

Abstract
One of the hallmarks of cardiomyopathy and heart failure is pronounced and progressive cardiomyocyte death. Understanding the mechanisms involved in cardiomyocyte cell death is a topic of great interest for treatment of cardiac disease. Mice null for desmin, the muscle-specific member of the intermediate filament gene family, develop cardiomyopathy characterized by extensive cardiomyocyte death, fibrosis, calcification, and eventual heart failure. The earliest ultrastructural defects are observed in mitochondria. In the present study, we have demonstrated that these mitochondrial abnormalities are the primary cause of the observed cardiomyopathy and that these defects can be ameliorated by overexpression of bcl-2 in desmin null heart. Overexpression of bcl-2 in the desmin null heart results in correction of mitochondrial defects, reduced occurrence of fibrotic lesions in the myocardium, prevention of cardiac hypertrophy, restoration of cardiomyocyte ultrastructure, and significant improvement of cardiac function. Furthermore, we have found that loss of desmin also diminishes the capacity of mitochondria to resist exposure to calcium, a defect that can be partially restored by bcl-2 overexpression. These results point to a unique function for desmin in protection of mitochondria from calcium exposure that can be partially rescued by overexpression of bcl-2. We show that bcl-2 cardiac overexpression has provided significant improvement of an inherited form of cardiomyopathy, revealing the potential for bcl-2, and perhaps other genes in the family, as therapeutic agents for heart disease of many types, including inherited forms.
Cardiomyocyte death commonly contributes to development of many cardiac disease states, including cardiomyopathy and heart failure. Recent efforts toward treatment of cardiac disease have focused on prevention of cell death (1). The antiapoptotic bcl-2 gene (2) is of particular interest for protection against cardiomyocyte death. Although bcl-2 is expressed at low levels in cardiomyocytes, this potent modulator of cell death is a promising therapeutic agent for heart disease, because it has been shown to reduce cell death during ischemia (3, 4), block p53-mediated apoptosis in cardiomyocytes (5), and increase the ability of mitochondria to resist high levels of calcium (6). Members of the bcl-2 family are thought to function at the contact sites (CS) of the mitochondrial membrane (7–9) and influence the membrane potential (ψΔ) through mechanisms that are not completely understood.
Previous studies have shown that desmin serves an important role in the maintenance of cellular integrity of muscle tissues (10). Multiple mutations within the human desmin gene lead to myopathies of varying intensity (11). Desmin null mice (des–/–) develop multiple defects in all muscle types (12, 13), particularly within cardiac muscle where a dilated cardiomyopathy develops with age (14). The initiating event in desmin null cardiomyopathy appears to be cell death occurring in focal areas, resulting in formation of fibrotic lesions variably associated with large calcium deposits. Mitochondrial abnormalities are the first defects visible in desmin null cardiomyocytes and include loss of shape and positioning, proliferation, aggregation, swelling, and degeneration. Desmin null mitochondria also display diminished ADP-stimulated respiratory function and loss of creatine kinase coupling (15, 16).
Although the above data strongly link desmin to mitochondrial behavior and function (16), the mechanism by which the absence of desmin leads to the observed mitochondrial abnormalities and cell death remains elusive. With the central role of bcl-2 in mitochondrial-regulated cell death and recent linkage of intermediate filaments, including desmin, to cell death events (17, 18), the des–/– mouse is an excellent model to assess both the protective impact of bcl-2 overexpression on cardiomyopathy and the mechanism by which desmin regulates mitochondrial behavior and cell survival. For this purpose, transgenic mice were generated with cardiac-specific overexpression of a murine bcl-2 cDNA and crossed into a desmin null background. Bcl-2 overexpression in the des–/– heart ameliorates cardiomyopathy and corrects the observed mitochondrial defects. All aspects of desmin null cardiomyopathy display reduced pathology, and bcl-2 overexpression significantly improves the diminished capacity of des–/– mitochondria to resist exposure to calcium. This finding points to a unique role for desmin in mitochondria buffering of calcium release in cardiomyocytes and presents a mechanism by which desmin deficiency would lead to cardiomyocyte cell death.
Methods
Transgenic Animal Production and Care. Desmin null mice (Destm1Cap/tm1Cap) were generated in the 129SV inbred background through standard methods (12). Two bcl-2 transgenic lines [Tg(bcl2)Acap and Tg(bcl2)Bcap] were generated by using the α-myosin heavy chain promoter (19) to drive a murine bcl-2 cDNA (20) in a C57BLK/6 background. Bcl-2 lines were assessed for expression by Western blot using a bcl-2 rabbit polyclonal antibody (N-19, Santa Cruz Biotechnology) at 1:200 dilution. Mice were maintained in a manner prescribed by National Institutes of Health regulations.
Heart-to-Body Weight Ratio (HW/BW) and Northern Blotting. HW/BW was determined as described (14), and a Fischer's ANOVA test was used to establish significance. Heart weights (mean ± SD) for des+/+ Tg(bcl2)BCap–/–, des–/– Tg(bcl2)BCap–/–, des–/– Tg(bcl2)BCap+/–, and des–/– Tg(bcl2)BCap+/+ mice were 94.6 ± 11.7, 113 ± 5.94, 78.3 ± 10.6, and 91.6 ± 10.1 mg at 2 months and 126 ± 17.6, 129 ± 23.1, 88.6 ± 6.87, and 102 ± 10.1 mg at 6 months.
For RNA isolation, five to seven animals of each age and genotype were killed, and dissected ventricles were frozen and ground into powder under liquid nitrogen. Total RNA was isolated from the pooled frozen tissues by using the TotallyRNA kit (Ambion, Austin, TX) per the manufacturer's instructions. Northern blotting was completed by using standard techniques with 8 μg of RNA loaded for each sample. Probes were used for α-skeletal actin and atrial natriuretic factor as described (14).
Electron Microscopy. Samples from two des+/+ Tg(bcl2)BCap–/–, three des–/– Tg(bcl2)BCap–/–, and four des–/– Tg(bcl2)BCap+/+ animals were processed for transmission electron microscopy as described (15). Briefly, hearts were perfused with cold 2.5% gluteraldehyde, then dissected ventricles were sliced into 1-mm cubes and fixed for 16 h at 4°C in the same fixative followed by postfixation in 1% osmium tetroxide for 2 h at room temperature. Samples were then dehydrated through an ethanol series and passed into Embed 812 (EMS, Fort Washington, PA) by using propylene oxide as a transitional solvent. Embedded blocks were cured at 55°C for 48 h and sectioned at 80 nm.
Doppler Analysis. Aortic outflow and transmitral inflow measurements were performed on 8-month-old animals as described (14). In brief, a 10-MHz pulsed doppler probe was placed at the xiphoid of mice of each genotype, and an ECG timing signal was superimposed on the doppler display by using a R-wave trigger. Calculation of systolic and diastolic parameters required the use of dspw software (Indus Instruments, Houston). Significance was determined by ANOVA.
Calcium-Mediated Mitochondria Swelling Assay. Mitochondria were isolated from whole hearts as described (21) with slight modification. Individual heart tissue was minced in ice-cold MSE+I (225 mM mannitol/75 mM sucrose/1 mM EGTA/1 mM Tris·HCl, pH 7.4/0.2 mM PMSF/37 μg/ml Na-p-tosyl-l-lysine chloromethylketone) and homogenized with five to seven strokes of Teflon head at 500 rpm on a Glas-Col (Terre Haute, IN) homogenizer. Homogenized tissue was centrifuged at 700 × g at 4°C for 5 min. The resulting supernatant was centrifuged at 7,700 × g for 10 min at 4°C. Mitochondria were diluted to 1 μg/μl in MSE+I with 100 μM calcium or without calcium at room temperature. Spectrophotometer readings were taken at 540 nM. Significance was determined by ANOVA.
Results
Generation of Desmin Null Mice Overexpressing Bcl-2 in the Heart. To examine the effect of bcl-2 overexpression on desmin null cardiomyopathy we generated two transgenic mouse lines [Tg(bcl2)ACap and Tg(bcl2)BCap] by using the cardiac-specific α-myosin heavy chain promoter (19) to drive expression of a murine bcl-2 cDNA (20). Each line strongly expresses bcl-2 protein of appropriate size; however, Tg(bcl2)BCap expressed at a higher level (Fig. 1a). Quantitative estimation of transgenic bcl-2 expression levels relative to the endogenous was not possible because the latter was undetectable. Both lines were crossed into the desmin null background (des–/–) and assayed for the level of rescue of desmin null cardiomyopathy.
Fig. 1.

Expression of a bcl-2 transgene prevents hypertrophy in desmin null hearts. (a) Whole heart protein from 2-month-old animals indicates endogenous bcl-2 expression is undetectable in the WT and des–/– heart. The Tg(bcl2)ACap transgenic line expresses high levels of bcl-2. A second transgenic line, Tg(bcl2)BCap, expresses bcl-2 at an even higher level. (b) Levels of hypertrophy markers atrial natriuretic factor (ANF) and α-skeletal actin (SKA) indicate both are up-regulated in des–/– ventricles at 2 and 6 months of age. Expression of the bcl-2 transgene reduces markers back to WT levels. Two alleles of the bcl-2 transgene are capable of reducing ANF and SKA expression below WT levels in 6-month-old animals. (c–e) Although bcl-2 expression does not significantly alter HW/BW in a WT background (c), it does return the increased HW/BW ratio in des–/– animals back to WT levels at both 2 (d)and6(e) months of age. *, P < 0.05 vs. desmin null at the same age; †, P < 0.005 vs. desmin null at the same age; ‡, P < 0.05 vs. WT at 6 months.
Decreased Appearance of Fibrotic Lesions and Calcification in Desmin Null Animals Overexpressing Bcl-2. Initial morphological observations of des–/– Tg(bcl2)ACap+/– hearts showed little difference from des–/– hearts. A similar degree of calcification on the surface of the heart appeared in both genotypes (data not shown). However, hearts with higher levels of bcl-2 expression, such as des–/– Tg(bcl2)BCap+/+ (Fig. 2d), display either no or significantly less calcification than that seen in the des–/– mice (Fig. 2b). These results were mirrored by histological analysis, with des–/– Tg(bcl2)BCap+/+ mice showing fewer fibrotic lesions that usually have a smaller surface area than that seen in des–/– or des–/– Tg(bcl2)ACap+/+ hearts (Fig. 2 e–j). Fewer lesions contain calcification in des–/– Tg(bcl2)ACap+/+ (Fig. 2 g, k, and o) and even fewer in des–/– Tg(bcl2)BCap+/+ hearts (Fig. 2 h, l, and p) when compared to des–/– hearts (Fig. 2n). These results demonstrate that overexpression of bcl-2 in the des–/– heart can prevent lesion formation in a dose-dependent manner.
Fig. 2.

Decreased fibrotic lesions and calcification in desmin null animals overexpressing bcl-2. Whole-mount heart from 2-month-old WT (a), des–/– (b), des–/– Tg(bcl2)ACap+/+ (c), and des–/– Tg(bcl2)BCap+/+ (d) animals. (e–h) Hemotoxylin and eosin-stained sections from the same genotype animals. (i–p) Fibrosis associated with calcium deposits is found throughout the left ventricle of des–/– hearts, as revealed by Mason's trichrome staining (j and n). Compared to des–/–, fibrotic lesions are less frequent and smaller in des–/– Tg(bcl2)ACap+/+ (k) and des–/– Tg(bcl2)BCap+/+ myocardium (l). Although calcification is obvious in Von Kossa staining of des–/– myocardium (n), there is less calcification of lesions in des–/– Tg(bcl2)BCap+/+ hearts (p). (Magnification: ×200.)
Expression of Bcl-2 Prevents Hypertrophy in Desmin Null Hearts. A hallmark of cardiomyopathy and heart failure is compensatory hypertrophy during the early stages of pathology. In desmin null cardiomyopathy, hypertrophy occurs in response to cardiac damage, with significant increase in heart size and distortion of the ventricles (Fig. 2b). Introduction of bcl-2 into the des–/– heart was able to prevent structural dilation of the heart (Fig. 2 c and d) and rescue the elevated HW/BW of des–/– animals (Fig. 1 d and e). Bcl-2 overexpression in the WT background does not appear to alter the HW/BW (Fig. 1c). We also find that the levels of molecular markers of hypertrophy such as α-skeletal actin and atrial natriuretic factor are reduced in all des–/– animals carrying a bcl-2 transgene (Fig. 1b). Again, we see a dose-dependent response as increased bcl-2 levels produce reduced HW/BW and diminished expression of hypertrophy markers that approach WT levels.
Restoration of Ultrastructural Abnormalities in des–/– Cardiomyocytes. Examination of the ultrastructure of des–/– Tg(bcl2)BCap+/+ cardiomyocytes shows a significant recovery from the damage seen in des–/– hearts. Electron microscopy of des–/– cardiomyocytes reveals multiple defects ranging from disrupted contractile apparatus to numerous mitochondrial deficiencies (Fig. 3b). These mitochondrial abnormalities include loss of positioning, proliferation, clumping, and formation of cleared areas of cytoplasm around mitochondria. In addition, individual mitochondria appear swollen and misshapen. Although such defects can be occasionally detected in des–/– Tg(bcl2)BCap+/+ cardiomyocytes, they are not as common and appear less intense when compared to des–/– animals. The most frequent mitochondrial defect observed in des–/– Tg(bcl2)BCap+/+ cardiomyocytes involves a loss of mitochondria alignment. These mitochondria fail to form the well-defined rows in between myofibrils that are seen in WT cardiomyocytes (Fig. 3c), illustrating the importance of desmin in maintenance of mitochondrial positioning. However, many des–/– Tg(bcl2)BCap+/+ cardiomyocytes appear identical to WT cardiomyocytes. When compared to WT, the most striking similarities are in myofibril integrity and alignment, which in des–/– Tg(bcl2)BCap+/+ cardiomyocytes appears quite normal, suggesting a minimal role for desmin in myofibril alignment.
Fig. 3.
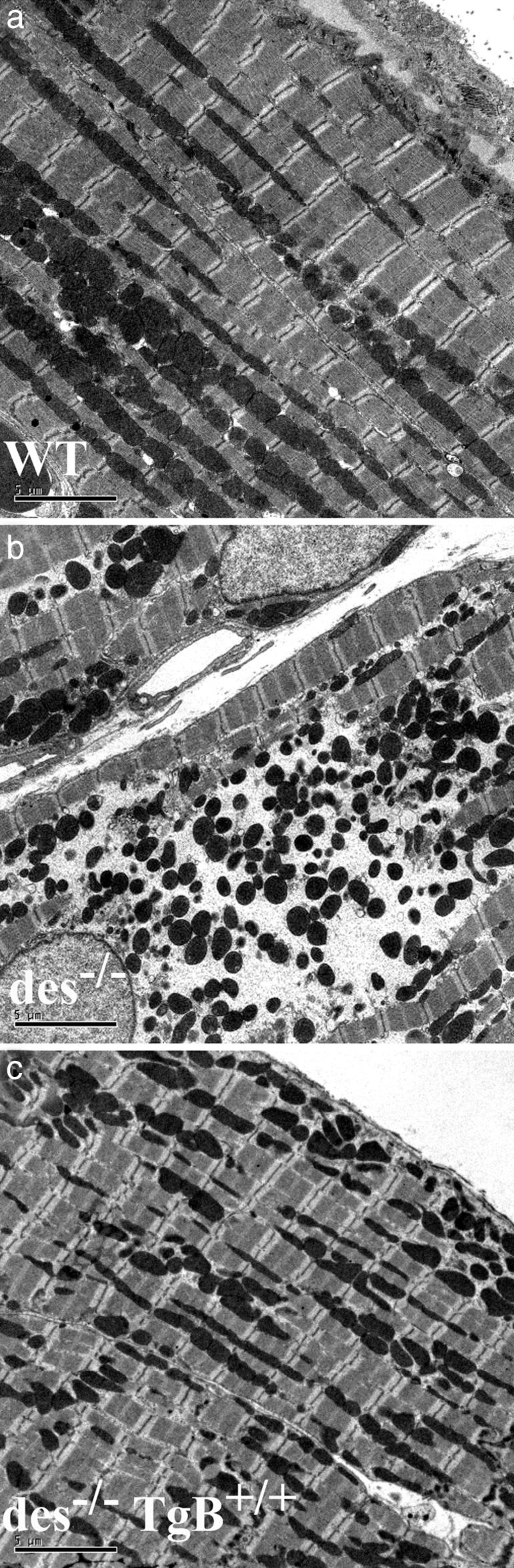
Reduction of damage to mitochondria and contractile apparatus. Transmission electron microscopy was performed in hearts from WT (a), des–/– (b), and des–/– Tg(bcl2)BCap+/+ (c) mice. Most sections from des–/– Tg(bcl2)BCap+/+ hearts (c) appear very similar to WT sections (a), in contrast to des–/– sections that display typical mitochondrial swelling and rupture and broken contractile apparatus (b). Occasionally, some areas within des–/– Tg(bcl2)BCap+/+ hearts display some degree of pathology, but usually not to the extent seen commonly in des–/– tissue. (Scale bar: 5 μm.)
Significant Improvement of Systolic Cardiac Function in des–/– Hearts with Bcl-2 Overexpression. Diminished cardiac systolic function with age is characteristic of desmin null cardiomyopathy (14). Although cardiac bcl-2 overexpression does not cause significant changes in cardiac function itself, it has prevented decreased cardiac function caused by ischemia (3, 4). Des–/– animals display decreased cardiac systolic function with age as measured by doppler echocardiography (14). Similar experiments with 8-month-old animals show that diastolic function was unchanged in all genotypes and no animals display arrhythmia (Table 1). Assessment of systolic function reveals that des–/– Tg(bcl2)BCap+/+ hearts do not experience decreased peak aortic velocity like that seen in des–/– animals (Table 1). Bcl-2-expressing des–/– animals do not completely maintain a WT level of function. This observation mirrors the trend that bcl-2 overexpression in the des–/– heart can diminish the degree of pathology but not completely rescue cardiac function. Consistent observation of this trend implies that multiple mechanisms of cardiac damage may be at work in the desmin null heart. Thus, both cell death and contractile dysfunction caused by altered mechanical force transduction in the absence of desmin could contribute to development of compromised cardiac function, even if cardiomyocyte death is the dominant mechanism of damage.
Table 1. Bcl-2 overexpression prevents decline in cardiac function in desmin null animals.
| Measurement | des+/+ (n = 8) | des-/- (n = 10) | des-/-TgB+/+ (n = 6) |
|---|---|---|---|
| Body weight, g | 23.7 ± 0.4 | 25.2 ± 0.7 | 21.7 ± 0.9 |
| Heart rate, min-1 | 458 ± 14 | 431 ± 13 | 441 ± 15 |
| Peak aortic velocity, cm/s | 88.3 ± 3.1 | 78.3 ± 1.5* | 83.0 ± 1.4 |
| Mean acceleration, cm/s2 | 6,800 ± 300 | 5,200 ± 200* | 6,400 ± 300 |
| Peak E velocity, cm/s | 65.2 ± 3.6 | 65.8 ± 2.7 | 60.7 ± 2.9 |
| Peak A velocity, cm/s | 46.7 ± 4.2 | 35.1 ± 2.8 | 38.9 ± 4.4 |
| E/A ratio, peak velocities | 1.41 ± 0.06 | 2.00 ± 0.22 | 1.68 ± 0.20 |
| IVRT, ms | 17.0 ± 0.9 | 19.8 ± 0.6 | 21.9 ± 1.2 |
| SD of RR intervals, cm | 2.65 ± 0.13 | 3.07 ± 1.43 | 1.87 ± 0.6 |
At 8 months of age des-/- Tg(bcl2)BCap+/+ peak aortic velocity (PAV) was not significantly different from WT. In contrast, PAV was depressed in the desmin null heart. Data are expressed as means ± SE. Each n represents an individual animal. *, P < 0.05 vs. des+/+. E/A, peak E velocity over peak A velocity; IVRT, isovolumic relaxation time; RR, interval between two consecutive R waves.
Increased Sensitivity of des–/– Mitochondria to Calcium Improved by Bcl-2 Overexpression. Mitochondria influence intracellular Ca2+ regulation within myocytes by sequestration of Ca2+ released from the endoplasmic reticulum. Isolated mitochondria from des–/– cardiomyocytes were treated with a high Ca2+ buffer and assessed for their degree of swelling by measurement of absorbance at 540 nm. Isolated des–/– mitochondria swell more than WT mitochondria at the same Ca2+ concentration, revealing des–/– mitochondria are more sensitive than WT mitochondria to Ca2+ (Table 2). Expression of bcl-2 in cardiomyocytes decreases this mitochondrial swelling, indicating that bcl-2 can improve the compromised ability of mitochondria to resist Ca2+ in des–/– cardiomyocytes. This finding suggests that one role of bcl-2 in the prevention of cardiomyocyte death in the des–/– heart is improvement of mitochondria ability to resist Ca2+ within the cell.
Table 2. Desmin null mitochondria display increased sensitivity to calcium induced swelling that can be reversed by bcl-2 expression.
| Mice | n | OD540 decrease | Minimum | Maximum |
|---|---|---|---|---|
| des+/+ | 7 | 0.993 ± 0.024* | 0.972 | 1.033 |
| des-/- | 9 | 1.008 ± 0.021* | 0.979 | 1.038 |
| des-/-TgB+/- | 2 | 0.980 ± 0.002* | 0.978 | 0.981 |
| des-/-TgB+/+ | 14 | 0.999 ± 0.090* | 0.801 | 1.211 |
| des+/+ + Ca2+ | 8 | 0.856 ± 0.050† | 0.758 | 0.910 |
| des-/- + Ca2+ | 9 | 0.780 ± 0.032 | 0.738 | 0.821 |
| des-/-TgB+/- + Ca2+ | 2 | 0.831 ± 0.007 | 0.826 | 0.836 |
| des-/-TgB+/+ + Ca2+ | 14 | 0.836 ± 0.050† | 0.742 | 0.931 |
Both des+/+ and des-/- Tg(bcl2)BCap+/+ mice display significantly improved ability to resist Ca2+ compared with des-/- Tg(bcl2)Bcap-/- animals. Data are expressed as mean percentage decrease in OD540 absorbance from time 0 to 30 min ± SE. Each n represents a mitochondria fraction isolated from a single 2-month-old heart. *, P < 0.005 vs. with Ca2+; †, P < 0.05 vs. des-/- Tg(bcl2)BCap-/- +Ca2+.
Discussion
We have found that bcl-2 overexpression in the des–/– heart corrects the mitochondrial defects and ameliorates cardiomyopathy caused by the absence of desmin. These data suggest that lack of desmin leads to mitochondrial dysfunction of a nature that can be corrected by high, but not lower, levels of bcl-2. The mechanism by which high levels bcl-2 can compensate for the absence of desmin in the present case can only be speculated. Several different proposed functions for bcl-2 in mitochondria could lead to prevention of cell death. It is clear that the ratio of antiapoptotic to proapoptotic members of the bcl-2 family alters the cell fate either toward or away from death (22). With the low endogenous level of bcl-2 in the heart it is possible that an increase in bcl-2 could provide additional protection over what would be seen from overexpression of other members of the bcl-2 family. Observation of a dose-dependent protective effect in des–/– mice indicates that these notions apply in the present case. Increase in bcl-2 expression in des–/– cardiomyocytes has little effect until the level reaches a threshold where the ratio of bcl-2 family members is sufficient to favor cell survival. However, it is unclear what function this additional bcl-2 plays in the cell. Although it is established that bcl-2 family members interact at the mitochondrial membrane and influence ψΔ, the mechanism by which this effect takes place has yet to be determined. Whether the cytoskeleton in general or desmin intermediate filaments in particular facilitate such function directly cannot be defined, given the fact that desmin associates with mitochondria either directly or indirectly (15, 16) and this association can stabilize mitochondrial membrane structure and function.
One mechanism through which bcl-2 could influence mitochondria function and cell survival is by altering the calcium handling dynamics of the cell. Bcl-2 and family members Bax and Bak have been shown to alter cellular response to Ca2+, by affecting either mitochondria or the endoplasmic reticulum (23–25). Mitochondria buffer the cytosolic Ca2+ level by sequestration of Ca2+ released from the endoplasmic reticulum. Destabilization of mitochondrial membranes in des–/– cardiomyocytes could result in compromised tolerance to normal Ca2+ levels, resulting in mitochondrial dysfunction and cell death. Bcl-2 can increase the Ca2+ buffering capacity of mitochondria from neurons (26) and cardiomyocytes (6) and prevent cell death induced by Ca2+ in cultured neurons (27). To test this hypothesis, we treated isolated mitochondria from des–/– cardiomyocytes with a high Ca2+ buffer and measured the degree of mitochondria swelling. Des–/– mitochondria have reduced ability to resist Ca2+, which can be improved by overexpression of bcl-2, suggesting that a mechanism by which bcl-2 prevents cardiomyocyte death in the des–/– heart is by improving of mitochondria ability to resist Ca2+ levels within the cell.
There are several potential mechanisms through which desmin could improve the ability of mitochondria to resist intracellular Ca2+ and prevent cell death. These include an indirect mechanism, perhaps by aligning the connection of mitochondria to the endoplasmic reticulum, thus allowing the creation of Ca2+ microdomains (28). Our results in an isolated mitochondria system suggest that desmin is directly involved in maintaining mitochondrial membrane integrity, particularly in the presence of Ca2+, perhaps by interacting with and stabilizing protein complexes associated with the CS of mitochondria (29). CS are dynamic structures in which the inner and outer mitochondrial membranes fuse. They are key participants in protein transport (29), energy coupling via formation of creatine phosphate (30), and fatty acids uptake for oxidative metabolism (31). Porin (voltage-dependent anion channel, VDAC) channels, which control mitochondrial permeability for ADP, and adenine nucleotide translocator, which in turn controls entry of ADP into the matrix, function at the CS (32). These molecules also function in pore formation in cell death events. CS may also participate in mitochondrial Ca2+ handling as VDAC is thought to function in import of calcium into mitochondria (33, 34). Thus, desmin interaction with CS could contribute to the efficiency of these varied mitochondrial functions, many of which are altered in the des–/– mouse.
Desmin may interact with CS by binding voltage-dependent anion channel (VDAC) either directly or through a desmin-associated protein. The latter possibility is supported by the demonstration that the microtubule binding protein MAP2 binds to VDAC (35). Interestingly, intermediate filaments seem to share common binding sites with mitogen-activated proteins on the brain mitochondrial membrane (36). Determination of the molecular interaction between desmin and mitochondria will reveal a mechanism by which the intermediate filament cytoskeleton can regulate various mitochondrial functions.
Bcl-2 family members are thought to function at mitochondria CS (7–9). A potential mechanism by which elevated bcl-2 could improve pathology in des–/– heart would involve additional bcl-2 at CS restoring a degree of the diminished CS functions in des–/– mitochondria, perhaps by providing additional stability to the protein complexes of CS. Although the specific defects that arise within CS of des–/– mitochondria have not yet been determined, it is probable that these alterations resemble mitochondrial defects that result from changes in bcl-2 family member levels within the cell. Thus, a direct link between desmin and bcl-2 member function is possible. Our studies did not identify any difference in bcl-2 levels between WT and des–/– heart (data not shown), indicating that the des–/– phenotype is not caused by altered bcl-2 expression levels. However, potential changes in bcl-2 mitochondrial localization caused by the absence of desmin have not been excluded. On the contrary, an independent study suggested, although not strongly, that such change might take place in desmin null mice (37). It will be important to verify these data, a prospect complicated because of the low endogenous level of bcl-2 in the heart. The use of bcl-2-overexpressing mice will help achieve this goal.
Although in the past bcl-2 has been shown to reduce cardiomyocyte death in cardiac ischemia models (4), we present an inherited model of cardiomyopathy that was significantly improved by overexpression of bcl-2. The role of bcl-2 members in human heart failure has not been thoroughly evaluated, but it is known that both bcl-2 and bax are expressed at high levels in the failing human heart (38). Because the bcl-2 family has the potential to affect multiple mechanisms of cardiac damage, including ischemia, calcium dysregulation, and increased oxidative stress, it is clear the bcl-2 family members are very attractive therapeutics that can benefit a broad range of cardiac diseases.
Acknowledgments
We thank Thuy Pham and Hank Adams for exceptional technical expertise and assistance. This work was supported by National Institutes of Health Grants AR39617 (to Y.C.) and AG17899 (to G.E.T.).
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: HW/BW, heart-to-body weight ratio; CS, contact sites.
References
- 1.Gill, C., Mestril, R. & Samali, A. (2002) FASEB J. 16, 135–146. [DOI] [PubMed] [Google Scholar]
- 2.McDonnell, T. J., Deane, N., Platt, F. M., Nunez, G., Jaeger, U., McKearn, J. P. & Korsmeyer, S. J. (1989) Cell 57, 79–88. [DOI] [PubMed] [Google Scholar]
- 3.Brocheriou, V., Hagege, A. A., Oubenaissa, A., Lambert, M., Mallet, V. O., Duriez, M., Wassef, M., Kahn, A., Menasche, P. & Gilgenkrantz, H. (2000) J. Gene Med. 2, 326–333. [DOI] [PubMed] [Google Scholar]
- 4.Chen, Z., Chua, C. C., Ho, Y. S., Hamdy, R. C. & Chua, B. H. (2001) Am. J. Physiol. 280, H2313–H2320. [DOI] [PubMed] [Google Scholar]
- 5.Kirshenbaum, L. A. & de Moissac, D. (1997) Circulation 96, 1580–1585. [DOI] [PubMed] [Google Scholar]
- 6.Zhu, L., Yu, Y., Chua, B. H., Ho, Y. S. & Kuo, T. H. (2001) J. Mol. Cell. Cardiol. 33, 2135–2144. [DOI] [PubMed] [Google Scholar]
- 7.Nechushtan, A., Smith, C. L., Lamensdorf, I., Yoon, S. H. & Youle, R. J. (2001) J. Cell Biol. 153, 1265–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vyssokikh, M. Y., Zorova, L., Zorov, D., Heimlich, G., Jurgensmeier, J. J. & Brdiczka, D. (2002) Mol. Biol. Rep. 29, 93–96. [DOI] [PubMed] [Google Scholar]
- 9.He, L., Perkins, G. A., Poblenz, A. T., Harris, J. B., Hung, M., Ellisman, M. H. & Fox, D. A. (2003) Proc. Natl. Acad. Sci. USA 100, 1022–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Capetanaki, Y. & Milner, D. J. (1998) Subcell. Biochem. 31, 463–495. [PubMed] [Google Scholar]
- 11.Dalakas, M. C., Dagvadorj, A., Goudeau, B., Park, K. Y., Takeda, K., Simon-Casteras, M., Vasconcelos, O., Sambuughin, N., Shatunov, A., Nagle, J. W., et al. (2003) Neuromuscul. Disord. 13, 252–258. [DOI] [PubMed] [Google Scholar]
- 12.Milner, D. J., Weitzer, G., Tran, D., Bradley, A. & Capetanaki, Y. (1996) J. Cell Biol. 134, 1255–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li, Z., Colucci-Guyon, E., Pincon-Raymond, M., Mericskay, M., Pournin, S., Paulin, D. & Babinet, C. (1996) Dev. Biol. 175, 362–366. [DOI] [PubMed] [Google Scholar]
- 14.Milner, D. J., Taffet, G. E., Wang, X., Pham, T., Tamura, T., Hartley, C., Gerdes, A. M. & Capetanaki, Y. (1999) J. Mol. Cell. Cardiol. 31, 2063–2076. [DOI] [PubMed] [Google Scholar]
- 15.Milner, D. J., Mavroidis, M., Weisleder, N. & Capetanaki, Y. (2000) J. Cell Biol. 150, 1283–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Capetanaki, Y. (2002) Trends Cardiovasc. Med. 12, 339–348. [DOI] [PubMed] [Google Scholar]
- 17.Oshima, R. G. (2002) Cell Death Differ. 9, 486–492. [DOI] [PubMed] [Google Scholar]
- 18.Chen, F., Chang, R., Trivedi, M., Capetanaki, Y. & Cryns, V. L. (2003) J. Biol. Chem. 278, 6848–6853. [DOI] [PubMed] [Google Scholar]
- 19.Subramaniam, A., Jones, W. K., Gulick, J., Wert, S., Neumannli, J. & Robbins, J. (1991) J. Biol. Chem. 266, 24613–24620. [PubMed] [Google Scholar]
- 20.Nunez, G., London, L., Hockenbery, D., Alexander, M., McKearn, J. P. & Korsmeyer, S. J. (1990) J. Immunol. 144, 3602–3610. [PubMed] [Google Scholar]
- 21.Mela, L. & Seitz, S. (1979) Methods Enzymol. 55, 39–46. [DOI] [PubMed] [Google Scholar]
- 22.Korsmeyer, S. J., Shutter, J. R., Veis, D. J., Merry, D. E. & Oltvai, Z. N. (1993) Semin. Cancer Biol. 4, 327–332. [PubMed] [Google Scholar]
- 23.Pinton, P., Ferrari, D., Magalhaes, P., Schulze-Osthoff, K., Di Virgilio, F., Pozzan, T. & Rizzuto, R. (2000) J. Cell Biol. 148, 857–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Foyouzi-Youssefi, R., Arnaudeau, S., Borner, C., Kelley, W. L., Tschopp, J., Lew, D. P., Demaurex, N. & Krause, K. H. (2000) Proc. Natl. Acad. Sci. USA 97, 5723–5728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Scorrano, L., Oakes, S. A., Opferman, J. T., Cheng, E. H., Sorcinelli, M. D., Pozzan, T. & Korsmeyer, S. J. (2003) Science 300, 135–139. [DOI] [PubMed] [Google Scholar]
- 26.Murphy, A. N., Bredesen, D. E., Cortopassi, G., Wang, E. & Fiskum, G. (1996) Proc. Natl. Acad. Sci. USA 93, 9893–9898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Murphy, A. N. & Fiskum, G. (1999) Biochem. Soc. Symp. 66, 33–41. [DOI] [PubMed] [Google Scholar]
- 28.Hajnóczky G., Csordás, G., Madesh, M. & Pacher, P. (2000) J. Physiol. (London) 529, 69–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van der Klei, I. J., Veenhuis, M. & Neupert, W. (1994) Microsc. Res. Tech. 27, 284–293. [DOI] [PubMed] [Google Scholar]
- 30.Brdiczka, D. & Wallimann, T. (1994) Mol. Cell. Biochem. 133–134, 69–83. [DOI] [PubMed] [Google Scholar]
- 31.Fraser, F. & Zammit, V. A. (1998) Biochem. J. 329, 225–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Crompton, M., Barksby, E., Johnson, N. & Capano, M. (1997) Biochimie 84, 143–152. [DOI] [PubMed] [Google Scholar]
- 33.Gincel, D., Zaid, H. & Shoshan-Barmatz, V. (2001) Biochem. J. 328, 147–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rapizzi, E., Pinton, P., Szabadkai, G., Wieckowski, M. R., Vandecasteele, G., Baird, G., Tuft, R. A., Fogarty, K. E. & Rizzuto, R. (2002) J. Cell Biol. 159, 613–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Linden, M. & Karlsson, G. (1996) Biochem. Biophys. Res. Commun. 218, 833–836. [DOI] [PubMed] [Google Scholar]
- 36.Leterrier, J. F., Rusakov, D. A., Nelson, B. D. & Linden, M. (1994) Microsc. Res. Tech. 27, 233–261. [DOI] [PubMed] [Google Scholar]
- 37.Linden, M., Li, Z., Paulin, D., Gotow, T. & Leterrier, J. F. (2001) J. Bioenerg. Biomembr. 33, 333–341. [DOI] [PubMed] [Google Scholar]
- 38.Latif, N., Khan, M. A., Birks, E., O'Farrell, A., Westbrook, J., Dunn, M. J. & Yacoub, M. H. (2000) J. Am. Coll. Cardiol. 35, 1769–1777. [DOI] [PubMed] [Google Scholar]