

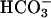
Abstract
The Cu,Zn superoxide dismutase catalyzes 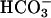 -dependent oxidations by H2O2. This activity has been shown to depend on the creation of a bound oxidant at the Cu(II) by interactions with H2O2. The bound oxidant was then thought to oxidize
-dependent oxidations by H2O2. This activity has been shown to depend on the creation of a bound oxidant at the Cu(II) by interactions with H2O2. The bound oxidant was then thought to oxidize 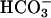 to
to 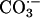 , which diffuses into the bulk solution and there oxidizes diverse substrates. We now find that CO2 rather than
, which diffuses into the bulk solution and there oxidizes diverse substrates. We now find that CO2 rather than 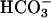 facilitates the peroxidations catalyzed by Cu,Zn superoxide dismutase. This fact was shown by a lag in the rate of peroxidation of NADPH when
facilitates the peroxidations catalyzed by Cu,Zn superoxide dismutase. This fact was shown by a lag in the rate of peroxidation of NADPH when 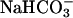 was added last and by a burst in the rate when aqueous CO2 was added last. Both the lag and the burst were eliminated by carbonic anhydrase.
was added last and by a burst in the rate when aqueous CO2 was added last. Both the lag and the burst were eliminated by carbonic anhydrase.
Keywords: carbon dioxide; bicarbonate; carbonic anhydrase; Cu,Zn SOD; hydrogen peroxide
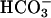 dramatically increases the rate of oxidation of diverse substrates by Cu,Zn superoxide dismutase (SOD1) plus H2O2, while only modestly increasing the intrinsically slow inactivation of this enzyme by H2O2 (1–4). The following reactions have been advanced as part of a scheme to explain the behavior of this system (1).
dramatically increases the rate of oxidation of diverse substrates by Cu,Zn superoxide dismutase (SOD1) plus H2O2, while only modestly increasing the intrinsically slow inactivation of this enzyme by H2O2 (1–4). The following reactions have been advanced as part of a scheme to explain the behavior of this system (1).
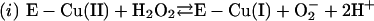 |
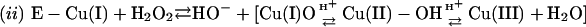 |
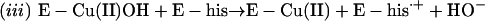 |
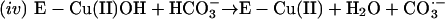 |
Thus, the cuprous enzyme produced in reaction i is oxidized in reaction ii by a second H2O2 to yield a strong bound oxidant, which can be written as Cu(I)O, Cu(II)OH, or Cu(III). This bound oxidant can either oxidize an adjacent histidine residue and in so doing inactivate the enzyme, or it can oxidize 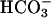 to the carbonate radical
to the carbonate radical 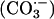 that is free to diffuse into the bulk solution and cause further oxidations. Before leaving the active site, the carbonate radical can oxidize residues in the ligand field of the copper and thus cause inactivation.
that is free to diffuse into the bulk solution and cause further oxidations. Before leaving the active site, the carbonate radical can oxidize residues in the ligand field of the copper and thus cause inactivation.
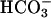 was assumed to be the species in the carbonate buffer that facilitated the oxidations observed. This was the case because, at neutral to mildly alkaline pH, it is the major species. It also seemed reasonable that an active site evolved to deal with the monovalent anion
was assumed to be the species in the carbonate buffer that facilitated the oxidations observed. This was the case because, at neutral to mildly alkaline pH, it is the major species. It also seemed reasonable that an active site evolved to deal with the monovalent anion 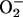 would preferentially react with other monovalent anions. Nevertheless, the experiments described below establish that at pH 7.4, it is CO2 rather than
would preferentially react with other monovalent anions. Nevertheless, the experiments described below establish that at pH 7.4, it is CO2 rather than 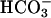 that profoundly facilitates the oxidation of NADPH by SOD1 plus H2O2.
that profoundly facilitates the oxidation of NADPH by SOD1 plus H2O2.
Materials and Methods
SOD1 was from Chemie Grünenthal (Aachen, Germany); H2O2, from Mallinckrodt; and NaHCO3, from Fisher; whereas NADPH and carbonic anhydrase were from Sigma. NADPH at 0.1 mM was reacted with 0.1 or 0.2 mg/ml SOD1 and 10 mM H2O2 in 100 mM potassium phosphate at pH 7.4. When 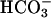 was used to facilitate the oxidation of NADPH (followed at 340 nm), it was added to 20 mM from a 1.0 M stock of NaHCO3 in water. When CO2 was used, it was taken from ice-cold water saturated with CO2 gas at 1.0 atmosphere (atm) (1 atm = 101.3 kPa) that would contain 76 mM CO2. Carbonic anhydrase when used was present at 0.1 mg/ml. Additional experimental details are presented in the legends to Figs. 1 and 2.
was used to facilitate the oxidation of NADPH (followed at 340 nm), it was added to 20 mM from a 1.0 M stock of NaHCO3 in water. When CO2 was used, it was taken from ice-cold water saturated with CO2 gas at 1.0 atmosphere (atm) (1 atm = 101.3 kPa) that would contain 76 mM CO2. Carbonic anhydrase when used was present at 0.1 mg/ml. Additional experimental details are presented in the legends to Figs. 1 and 2.
Fig. 1.

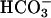 requires dehydration before facilitating. Shown is the oxidation of NADPH by SOD1 plus H2O2. The final reaction mixtures contained 0.1 mM NADPH, 0.1 mg/ml SOD1, 20 mM
requires dehydration before facilitating. Shown is the oxidation of NADPH by SOD1 plus H2O2. The final reaction mixtures contained 0.1 mM NADPH, 0.1 mg/ml SOD1, 20 mM 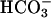 , and 10 mM H2O2, plus 100 mM sodium phosphate at pH 7.4 and at 23°C. Line 1, SOD or H2O2 added as the last component at the arrow; line 2, NaHCO3 added last at the arrow; line 3, as in line 2, except that 0.1 mg/ml carbonic anhydrase was present in the reaction mixture. Note the slow oxidation before the addition of
, and 10 mM H2O2, plus 100 mM sodium phosphate at pH 7.4 and at 23°C. Line 1, SOD or H2O2 added as the last component at the arrow; line 2, NaHCO3 added last at the arrow; line 3, as in line 2, except that 0.1 mg/ml carbonic anhydrase was present in the reaction mixture. Note the slow oxidation before the addition of 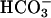 .
.
Fig. 2.

CO2 added as such supports the peroxidation activity of SOD1. Reaction conditions shown are essentially as in Fig. 1, except that water at 0°C saturated with CO2 gas was the last component added in place of 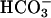 . Line 1, CO2 added at the arrow to reaction mixture as in Fig. 1; line 2, CO2 added at the arrow to a reaction mixture as in Fig. 1 but containing 0.2 mg/ml SOD1; line 3, as in line 2 but with 0.1 mg/ml carbonic anhydrase in the reaction mixture.
. Line 1, CO2 added at the arrow to reaction mixture as in Fig. 1; line 2, CO2 added at the arrow to a reaction mixture as in Fig. 1 but containing 0.2 mg/ml SOD1; line 3, as in line 2 but with 0.1 mg/ml carbonic anhydrase in the reaction mixture.
Results
Line 1 in Fig. 1 demonstrates an immediately linear rate of NADPH oxidation that was seen when the HCO3-dependent oxidation by SOD1 plus H2O2 reaction was started by adding either SOD1 or H2O2. However, when NaHCO3 was the last component added, there was a distinct lag before the linear rate was achieved, shown by line 2. Because NaHCO3 was added from an alkaline stock solution into a reaction mixture buffered at pH 7.4, it seemed possible that this lag was due to the perceptibly slow dehydration of H2CO3 to CO2. That this was the case was shown by the effect of carbonic anhydrase (line 3), which eliminated this lag. These results indicated that CO2 rather than 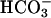 was the essential reactant.
was the essential reactant.
In that case, it could be predicted that starting the reaction with CO2-saturated water would yield an initially fast reaction that would slow to a linear rate as the hydration and ionization to 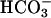 reached an equilibrium. Moreover, carbonic anhydrase should eliminate the burst seen when CO2 was added last, because it had eliminated the lag when
reached an equilibrium. Moreover, carbonic anhydrase should eliminate the burst seen when CO2 was added last, because it had eliminated the lag when 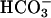 was added last. Fig. 2 demonstrates that these predictions were correct. Thus line 1 was obtained when 150 μl of CO2 solution was added to a 3-ml reaction mixture otherwise identical to that used in Fig. 1. The very evident initial burst was made even more pronounced when the concentration of SOD1 was doubled (line 2). Finally, carbonic anhydrase eliminated the initial burst (line 3). It should be emphasized that the initial CO2 concentration in the reaction mixture would have been 3.8 mM, substantially lower than the concentration of
was added last. Fig. 2 demonstrates that these predictions were correct. Thus line 1 was obtained when 150 μl of CO2 solution was added to a 3-ml reaction mixture otherwise identical to that used in Fig. 1. The very evident initial burst was made even more pronounced when the concentration of SOD1 was doubled (line 2). Finally, carbonic anhydrase eliminated the initial burst (line 3). It should be emphasized that the initial CO2 concentration in the reaction mixture would have been 3.8 mM, substantially lower than the concentration of 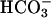 (20 mM) used in Fig. 1, yet the initial rate was greater than that seen with
(20 mM) used in Fig. 1, yet the initial rate was greater than that seen with 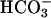 .
.
Discussion
These results establish that, at pH 7.4, the peroxidase activity of SOD1 depends on CO2 rather than on 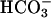 . The methods used to demonstrate this were virtually identical to those Cooper et al. (5) used to show that CO2 is the substrate for phosphoenol pyruvate carboxy kinase and carboxy transphosphorylase. In the case of the peroxidase activity of SOD1, we can envision two routes for the oxidation of CO2 to
. The methods used to demonstrate this were virtually identical to those Cooper et al. (5) used to show that CO2 is the substrate for phosphoenol pyruvate carboxy kinase and carboxy transphosphorylase. In the case of the peroxidase activity of SOD1, we can envision two routes for the oxidation of CO2 to 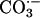 by the bound oxidant. Thus,
by the bound oxidant. Thus,
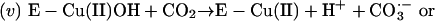 |
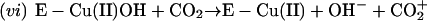 |
and then
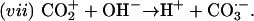 |
Elam et al. (6) have proposed still another route to 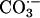 ; thus,
; thus,
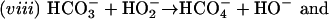 |
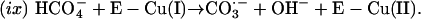 |
However, the rate of formation of 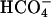 from CO2 plus
from CO2 plus 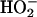 is slow (7), yet we saw the same initial rate of oxidation of NADPH when the
is slow (7), yet we saw the same initial rate of oxidation of NADPH when the 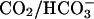 and H2O2 were preincubated before the addition of SOD1 as we did when
and H2O2 were preincubated before the addition of SOD1 as we did when 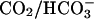 and SOD1 were preincubated before the addition of H2O2, rendering the mechanism defined by reactions viii and ix less likely.
and SOD1 were preincubated before the addition of H2O2, rendering the mechanism defined by reactions viii and ix less likely.
CO2 is a linear molecule that exists partially as a zwitterion, i.e., –O—C O+ (8). This structure is also supported by its considerable solubility in water (9), which results in 76 mM CO2 in water equilibrated with 1-atmosphere CO2 at 0°C compared with only 2.18 mM for O2 under the same conditions. Perhaps the entry of CO2 into the active site of SOD1 is facilitated by this zwitterionic form.
O+ (8). This structure is also supported by its considerable solubility in water (9), which results in 76 mM CO2 in water equilibrated with 1-atmosphere CO2 at 0°C compared with only 2.18 mM for O2 under the same conditions. Perhaps the entry of CO2 into the active site of SOD1 is facilitated by this zwitterionic form.
Carbonic anhydrase contains Zn(II) at its active site, and SOD1 contains Zn(II) joined to Cu(II) by the imidazolate moiety of a histidine residue. Here, it is possible that the first point of interaction of CO2 with SOD1 is at the Zn(II) rather than at the Cu center. The proximity of the Zn and Cu would allow CO2 interacting with the Zn to be oxidized by the Cu(II)—OH. While considering carbonic anhydrases, it is reasonable to ask whether this ubiquitous family of enzymes might play a defensive role by catalyzing the hydration of CO2, which can be oxidized to 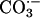 , in contrast to
, in contrast to 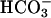 , which cannot. In support of this supposition is the report by Götz et al. (10) that deletion of the putative carbonic anhydrase from Saccharomyces cerevisiae caused an imposed oxygen sensitivity. Although it is certainly CO2 that is oxidized to
, which cannot. In support of this supposition is the report by Götz et al. (10) that deletion of the putative carbonic anhydrase from Saccharomyces cerevisiae caused an imposed oxygen sensitivity. Although it is certainly CO2 that is oxidized to 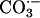 by SOD1 plus H2O2 at pH 7.4, there may be entities in cells that can similarly oxidize
by SOD1 plus H2O2 at pH 7.4, there may be entities in cells that can similarly oxidize 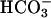 . In fact, it cannot be excluded that, at high pH, SOD1 plus H2O2 may be able to oxidize
. In fact, it cannot be excluded that, at high pH, SOD1 plus H2O2 may be able to oxidize 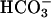 or
or 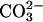 .
.
The results and discussion presented above preclude some hypotheses presented earlier, including the notions that 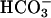 facilitates oxidations by SOD1 plus H2O2 by creating a binding site for H2O2 (4) and that
facilitates oxidations by SOD1 plus H2O2 by creating a binding site for H2O2 (4) and that 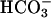 specifically binds in the solvent access channel of SOD1 and, while so bound, becomes oxidized, at which time the resulting
specifically binds in the solvent access channel of SOD1 and, while so bound, becomes oxidized, at which time the resulting 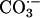 protrudes sufficiently to oxidize substrates in the bulk solution (6).
protrudes sufficiently to oxidize substrates in the bulk solution (6).
Acknowledgments
This work was supported by research grants from the Amyotrophic Lateral Sclerosis Association and by Grant R01 DK 59868 from the National Institute of Diabetes and Digestive and Kidney Diseases.
Abbreviation: SOD1, Cu,Zn superoxide dismutase.
References
- 1.Liochev, S. I. & Fridovich, I. (2002) J. Biol. Chem. 277, 34674–34678. [DOI] [PubMed] [Google Scholar]
- 2.Goss, S. P. A., Singh, R. J. & Kalyanaraman, B. (1999) J. Biol. Chem. 274, 28333–28339. [DOI] [PubMed] [Google Scholar]
- 3.Zhang, H., Joseph, J., Gurney, M., Becker, D. & Kalyanaraman, B. (2002) J. Biol. Chem. 277, 1013–1020. [DOI] [PubMed] [Google Scholar]
- 4.Sankarapandi, S. & Zweier, J. L. (1999) J. Biol. Chem. 274, 1226–1232. [DOI] [PubMed] [Google Scholar]
- 5.Cooper, T. G., Tchen, T. T., Wood, H. G. & Benedict, C. R. (1968) J. Biol. Chem. 243, 3857–3863. [PubMed] [Google Scholar]
- 6.Elam, J. S., Malek, K., Rodriquez, J. A., Doucette, P. A., Taylor, A. B., Hayward, L. J., Cabelli, D. E., Valentine, J. S. & Hart, P. J. (2003) J. Biol. Chem. 278, 21032–21039. [DOI] [PubMed] [Google Scholar]
- 7.Richardson, D. E., Yao, H., Frank, K. M. & Bennett, D. A. (2000) J. Am. Chem. Soc. 122, 1729–1739. [Google Scholar]
- 8.Moeller, T. (1954) in Inorganic Chemistry: An Advanced Textbook (Wiley, New York), p. 689.
- 9.Lange, N. A. (1961) in Lange's Handbook of Chemistry, ed. Lange, N. A. (McGraw–Hill, New York), p. 1100.
- 10.Götz, R., Gnan, A. & Zimmermann, F. K. (1999) Yeast 15, 855–864. [DOI] [PubMed] [Google Scholar]