52–94) that is predicted to have several palmitoylation sites (Cys74, Cys75, Cys77, Cys79, and Cys82). Our mass spectrometry analysis revealed both previously reported and new post-translational modifications (Figure 3A).
Tyrosine Nitration
We detected nitration in 6 of 11 surface accessible tyrosine residues, three in the C2A domain (Tyr151, Tyr216, and Tyr229) and three in the C2B domain (Tyr311, Tyr364, and Tyr380) (Figure 3B). Tyrosine nitration is a new post-translational modification of Syt1 that has not been previously reported for any synaptotagmin isoform. Integration of the peak intensity for the individual Syt1 peptides suggests a stoichiometry for the nitrotyrosine modifications of 1–10% depending on the site, with the exception of Tyr151, which appears to be ∼100% modified because an unmodified form was not identified. The estimated 1–10% modification for the other sites is a lower limit as the modification is likely to decrease the ionization potential of the respective peptides. Despite the low level of modification of individual sites within the protein population, low-level modifications can have significant signaling and other bioactive effects on a system.24−26
To test whether tyrosine nitration is present in vivo, we purified Rattus synaptic vesicles, performed SDS–PAGE, and performed Western blotting with an anti-nitrotyrosine antibody. Although synaptic vesicles contain many different proteins,(19) we observed five distinct nitrated tyrosine bands (Figure 3C) whose identities are unknown. However, the location of one of these bands is consistent with Syt1 (Figure 3C), suggesting that tyrosine nitration reported here might occur in vivo. Sequence alignment showed that Tyr151 is conserved among all Rattus synaptotagmin isoforms, Tyr216 is conserved in all but Syt3, Syt8, Syt12, and Syt13 isoforms, Tyr229 is conserved in Syt1–Syt3, Syt5, Syt9 and Syt10, Tyr311 is conserved in all isoforms apart from Syt12 and Syt13, Tyr364 is conserved in Syt1–Syt3, Syt5, Syt6, Syt9, and Syt10, and Tyr380 is present in only Syt1 (Figure 3B). Interestingly, R. norvegicus Syt1 Tyr311 corresponds to the same residue as the tyrosine characterized in Drosophila melanogaster’s Syt AD3 mutant (D. melanogaster Tyr364Asn).(27) Mutation of this Syt1 residue, Tyr311Asn, has an effect on both Syt1-mediated Ca2+-triggered exocytosis(27) and Syt1 endocytosis.(28)
Syt1 Tyr364 is a part of the Ca2+-binding region of the C2B domain and is only partially conserved (Figure 3B). Although Tyr364 participates in Ca2+ coordination via its backbone oxygen, it is possible that nitration of its side chain will influence the conformation of the Ca2+-binding loops. Sequence comparison of Syt isoforms (Figure 3B) shows that Syt4, Syt8, Syt11–Syt13, and Syt15 do not have a tyrosine residue at this position. Interestingly, these same isoforms have been either shown or predicted not to bind Ca2+ via their Ca2+-binding loops.29,30 Furthermore, the same isoforms bind anionic lipids and the syntaxin–SNAP-25 binary complex in a Ca2+-independent manner and inhibit SNARE-mediated, Ca2+-triggered lipid mixing.(30)
Phosphorylation
We detected phosphorylation of the previously reported Thr128 site(8) and of a new Ser30 site (Figure 3A). Ser30 is the first reported phosphorylation site in the Syt1 luminal domain. However, we did not observe the previously characterized phosphorylation sites, Thr125 (detected in vitro after reaction with casein kinase II) and Thr112 (detected in vitro after reaction with Ca2+/calmodulin-dependent protein kinase II and protein kinase C).(7) Phosphorylation sites Thr112, Thr125, Thr128, and Ser30 are not conserved among different Rattus synaptotagmin isoforms (Figure 3B) and thus are likely isoform specific. The lack of detected phosphorylation of Thr112 and Thr125 is likely due to the lack of required kinases in insect cells and/or a low fraction of insect cell-expressed Syt1 phosphorylated at these sites.
Glycosylation
We detected O-glycosylation of Thr26 (Figure 3A), which is a new O-glycosylation site. We did not detect O-glycosylation at the previously reported sites, Thr15 and Thr16,(3) which is not surprising because O-glycosylation of Thr15 and Thr16 requires the presence of synaptobrevin.(3) Analogous to the previously reported O-glycosylation sites Thr15 and Thr16 and N-glycosylation site Asn24, the newly detected O-glycosylation site Thr26 is not conserved among Syt isoforms (Figure 3B). Our mass spectrometry data did not show N-glycosylation of the previously reported Asn24 site that was detected for Syt1 obtained from synaptic vesicles, PC12 cells, and COS-7 cells, with N-glycan complexity varying between expression sources.1−3 The presence of some form of N-glycans on Hi5 insect cell-expressed Syt1 cannot be excluded because we have not used mass spectrometry methods that would allow detection of complex N-glycans. To test whether N-glycosylation is present in Hi5 insect cell-expressed Syt1, we treated purified Syt1 with PGNase F (Figure 3D). Purified Syt1 shows three bands in the ∼65 kDa region, and only the faint top band disappears after incubation with PGNase F (Figure 3D), suggesting that a small fraction of Syt1 could be N-glycosylated. Interestingly, analogous to the observed differences in N-glycosylation of Syt1 among different mammalian expression sources,1−3 the level of N-glycosylation of Syt1 in insect cells also varies with the expression source: for Sf9 cell expression, approximately half of Syt1 is N-glycosylated (compare Figure S6C and Figure S6D of ref (31)). Furthermore, differences in glycosylation pathways between insect and mammalian cell lines have been previously described.(32)
Comparison of the Lipid Binding Profile of Insect Cell-Expressed Syt1 and E. coli-Expressed Syt1-C2AB and Syt3-C2AB Fragments
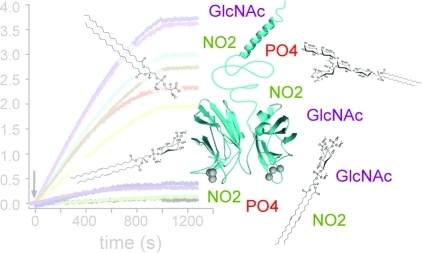
Synaptotagmin isoforms Syt1 and Syt3 bind anionic lipids, phosphatidylserine (PS) and phosphatidylinositol 4,5-bisphosphate [PtdIns(4,5)P2], in a Ca2+-dependent manner via the Ca2+-binding loops of the C2 domains.10,12,14 In addition, the Syt1-C2AB fragment binds PtdIns(4,5)P2 in a Ca2+-independent manner via its lysine rich region of the C2B domain.10,33,34 Both PS and PtdIns(4,5)P2 are enriched in the synaptic plasma membrane and are therefore poised to interact with Syt1 and participate in synaptic vesicle–plasma membrane interaction and fusion. In light of reports about the role of Syt1 in synaptic vesicle35,36 and Syt3 in multivesicular body(37) endocytosis and recycling, processes in which a different set of anionic lipids is likely involved,(38) we screened for other potential lipid binding partners of synaptotagmin isoforms Syt1 and Syt3 (Figure 4). In addition, to determine if the previously uncharacterized full-length Syt1 binds lipids different from the isolated C2AB fragment (residues 96–421), we compared lipid binding profiles of insect cell-expressed full-length Syt1 and E. coli-expressed Syt1-C2AB and Syt3-C2AB fragments using two different lipid binding assays, the lipid overlay assay (Figure 4) and the biosensor assay (Figure 5). Using a lipid overlay assay, an array of single-lipid species was simultaneously tested for interaction with a protein of interest (Figure 4A), thus allowing rapid screening of protein–lipid interaction with a large number of different classes of lipids. We tested the interaction of full-length Syt1 and Syt1-C2AB and Syt3-C2AB fragments with 26 lipids from different lipid classes (sterols, sphingolipids, and phospholipids) in the presence and absence of Ca2+. Using a biosensor assay, a protein of interest tethered to an array of biosensors was simultaneously assayed for its interaction with an array of liposomes containing a single target lipid species and lipids found in plasma and synaptic vesicle membranes, that is, lipids that are the in vivo lipid membrane milieu of Syt (Figure 5A).
Figure 4.

Lipid binding screen using a lipid overlay assay. (A) Schematic of the lipid overlay assay (left). Full-length Syt1 (cyan) is shown bound to a lipid spot composed of a single lipid species (gray). The right panel shows a representative example of a lipid strip from a lipid overlay assay showing the lipid binding profile of the biotin-tagged Syt1-C2AB fragment (for full names of lipid acronyms, see below). Each dark spot on the strip corresponds to a Syt1-C2AB fragment bound to a single lipid species. (B) Lipid binding profile of full-length Syt1 and Syt1-C2AB and Syt3-C2AB fragments. After incubation with Syt in the presence (gray bars) or absence (striped white bars) of Ca2+, chromogenic or fluorescence intensities of all spots on Lipid Strips were integrated and corrected for background intensities. The shown intensities are in arbitrary units (AU). The reproducibility of lipid binding was tested for a subset of lipids: error bars represent standard deviations from means of two or three independent experiments. Abbreviations: S1P, sphingosine 1-phosphate; SM, sphingomyelin; SPC, sphingosylphosphorylcholine; Sulfatide, 3-sulfogalactosyl ceramide; Psychosine, galactosylsphingosine; GM1, monosialoganglioside; GD3, disialoganglioside; PtdIns, phosphatidylinositol; PtdIns3P, phosphatidylinositol 3-phosphate; PtdIns4P, phosphatidylinositol 4-phosphate; PtdIns5P, phosphatidylinositol 5-phosphate; PtdIns(3,4)P2, phosphatidylinositol 3,4-bisphosphate; PtdIns(3,5)P2, phosphatidylinositol 3,5-bisphosphate; PtdIns(4,5)P2, phosphatidylinositol 4,5-bisphosphate; PtdIns(3,4,5)P3, phosphatidylinositol 3,4,5-trisphosphate; PS, phosphatidylserine; PA, phosphatidic acid; LPA, lysophosphatidic acid; PC, phosphatidylcholine; LPC, lysophosphatidylcholine; PE, phosphatidylethanolamine.
The lipid overlay assay showed that, in addition to the previously reported Ca2+-dependent binding of Syt1 to anionic lipids PS and PtdIn(4,5)P2, both full-length insect cell-expressed Syt1 and the E. coli-expressed Syt1-C2AB fragment bind to PtdIns3P, PtdIns4P, PtdIns5P, PtdIns(3,5)P2, and sulfatide in a Ca2+-dependent fashion (Figure 4B). The main differences in anionic lipid biding between the Syt1-C2AB fragment and full-length Syt1 are in the Ca2+-dependent binding of full-length Syt1 to gangliosides (GM1 and GD3) and PtdIns(3,4,5)P3. Thus, interaction of full-length Syt1 with GM1, GD3, and PtdIns(3,4,5)P3 lipids might occur through Syt1’s luminal, transmembrane, and/or membrane proximal regions, through the linker (residues 69–95), or it might be due to the post-translational modifications in the C2AB domain. The amount of Syt1 bound to PtdIns3P, PtdIns4P, and PtdIns5P in the presence of Ca2+ using the lipid overlay assay exceeded the amount bound to PS and PtdIns(4,5)P2.
We compared the lipid binding profile of the Syt1-C2AB fragment with that of Syt3; Syt3's physiological function is yet unknown, but Syt3 shares many biochemical properties15,30,39 with Syt1. With the exception of sulfatide, the C2AB fragments of Syt1 and Syt3 interact with the same lipids, although the extent of binding differs in several cases (Figure 4B). The Syt3-C2AB fragment binds both PtdIns3P and PtdIns5P in a Ca2+-dependent manner, but less well than Syt1.
Because the lipid overlay assay was performed at a single time point and at a single concentration of lipids, it is uncertain whether Syt–lipid interactions have reached equilibrium and if the observed difference in the extent of binding of Syt to different anions has different affinities or different on or off rates. In addition, the observed extent of binding of Syt to various lipids can be due to the spatial arrangement of lipid charges in this assay, which is likely different than the one in liposomes composed of multiple lipid species. For the lipid overlay assay, patches of lipid contain 100% of the lipid species of interest (Figure 4A), which results in dense packing of lipids and, consequently, dense packing of lipid charges. This spatial arrangement of charge is likely different from that in liposomes composed of multiple lipid species. Liposomes usually contain a few percent of anionic lipids that, in the absence of liquid–liquid or liquid–solid phase separation, diffuse in the plane of the bilayer and mix with other lipids. Consequently, the local concentration, in the absence of phase separation, of any individual lipid is typically relatively low, and in the case of charged lipids, the charges are likely dispersed.
To compare how the lipid binding profile shown in Figure 4 compares to a situation in which anionic charges are dispersed and present at physiological concentrations in liposomes containing other lipids, we repeated the lipid binding assay using liposomes (for a subset of anionic lipids identified in Figure 4). Anionic lipids, PS, PtdIns, and various phosphorylated PtdIns’s at physiologically comparable concentrations18,19 were mixed with other lipids at ratios and identities corresponding to the composition of synaptic vesicles18,19 and Table 1. The C2AB fragments of Syt1 and Syt3 were tethered to the biosensors in a site specific manner, and binding of liposomes to the biosensors in the presence and absence of Ca2+ was evaluated at a set time point. Syt1 and Syt3-C2AB domains interact with the same lipids as in the lipid overlay assay in the presence of Ca2+. The two lipid binding assays mostly differ in detection of Ca2+-independent interactions, especially in the case of PtdInsP2s and PtdInsP3 (Figure 5B,C) where binding to these lipids is prominent when they are packed less tightly in the context of liposomes.
We tested which previously identified lipid biding regions of Syt1 [the Ca2+-binding loops, the Lys rich region (residues 324–327) of the C2B domain, or the Arg398 and Arg399 residues of the C2B domain33,40] are involved in binding to the lipids used in our screen. We compared PS, PtdIns, PtdInsPs, PtdInsP2s, and PtdInsP3 binding profiles for the Syt1-C2AB fragment with the lipid biding profiles of the Syt1-C2AB Lys326Glu/Lys327Ala and Arg398Glu/Arg399Glu mutants (Figure 5D,E). In the absence of Ca2+, we detected binding of wild-type protein and both mutants only to phosphorylated PtdIns and not PS (Figure 5D). However, in the absence of Ca2+, the Lys326Glu/Lys327Ala mutant binds phosphorylated PtdIns's less well than the wild type while the Arg398Glu/Arg399Glu mutant binds them to the same degree or slightly better. In the presence of Ca2+, the wild-type protein and both mutants bind to both PS and phosphorylated PtdIns's with the exception of PtdIns(3,4,5)P3. Furthermore, both the Lys326Glu/Lys327Ala and Arg398Glu/Arg399Glu mutants bind PS, PtdInsP, and PtdInsP2 as well as the wild type (Figure 5E). Thus, areas other than the Ca2+-binding loops are involved in Ca2+-independent binding of Syt1 and the phosphorylated phosphatidylinositides used in our screen (Figure 5D), while binding in the presence of Ca2+ to PS and phosphorylated phosphatidylinositides is predominantly mediated through Ca2+-binding loops (Figure 5E). Both observations are consistent with previously reported interactions of the Syt1-C2AB fragment with PtdIns(4,5)P2.10,13,34
Discussion
We showed that mammalian full-length Syt1 can be expressed and purified from insect cells and performed an extensive characterization of the post-translational modifications and lipid binding properties of the purified protein. We found both previously described and several new post-translational modifications and lipid binding partners. Syt1 binds anionic phospholipids in a Ca2+-dependent manner by coordinating Ca2+ between its Ca2+-binding loops and anionic phospholipid headgroups.(40) The two anionic phospholipids with which Syt1 has been previously shown to interact in a Ca2+-dependent manner, PS and PtdIns(4,5)P2,10,11 are both enriched in the inner leaflet of the synaptic plasma membrane. Syt1 has also been reported to interact in a Ca2+-independent manner with PtdIns(3,4,5)P3,(11) a lipid also enriched in the plasma membrane.(38)
We performed lipid binding assays in two contexts: one in which single lipid species were tightly packed and the other in which anionic lipids were dispersed among other lipids. Tight packing might be similar to a situation in which lipids would be located in lipid domains within cell membranes, while the latter lipid arrangement is similar to a situation in which anionic lipids are spaced far from one another. Interestingly, in both settings, binding of Syt1 to several phosphorylated phosphatidylinositides is stronger than binding to PS or PtdIns(4,5)P2. Phosphatidylinositol (PtdIns) and its phosphorylated derivatives [PtdIns3P, PtdIns4P, PtdIns5P, PtdIns(3,4)P2, PtdIns(3,5)P2, PtdIns(4,5)P2, and PtdIns(3,4,5)P3] comprise ∼10% of the total cell lipids. Their enrichment in cell organelles is highly specific and dynamic and is maintained by phosphorylation of inositol at positions −3, −4, and/or −5 by different classes of PtdIns kinases.(38) PtdIns(4,5)P2 and PtdIns(3,4,5)P3 have been shown to be enriched in the plasma membrane; PtdIns4P is enriched in the Golgi but can also be found in the plasma membrane, while PtdIns3P and PtdIns(3,5)P2 are enriched in early and late endosomes where they play a role in trafficking of these organelles.(38)
Binding of Syt1 to phosphorylated PtdIns enriched in the plasma membrane [PtdIns4P, PtdIns(3,4)P2, PtdIns(4,5)P2, and PtdIns(3,4,5)P3] likely reflects Syt1’s role in synaptic vesicle exocytosis. Binding to non-plasma membrane PtdIns's [PtdIns3P, PtdIns5P, PtdIns(3,4)P2, and PtdIns(3,5)P2] reported here may play a role in synapototagmin-mediated synaptic vesicle recycling. Syt1, in addition to being the primary Ca2+ sensor for fast synchronous neurotransmitter release, is also necessary for endocytosis of synaptic vesicles.(35) The role of synaptotagmin in endocytosis seems to be conserved across species because it is observed in plants,(41) nematodes,(42) insects,(43) and mammals.(35) The mechanism of Syt1-mediated endocytosis is poorly understood; it is sensitive to Ca2+ concentration(35) and thought to involve AP2–clathrin interactions(44) and/or AP2-independent interactions involving Syt1’s C-terminal domain.(28) While PtdIns(4,5)P2 is required for the formation of the clathrin coat, pinching of the recycled vesicle from the plasma membrane requires destruction of PtdIns(4,5)P2.(45) Thus, it is possible that Syt1’s association with endosomal phosphorylated PtdIns plays a role in the synaptic vesicle recycling mechanism.
Syt1 and Syt3 C2 domains share many biochemical properties with regard to the interaction with SNARE proteins, PS-containing lipid membranes, SNARE-mediated lipid mixing, and membrane bending.15,30,39,46 However, Syt3 is not involved in fast neurotransmitter release,(47) and its role in neurons is currently unknown. It is therefore important to expand the biochemical characterization of the Syt3-C2AB fragment and detect differences between lipid interactions of Syt1 and Syt3. Similar to those with Syt1, interactions of the Syt3-C2AB fragment with phosphorylated PtdIns likely point to its role in vesicle recycling. As a case in point, Syt3 is the only synaptotagmin isoform found in T cells where it is localized to multivesicular bodies and is reported to control recycling of CXCR4 through multivesicular bodies, thus controlling chemokine-induced T cell migration.(37) Interestingly, we find (Figure 5D,E) that the mode of interaction of the Syt1-C2AB fragment with phosphorylated PtdIns is conserved: in the absence of Ca2+, the primary site of interaction is the lysine rich region in the C2B domain (Lys324–Lys327) with a minor contribution from Arg398 and Arg399, while in the presence of Ca2+, the Ca2+-binding loops seem to primarily interact with singly and doubly phosphorylated PtdIns. Our data are consistent with previously characterized interaction of Syt1 with PtdIns(4,5)P2.10,13,34
Using a lipid overlay assay, we observed an interaction of full-length Syt1 but not of the Syt1-C2AB fragment with the gangliosides used in our screen, GM1 and GD3, suggesting that either the luminal region, the transmembrane domain, residues 69–95 of the unstructured linker, or post-translational modifications of the C2AB domain mediate this interaction. Although direct interaction between Syt1 and gangliosides has not been previously reported, the Syt1–ganglioside interaction is physiologically relevant because botulinum neurotoxin B (BoNT/B) gains entry into cells by binding Syt1 only in the presence of gangliosides.(48) Although gangliosides might be interacting with several regions on Syt1, the interaction is likely mediated via Syt1 luminal and/or transmembrane domains on the basis of the following. First, the Syt1-BoNT/B binding site is located on the luminal/transmembrane domain of Syt1, and gangliosides are necessary for that interaction.23,48 Second, the luminal domain of Syt1 faces the same membrane leaflet in which gangliosides are preferentially located (extracytoplasmic leaflet of the plasma membrane).
We have also identified an interaction of Syt1 with sulfatides. Sulfatides are highly enriched in myelin sheets and are not abundant in other tissues. During myelogenesis in oligodendrocytes, Syt7,(49) not Syt1, is a likely Ca2+ sensor during lysosomal exocytosis and endocytic recycling of the oligodendroglial plasma membrane. Our lipid binding assay shows that Syt1, not Syt3, binds sulfatide in a Ca2+-dependent manner. Because of the different localization of sulfatides relative to Syt1, sulfatide binding to Syt1 likely is not physiologically relevant and reflects charge architecture requirements for Syt1–anionic lipid binding.
We found that Syt1 can be nitrated at 6 of 11 surface-exposed tyrosine residues, three in the C2A domain (Tyr151, Tyr216, and Tyr229) and three in the C2B domain (Tyr311, Tyr364, and Tyr380), and that Syt might contain nitrated tyrosines in vivo. Tyrosine nitration has not been previously reported for any synaptotagmin isoform, while other proteins involved in synaptic vesicle fusion, syntaxin1A,(50) NSF,(51) and potentially SNAP25,50,52,53 but not synaptobrevin 2,(50) have been found to be S-nitrosylated.
Nitration and nitrosylation play a role in modulation of synaptic transmission/exocytosis under both normal and pathological conditions. Nitric oxide (NO) modulates long-term potentiation (LTP), a mechanism thought to be responsible for learning and memory formation in the brain, by acting as a retrograde secondary messenger from the postsynaptic to presynaptic neuron. Its role is still unclear because some studies suggest that NO’s presence is required for the induction of LTP54,55 while other reports suggest that its presence reduces the magnitude of LTP.(56) In addition to the potential role of nitric oxide in LTP, increased levels of protein nitration and nitrosylation have been observed in brain tissue from patients with neurodegenerative diseases (Alzheimer's and Parkinson’s) as well as diabetes. The molecular mechanism by which synaptic protein nitration and nitrosylation are connected with LTP and neurodegenerative diseases is unknown. Interestingly, S-nitrosylation of syntaxin at Cys145 disrupts its interaction with Munc18, but not with its SNARE complex partners, SNAP25 and synaptobrevin.53,57 Treatment with NO slightly increases the extent of formation of the SNARE complex both in vitro and in rat hippocampal synaptosomes,(57) while NSF nitrosylation has an inhibitory effect on SNARE disassembly and exocytosis.(51)
Tyr311 that we found to be nitrated is located close to the lysine rich region of the C2B domain (residues 324–327) that has been implicated in interaction with phosphorylated inositides.10,13,33,34 In D. melanogaster’s Syt1 (Tyr364Asn in D. melanogaster), the Tyr311Asn mutation decreases the amount of exocytosis at Drosophila’s neuromuscular junction. The decrease in the amount of exocytosis may be caused by affecting oligomerization of Syt,58,59 Syt1’s Ca2+ affinity,(60) and/or the Syt1–syntxin/SNAP-25 interaction.(61) In addition to its effect on Ca2+-triggered exocytosis and Syt1–syntaxin/SNAP-25 interaction, the Tyr311Asn mutant partially inhibits Syt1 endocytosis in PC12 cells;(28) thus, nitration of Tyr311 might have an effect on synaptic vesicle recycling.
The second nitrated residue in C2B, Tyr364, is a part of the Ca2+-binding motif and is partially conserved among synaptotagmin isoforms (Figure 3B). Nitration of the Tyr364 side chain could potentially change the conformation of the Ca2+-binding loop and alter Ca2+ binding. Tyr364 is located between Asp363 and Asp365, and mutation of these residues (corresponding to D. melanogaster Asp416Asn and Asp418Asn) abolished C2B domain-mediated Ca2+ triggering of synchronous synaptic vesicle fusion.(62) Sequence comparison of Syt isoforms (Figure 3B) shows that the same synaptotagmin isoforms (Syt4, Syt8, Syt11–Syt13, and Syt15) that do not have a tyrosine residue at position 364 (that have been either shown or predicted not to bind Ca2+ via the Ca2+-binding loops29,30) bind anionic lipids and the syntaxin/SNAP-25 complex in a Ca2+-independent manner and inhibit SNARE-mediated lipid mixing in vitro.(30)
The third nitrated C2B residue, Tyr380, is located in the loop leading to the proposed SNARE interaction helix(63) on the side of the C2B domain opposite from Ca2+-binding loops, thus potentially modulating the Syt1–SNARE interaction. Tyrosines that are nitrated in the C2A domain, Tyr151 and Tyr216, are clustered together opposite of the Ca2+-binding loops while Tyr229 is very close to the Ca2+-binding loop, and nitration could potentially change the conformation of the loop. Tyr229 is adjacent to Asp230 and in the vicinity of Asp232 (corresponding to D. melanogaster Asp282 and Asp284), mutations of which abolish C2A-mediated Ca2+-triggered suppression of asynchronous synaptic vesicle fusion.(62) Finally, the Syt1 tyrosine nitrations reported here could alter its Ca2+-mediated interactions with anionic lipid and/or SNARE and other proteins, thus modulating Ca2+-triggered synaptic vesicle exo- and endocytosis.
Several studies suggest that phosphorylation can modulate neurotransmitter release,64−66 although the role of Syt1 phosphorylation at the previously characterized in vitro sites (Thr112, Thr128, and Thr125) is unknown. Following stimulation with K+ depolarization or phorbol esters in PC12 cells and in rat synaptosomes, the fraction of Syt1 phosphorylated at Thr112 increased,(7) whereas in chromaffin cells, mutations of Thr112, Thr125, and Thr128 had no detectable effect on exocytosis.(67) However, the effect of phosphorylation on exocytosis may be subtle because only a small fraction of total Syt1 is phosphorylated in vivo.(7) We have detected phosphorylation at two sites in Syt1: the previously reported Thr128 site(8) and the new Ser30 site. The previously reported Thr112, Thr125, and Thr128 phosphorylation sites are located in the linker region between transmembrane and Ca2+-binding C2 domains, while the new Ser30 phosphorylation site is located in the luminal Syt1 domain. Consequently, the potential physiological role of Syt1 phosphorylation at Ser30 is likely different from that for the other phosphorylation sites.
The N-terminal luminal domain of Syt1, containing both N- and O-glycosylation sites, is highly conserved across species,(4) although it is not conserved among different synaptotagmin isoforms. For Syt1, both O-glycosylation and N-glycosylation regulate Syt1’s localization to exocytic vesicles.2,4 The newly identified O-glycosylation site at Thr26 might be specific for insect cell expression, so characterization of Syt1 purified from natural sources will be required.
In summary, we expressed and purified mammalian Syt1 in insect cells at levels amenable to biochemical studies and characterized the purified protein in unprecedented detail. We have shown that mammalian Syt1 expressed and purified in insect cells is properly folded and carries post-translational modifications already identified in mammalian cells along with a new post-translational modification of Syt1, tyrosine nitration, and new phosphorylation and O-glycosylation sites. In addition, we have expanded the panel of anionic lipids with which Syt1 and Syt3 interact. We found that interaction of the Syt1-C2AB fragment with different anionic lipids is conserved and mediated by the same regions in the C2AB fragment. Ca2+-independent interactions seem to be primarily mediated via a lysine rich region in the C2B domain with a minor contribution from Arg398 and Arg399, while Ca2+-dependent interactions are primarily mediated via the Ca2+-binding loops. In conclusion, insect cell-expressed full-length mammalian Syt1 should be a useful tool for further elucidation of the functional roles of Syt1, its post-translational modifications, and its interactions with various lipid and protein binding partners in the synaptic vesicle cycle.
Acknowledgments
We thank Chengbiao Wu for assistance with synaptic vesicle preparation, Henriette Macmillan for assistance with Western blots, and Elizabeth D. Mellins for access to the Octet system at the Stanford University School of Medicine, Immunology, and Pediatrics.
Author Present Address
⊥ Department of Energy, 1000 Independence Ave. SW, Washington, DC 20585.
We gratefully acknowledge the National Institutes of Health for support of A.T.B. (R37-MH63105).
Author Contributions
M.V. and P.S. expressed and purified full-length Syt1 and performed CD analysis. M.V. designed and purified Syt-C2AB proteins and collected presented data. R.C.H. and K.C.H. collected and analyzed mass spectrometry data. A.T.B., M.V., P.S., R.C.H., K.C.H., and S.C. performed experimental design and data analysis and wrote the manuscript.
Funding Statement
National Institutes of Health, United States
References
- Perin M. S.; Brose N.; Jahn R.; Sudhof T. C. (1991) Domain structure of synaptotagmin (p65). J. Biol. Chem. 266, 623–629. [PubMed] [Google Scholar]
- Han W.; Rhee J. S.; Maximov A.; Lao Y.; Mashimo T.; Rosenmund C.; Sudhof T. C. (2004) N-Glycosylation is essential for vesicular targeting of synaptotagmin 1. Neuron 41, 85–99. [DOI] [PubMed] [Google Scholar]
- Fukuda M. (2002) Vesicle-associated membrane protein-2/synaptobrevin binding to synaptotagmin I promotes O-glycosylation of synaptotagmin I. J. Biol. Chem. 277, 30351–30358. [DOI] [PubMed] [Google Scholar]
- Kanno E.; Fukuda M. (2008) Increased plasma membrane localization of O-glycosylation-deficient mutant of synaptotagmin I in PC12 cells. J. Neurosci. Res. 86, 1036–1043. [DOI] [PubMed] [Google Scholar]
- Veit M.; Sollner T. H.; Rothman J. E. (1996) Multiple palmitoylation of synaptotagmin and the t-SNARE SNAP-25. FEBS Lett. 385, 119–123. [DOI] [PubMed] [Google Scholar]
- Heindel U.; Schmidt M. F.; Veit M. (2003) Palmitoylation sites and processing of synaptotagmin I, the putative calcium sensor for neurosecretion. FEBS Lett. 544, 57–62. [DOI] [PubMed] [Google Scholar]
- Hilfiker S.; Pieribone V. A.; Nordstedt C.; Greengard P.; Czernik A. J. (1999) Regulation of synaptotagmin I phosphorylation by multiple protein kinases. J. Neurochem. 73, 921–932. [DOI] [PubMed] [Google Scholar]
- Davletov B.; Sontag J. M.; Hata Y.; Petrenko A. G.; Fykse E. M.; Jahn R.; Sudhof T. C. (1993) Phosphorylation of synaptotagmin I by casein kinase II. J. Biol. Chem. 268, 6816–6822. [PubMed] [Google Scholar]
- Brose N.; Petrenko A. G.; Sudhof T. C.; Jahn R. (1992) Synaptotagmin: A calcium sensor on the synaptic vesicle surface. Science 256, 1021–1025. [DOI] [PubMed] [Google Scholar]
- Bai J.; Tucker W. C.; Chapman E. R. (2004) PIP2 increases the speed of response of synaptotagmin and steers its membrane-penetration activity toward the plasma membrane. Nat. Struct. Mol. Biol. 11, 36–44. [DOI] [PubMed] [Google Scholar]
- Schiavo G.; Gu Q. M.; Prestwich G. D.; Sollner T. H.; Rothman J. E. (1996) Calcium-dependent switching of the specificity of phosphoinositide binding to synaptotagmin. Proc. Natl. Acad. Sci. U.S.A. 93, 13327–13332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrick D. Z.; Sterbling S.; Rasch K. A.; Hinderliter A.; Cafiso D. S. (2006) Position of synaptotagmin I at the membrane interface: cooperative interactions of tandem C2 domains. Biochemistry 45, 9668–9674. [DOI] [PubMed] [Google Scholar]
- Radhakrishnan A.; Stein A.; Jahn R.; Fasshauer D. (2009) The Ca2+ affinity of synaptotagmin 1 is markedly increased by a specific interaction of its C2B domain with phosphatidylinositol 4,5-bisphosphate. J. Biol. Chem. 284, 25749–25760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo W.; Herrick D. Z.; Cafiso D. S. (2011) Phosphatidylinositol 4,5-bisphosphate alters synaptotagmin 1 membrane docking and drives opposing bilayers closer together. Biochemistry 50, 2633–2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vrljic M.; Strop P.; Ernst J. A.; Sutton R. B.; Chu S.; Brunger A. T. (2010) Molecular mechanism of the synaptotagmin-SNARE interaction in Ca2+-triggered vesicle fusion. Nat. Struct. Mol. Biol. 17, 325–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ubach J.; Lao Y.; Fernandez I.; Arac D.; Sudhof T. C.; Rizo J. (2001) The C2B domain of synaptotagmin I is a Ca2+-binding module. Biochemistry 40, 5854–5860. [DOI] [PubMed] [Google Scholar]
- Bowen M. E.; Weninger K.; Brunger A. T.; Chu S. (2004) Single molecule observation of liposome-bilayer fusion thermally induced by soluble N-ethyl maleimide sensitive-factor attachment protein receptors (SNAREs). Biophys. J. 87, 3569–3584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westhead E. W. (1987) Lipid Composition and Orientation in Secretory Vesicles. Ann. N.Y. Acad. Sci. 493, 92–100. [DOI] [PubMed] [Google Scholar]
- Takamori S.; Holt M.; Stenius K.; Lemke E. A.; Gronborg M.; Riedel D.; Urlaub H.; Schenck S.; Brugger B.; Ringler P.; Muller S. A.; Rammner B.; Grater F.; Hub J. S.; De Groot B. L.; Mieskes G.; Moriyama Y.; Klingauf J.; Grubmuller H.; Heuser J.; Wieland F.; Jahn R. (2006) Molecular anatomy of a trafficking organelle. Cell 127, 831–846. [DOI] [PubMed] [Google Scholar]
- Hell J. W.; Maycox P. R.; Stadler H.; Jahn R. (1988) Uptake of GABA by rat brain synaptic vesicles isolated by a new procedure. EMBO J. 7, 3023–3029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig R.; Beavis R. C. (2004) TANDEM: Matching proteins with tandem mass spectra. Bioinformatics 20, 1466–1467. [DOI] [PubMed] [Google Scholar]
- Shin O. H.; Xu J.; Rizo J.; Sudhof T. C. (2009) Differential but convergent functions of Ca2+ binding to synaptotagmin-1 C2 domains mediate neurotransmitter release. Proc. Natl. Acad. Sci. U.S.A. 106, 16469–16474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin R.; Rummel A.; Binz T.; Brunger A. T. (2006) Botulinum neurotoxin B recognizes its protein receptor with high affinity and specificity. Nature 444, 1092–1095. [DOI] [PubMed] [Google Scholar]
- Jensen O. (2006) Interpreting the protein language using proteomics. Nat. Rev. Mol. Cell Biol. 6, 391–403. [DOI] [PubMed] [Google Scholar]
- Olsen J.; Vermeulen M.; Santamaria A.; Kumar C.; Miller M.; Jensen L.; Gnad F.; Cox J.; Jensen T.; Nigg E.; Brunak S.; Mann M. (2010) Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Sci. Signaling 3, ra3. [DOI] [PubMed] [Google Scholar]
- Wu R.; Haas W.; Dephoure N.; Huttlin E.; Zhai B.; Sowa M.; Gygi S. (2011) A large-scale method to measure absolute protein phosphorylation stoichiometries. Nat. Methods 8, 677–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiAntonio A.; Schwarz T. L. (1994) The effect on synaptic physiology of synaptotagmin mutations in Drosophila. Neuron 12, 909–920. [DOI] [PubMed] [Google Scholar]
- Jarousse N.; Kelly R. B. (2001) The AP2 binding site of synaptotagmin 1 is not an internalization signal but a regulator of endocytosis. J. Cell Biol. 154, 857–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai H.; Shin O. H.; Machius M.; Tomchick D. R.; Sudhof T. C.; Rizo J. (2004) Structural basis for the evolutionary inactivation of Ca2+ binding to synaptotagmin 4. Nat. Struct. Mol. Biol. 11, 844–849. [DOI] [PubMed] [Google Scholar]
- Bhalla A.; Chicka M. C.; Chapman E. R. (2008) Analysis of the synaptotagmin family during reconstituted membrane fusion. Uncovering a class of inhibitory isoforms. J. Biol. Chem. 283, 21799–21807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyoung M.; Srivastava A.; Zhang Y.; Diao J.; Vrljic M.; Grob P.; Nogales E.; Chu S.; Brunger A. T. (2011) In vitro system capable of differentiating fast Ca2+-triggered content mixing from lipid exchange for mechanistic studies of neurotransmitter release . PNAS Plus 108, E304–E313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison R. L.; Jarvis D. L. (2006) Protein N-glycosylation in the baculovirus-insect cell expression system and engineering of insect cells to produce “mammalianized” recombinant glycoproteins. Adv. Virus Res. 68, 159–191. [DOI] [PubMed] [Google Scholar]
- Fukuda M.; Kojima T.; Aruga J.; Niinobe M.; Mikoshiba K. (1995) Functional diversity of C2 domains of synaptotagmin family. Mutational analysis of inositol high polyphosphate binding domain. J. Biol. Chem. 270, 26523–26527. [DOI] [PubMed] [Google Scholar]
- Li L.; Shin O. H.; Rhee J. S.; Arac D.; Rah J. C.; Rizo J.; Sudhof T.; Rosenmund C. (2006) Phosphatidylinositol phosphates as co-activators of Ca2+ binding to C2 domains of synaptotagmin 1. J. Biol. Chem. 281, 15845–15852. [DOI] [PubMed] [Google Scholar]
- Nicholson-Tomishima K.; Ryan T. A. (2004) Kinetic efficiency of endocytosis at mammalian CNS synapses requires synaptotagmin I. Proc. Natl. Acad. Sci. U.S.A. 101, 16648–16652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung N.; Wienisch M.; Gu M.; Rand J. B.; Muller S. L.; Krause G.; Jorgensen E. M.; Klingauf J.; Haucke V. (2007) Molecular basis of synaptic vesicle cargo recognition by the endocytic sorting adaptor stonin 2. J. Cell Biol. 179, 1497–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masztalerz A.; Zeelenberg I. S.; Wijnands Y. M.; de Bruijn R.; Drager A. M.; Janssen H.; Roos E. (2007) Synaptotagmin 3 deficiency in T cells impairs recycling of the chemokine receptor CXCR4 and thereby inhibits CXCL12 chemokine-induced migration. J. Cell Sci. 120, 219–228. [DOI] [PubMed] [Google Scholar]
- Di Paolo G.; De Camilli P. (2006) Phosphoinositides in cell regulation and membrane dynamics. Nature 443, 651–657. [DOI] [PubMed] [Google Scholar]
- Martens S.; Kozlov M. M.; McMahon H. T. (2007) How synaptotagmin promotes membrane fusion. Science 316, 1205–1208. [DOI] [PubMed] [Google Scholar]
- Arac D.; Chen X.; Khant H. A.; Ubach J.; Ludtke S. J.; Kikkawa M.; Johnson A. E.; Chiu W.; Sudhof T. C.; Rizo J. (2006) Close membrane-membrane proximity induced by Ca2+-dependent multivalent binding of synaptotagmin-1 to phospholipids. Nat. Struct. Mol. Biol. 13, 209–217. [DOI] [PubMed] [Google Scholar]
- Lewis J. D.; Lazarowitz S. G. (2010) Arabidopsis synaptotagmin SYTA regulates endocytosis and virus movement protein cell-to-cell transport. Proc. Natl. Acad. Sci. U.S.A. 107, 2491–2496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen E. M.; Hartwieg E.; Schuske K.; Nonet M. L.; Jin Y.; Horvitz H. R. (1995) Defective recycling of synaptic vesicles in synaptotagmin mutants of Caenorhabditis elegans. Nature 378, 196–199. [DOI] [PubMed] [Google Scholar]
- Poskanzer K. E.; Marek K. W.; Sweeney S. T.; Davis G. W. (2003) Synaptotagmin I is necessary for compensatory synaptic vesicle endocytosis in vivo. Nature 426, 559–563. [DOI] [PubMed] [Google Scholar]
- Zhang J. Z.; Davletov B. A.; Sudhof T. C.; Anderson R. G. (1994) Synaptotagmin I is a high affinity receptor for clathrin AP-2: Implications for membrane recycling. Cell 78, 751–760. [DOI] [PubMed] [Google Scholar]
- Rohrbough J.; Broadie K. (2005) Lipid regulation of the synaptic vesicle cycle. Nat. Rev. Neurosci. 6, 139–150. [DOI] [PubMed] [Google Scholar]
- Tang J.; Maximov A.; Shin O. H.; Dai H.; Rizo J.; Sudhof T. C. (2006) A complexin/synaptotagmin 1 switch controls fast synaptic vesicle exocytosis. Cell 126, 1175–1187. [DOI] [PubMed] [Google Scholar]
- Xu J.; Mashimo T.; Sudhof T. C. (2007) Synaptotagmin-1, -2, and -9: Ca2+ sensors for fast release that specify distinct presynaptic properties in subsets of neurons. Neuron 54, 567–581. [DOI] [PubMed] [Google Scholar]
- Dong M.; Richards D. A.; Goodnough M. C.; Tepp W. H.; Johnson E. A.; Chapman E. R. (2003) Synaptotagmins I and II mediate entry of botulinum neurotoxin B into cells. J. Cell Biol. 162, 1293–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arantes R. M.; Andrews N. W. (2006) A role for synaptotagmin VII-regulated exocytosis of lysosomes in neurite outgrowth from primary sympathetic neurons. J. Neurosci. 26, 4630–4637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prior I. A.; Clague M. J. (2000) Detection of thiol modification following generation of reactive nitrogen species: Analysis of synaptic vesicle proteins. Biochim. Biophys. Acta 1475, 281–286. [DOI] [PubMed] [Google Scholar]
- Matsushita K.; Morrell C. N.; Cambien B.; Yang S. X.; Yamakuchi M.; Bao C.; Hara M. R.; Quick R. A.; Cao W.; O’Rourke B.; Lowenstein J. M.; Pevsner J.; Wagner D. D.; Lowenstein C. J. (2003) Nitric oxide regulates exocytosis by S-nitrosylation of N-ethylmaleimide-sensitive factor. Cell 115, 139–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess D. T.; Lin L. H.; Freeman J. A.; Norden J. J. (1994) Modification of cysteine residues within Go and other neuronal proteins by exposure to nitric oxide. Neuropharmacology 33, 1283–1292. [DOI] [PubMed] [Google Scholar]
- Palmer Z. J.; Duncan R. R.; Johnson J. R.; Lian L. Y.; Mello L. V.; Booth D.; Barclay J. W.; Graham M. E.; Burgoyne R. D.; Prior I. A.; Morgan A. (2008) S-Nitrosylation of syntaxin 1 at Cys(145) is a regulatory switch controlling Munc18-1 binding. Biochem. J. 413, 479–491. [DOI] [PubMed] [Google Scholar]
- Jacoby S.; Sims R. E.; Hartell N. A. (2001) Nitric oxide is required for the induction and heterosynaptic spread of long-term potentiation in rat cerebellar slices. J. Physiol. 535, 825–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuman E. M.; Madison D. V. (1991) A requirement for the intercellular messenger nitric oxide in long-term potentiation. Science 254, 1503–1506. [DOI] [PubMed] [Google Scholar]
- Murphy K. P.; Bliss T. V. (1999) Photolytically released nitric oxide produces a delayed but persistent suppression of LTP in area CA1 of the rat hippocampal slice. J. Physiol. 515, Part 2453–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meffert M. K.; Calakos N. C.; Scheller R. H.; Schulman H. (1996) Nitric oxide modulates synaptic vesicle docking fusion reactions. Neuron 16, 1229–1236. [DOI] [PubMed] [Google Scholar]
- Fukuda M.; Kabayama H.; Mikoshiba K. (2000) Drosophila AD3 mutation of synaptotagmin impairs calcium-dependent self-oligomerization activity. FEBS Lett. 482, 269–272. [DOI] [PubMed] [Google Scholar]
- Littleton J. T.; Bai J.; Vyas B.; Desai R.; Baltus A. E.; Garment M. B.; Carlson S. D.; Ganetzky B.; Chapman E. R. (2001) Synaptotagmin mutants reveal essential functions for the C2B domain in Ca2+-triggered fusion and recycling of synaptic vesicles in vivo. J. Neurosci. 21, 1421–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borden C. R.; Stevens C. F.; Sullivan J. M.; Zhu Y. (2005) Synaptotagmin mutants Y311N and K326/327A alter the calcium dependence of neurotransmission. Mol. Cell. Neurosci. 29, 462–470. [DOI] [PubMed] [Google Scholar]
- Rickman C.; Jimenez J. L.; Graham M. E.; Archer D. A.; Soloviev M.; Burgoyne R. D.; Davletov B. (2006) Conserved prefusion protein assembly in regulated exocytosis. Mol. Biol. Cell 17, 283–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshihara M.; Guan Z.; Littleton J. T. (2010) Differential regulation of synchronous versus asynchronous neurotransmitter release by the C2 domains of synaptotagmin 1. Proc. Natl. Acad. Sci. U.S.A. 107, 14869–14874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi U. B.; Strop P.; Vrljic M.; Chu S.; Brunger A. T.; Weninger K. R. (2010) Single-molecule FRET-derived model of the synaptotagmin 1-SNARE fusion complex. Nat. Struct. Mol. Biol. 17, 318–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llinas R.; McGuinness T. L.; Leonard C. S.; Sugimori M.; Greengard P. (1985) Intraterminal injection of synapsin I or calcium/calmodulin-dependent protein kinase II alters neurotransmitter release at the squid giant synapse. Proc. Natl. Acad. Sci. U.S.A. 82, 3035–3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols R. A.; Sihra T. S.; Czernik A. J.; Nairn A. C.; Greengard P. (1990) Calcium/calmodulin-dependent protein kinase II increases glutamate and noradrenaline release from synaptosomes. Nature 343, 647–651. [DOI] [PubMed] [Google Scholar]
- Hilfiker S.; Augustine G. J. (1999) Regulation of synaptic vesicle fusion by protein kinase C. J. Physiol. 515, Part 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy G.; Kim J. H.; Pang Z. P.; Matti U.; Rettig J.; Sudhof T. C.; Sorensen J. B. (2006) Different effects on fast exocytosis induced by synaptotagmin 1 and 2 isoforms and abundance but not by phosphorylation. J. Neurosci. 26, 632–643. [DOI] [PMC free article] [PubMed] [Google Scholar]