

Abstract
In vivo electroporation is a powerful method for delivering DNA expression plasmids, RNAi reagents, and morpholino anti-sense oligonucleotides to specific regions of developing embryos, including those of C. elegans, chick, Xenopus, zebrafish, and mouse 1. In zebrafish, in vivo electroporation has been shown to have excellent spatial and temporal resolution for the delivery of these reagents 2-7. The temporal resolution of this method is important because it allows for incorporation of these reagents at specific stages in development. Furthermore, because expression from electroporated vectors occurs within 6 hours 7, this method is more timely than transgenic approaches. While the spatial resolution can be extremely precise when targeting a single cell 2, 6, it is often preferable to incorporate reagents into a specific cell population within a tissue or structure. When targeting multiple cells, in vivo electroporation is efficient for delivery to a specific region of the embryo; however, particularly within the developing nervous system, it is difficult to target specific cell types solely through spatially discrete electroporation. Alternatively, enhancer trap transgenic lines offer excellent cell type-specific expression of transgenes 8. Here we describe an approach that combines transgenic Gal4-based enhancer trap lines 8 with spatially discrete in vivo electroporation 7, 9 to specifically target developing neurons of the zebrafish olfactory bulb. The Et(zic4:Gal4TA4,UAS:mCherry)hzm5 (formerly GA80_9) enhancer trap line previously described 8, displays targeted transgenic expression of mCherry mediated by a zebrafish optimized Gal4 (KalTA4) transcriptional activator in multiple regions of the developing brain including hindbrain, cerebellum, forebrain, and the olfactory bulb. To target GFP expression specifically to the olfactory bulb, a plasmid with the coding sequence of GFP under control of multiple Gal4 binding sites (UAS) was electroporated into the anterior end of the forebrain at 24-28 hours post-fertilization (hpf). Although this method incorporates plasmid DNA into multiple regions of the forebrain, GFP expression is only induced in cells transgenically expressing the KalTA4 transcription factor. Thus, by using the GA080_9 transgenic line, this approach led to GFP expression exclusively in the developing olfactory bulb. GFP expressing cells targeted through this approach showed typical axonal projections, as previously described for mitral cells of the olfactory bulb 10. This method could also be used for targeted delivery of other reagents including short-hairpin RNA interference expression plasmids, which would provide a method for spatially and temporally discrete loss-of-function analysis.
Keywords: Neuroscience, Issue 54, electroporation, zebrafish, olfactory bulb, Gal4 enhancer trap
Protocol
1. Transgenic embryos
Here we are using a transgenic line of zebrafish created through a transposon-mediated enhancer trap method 8. The transgenic insert includes two coding sequences: KalTA4, a zebrafish-optimized version of the Gal4 transcriptional activator, and the red fluorescent protein, mCherry. The enhancer trap method yields lines of transgenic zebrafish that express this transgenic cassette under control of an endogenous enhancer sequence. Thus, depending on the identity of the specific enhancer sequence, expression of both KalTA4 and mCherry is localized to specific regions of the developing brain. For the Et(zic4:Gal4TA4,UAS:mCherry)hzm5 line that we are using here, expression is localized to several sites in the hindbrain, forebrain, and, of particular importance here, the olfactory bulb 8 (see also Fig. 1). Given this pattern of expression, and the localization of the cassette within the genome, the transgenes are predicted to be under control of the zic4 promoter 8 (see also http://www.helmholtz-muenchen.de/en/idg/groups/neuroimaging/lines_distel/main.).
Adult Et(zic4:Gal4TA4,UAS:mCherry)hzm5 fish heterozygous for the transgenic insert are typically mated with WT (AB) fish, leading to approximately 50% transgenic offspring. (For a higher percentage of transgenic offspring, two heterozygotes can be mated.)
In order to block pigment formation and facilitate fluorescent imaging, treat transgenic embryos with 100 μM N-phenylthiourea (PTU) beginning at 6-8 hpf.
2. Mounting embryos
At 24-28 hpf, transfer embryos to egg water without PTU. Identify transgenic embryos by expression of mCherry using a fluorescent microscope. Remove the chorionic membrane from transgenic embryos using fine forceps. Transfer embryos freed from their chorion to a Petri dish containing electroporation buffer plus 0.02% tricaine [Electroporation Buffer: 180 mM NaCl, 5 mM KCl, 1.8 mM CaCl2 , 5 mM HEPES, pH 7.2] 4. Allow 2 minutes for the anesthetic to take effect before proceeding to the mounting step.
Prepare a 0.5% solution of low-melting-point agarose in electroporation buffer plus tricaine by microwaving the solution, and then placing the solution into a 34-37°C incubator. Once the temperature of the agarose solution has equilibrated, place a large drop (˜500 μl) of the agarose solution to the center of a 60 mm Petri dish. Add 6-8 anesthetized embryos to this drop of agarose.
Using fine forceps (or the fine ends of cut microloader pipet tips), position the embryos such that they are all facing in the same direction and aligned in a vertical row. To solidify the agarose, and trap the embryos in position, place the dish onto a large Petri dish with 2-3 mm of ice.
Once the agarose has solidified, flood the dish with electroporation buffer plus tricaine, ensuring that the solution completely covers the solidified agarose drop.
3. Micro-injection of DNA expression plasmid
Prepare the GFP expression plasmid using a midi- or maxi-prep plasmid isolation kit (e.g. Qiagen). Suspend the plasmid to a final concentration of 0.5 μg/μl in sterile water plus 0.03% phenol red. For the olfactory bulb targeting experiment described here, we are using an expression plasmid with the coding sequence of EGFP under the control of multiple Gal4 binding sites (14 tandem UAS sequences and the basal fish promoter, E1b). Using this plasmid, only cells transgenically expressing the KalTA4 transcription factor will be able to express GFP; thus, mediating targeted expression of GFP. Note: here we are using phenol red for visualization of DNA injections, which should be applicable for targeting other regions of the developing brain, as long as the region can be accessed from the ventricles. For targeting of other cell types that require fluorescent visualization, Bodipy or Quantam Dot dyes could potentially be used to visualize injections under fluorescent illumination.
Microinjection pipettes can be fabricated using either a horizontal or vertical pipette puller. Heat and pull-strength settings will vary depending on the type of puller, the type of heating element, and the type of glass capillary tubes. Using a Sutter P-30 vertical puller, use a heat setting of 980 and a pull-force of 960 to produce long, tapered, sharp pipettes from thin-walled borosilicate capillary glass with filaments [Sutter Instrument #BF100-78-10]. Microinjetion pipettes pulled with these settings are sealed and must be broken back before injection.
Use a microloader pipette tip to add 1-2 μl of DNA plasmid solution to the injection pipette.
Mount and secure the loaded pipette into the pressure injection system pipette holder.
Break back the loaded pipette with fine forceps until pressure pulses produce puffed release of the red DNA solution. Alternatively, tips can be broken back by quickly penetrating a submerged, folded laboratory Kimwipe. Note: because the injection pipettes are initially sealed and must be manually broken back, the tip size and injection volume are variable. Ideal injection pipettes should require 3-5 injection pulses to completely fill the developing forebrain ventricle of 24 hpf embryos (100 ms injection pulses at 40 psi).
Inject the plasmid DNA solution into the ventricles of the developing brain such that DNA is forced adjacent to, and intercalated into, the region of brain destined to form the olfactory bulb. The olfactory bulb is derived from cells in the most anterior wall of the forebrain ventricle. Thus, by inserting the injection pipette into the ventricles such that the pipette is pointing toward the anterior end of the developing brain, pressure injection forces the DNA into and adjacent to the prospective olfactory bulb. Inject DNA until red dye is clearly visible at the anterior end of the forebrain ventricle. Because the DNA begins to diffuse immediately, diluting the DNA, it is important to proceed to the electroporation step as soon as possible after injection (e.g., within 10 sec).
4. Electroporation
Electroporation is carried out using self-constructed electrodes made from Grass platinum subdermal electrodes (Grass Technology #E2). The electrodes are attached to a hand-held probe, and the final distance between the platinum electrodes should be 1 mm (for details on construction see 9). Square-wave electroporation pulses are produced by connecting the electrodes to a Grass SD-9 Stimulator (Grass Technology, Model SD-9).
Set the SD-9 stimulator to produce single, 5 ms electroporation pulses at 70 volts. Position the electrodes such that the positive electrode is positioned adjacent to the anterior end of the embryo, while the negative electrode is posterior to the embryo head. Manually initiate ˜7-8 electroporation pulses using the single mode switch of the SD-9 stimulator, separating each pulse by ˜1 sec. Appropriate voltages will vary depending on the age of the embryos and the voltage source used. Younger embryos require lower voltages to avoid embryo damage, whereas older embryos require higher voltages to initiate electroporation 7.
Allow the embryos to recover for at least 10 min after electroporation before freeing them from the agarose.
Using fine forceps, free the embryos from the agarose by gently tracing the outline of the embryos while avoiding actual contact with the embryos themselves.
Once freed, return the embryos to egg water (with PTU if needed). Keep them at 28°C until they are to be imaged for GFP expression.
5. Mounting embryos for imaging
Transfer embryos from egg water plus PTU to egg water plus 0.02% tricaine. Allow several minutes for the anesthetic to take effect before proceeding to the mounting step.
Prepare a 0.5% solution of low-melting-point agarose in egg water plus tricaine by microwaving the solution, and then placing the solution into a 34-37°C incubator. Once the temperature of the agarose solution has equilibrated, place a large drop (˜300 μl) of the agarose solution to the center of a 35 mm Petri dish. Add one anesthetized embryo to this drop of agarose.
Using fine forceps (or the fine ends of cut microloader pipet tips), position the embryos such that they are upright, making it possible to visualize a dorsal view of the developing brain with an upright microscope (inverted scopes will require a different embryo geometry, and a different dish allowing visualization from below; see 12). Solidify the agarose by placing the dish onto a large Petri dish with 2-3 mm of ice. Re-orientation with fine forceps will likely be required during the solidification to ensure that the embryo does not fall over onto its side.
Once the agarose has solidified, flood the dish with egg water plus tricaine ensuring that the solution completely covers the solidified agarose drop.
6. Representative Results:
Although GFP expression can be observed as early as 6 hours after electroporation, here we are showing images from an embryo 4 days after electroporation such that the neurite structure of the targeted cells has sufficiently developed. The Et(zic4:Gal4TA4,UAS:mCherry)hzm5 transgenic enhancer trap line displays constitutive expression of mCherry (and KalTA4) in the hindbrain, cerebellum, forebrain, and olfactory bulb in embryos at 5 days post-fertilization (Fig. 1A and 1C). Embryos that have had a UAS-GFP expression plasmid incorporated by targeted electroporation (as described above), show GFP expression that is restricted to cells of the developing olfactory bulb (Fig. 1B, 1D, and 1E). Only cells transgenically-expressing the KalTA4 transcription factor are able to activate expression from this UAS-GFP plasmid. Incorporation of this plasmid into non-transgenic embryos does not lead to any detectable GFP expression. Thus, it is the combined action of targeted electroporation of UAS-GFP and localized transgenic expression of the KalTA4 driver that leads to exclusive expression of GFP in cells of the developing olfactory bulb. The GFP-expressing cells show typical axonal projections of olfactory bulb mitral cells (Fig. 1B and 1D) 10. In order to visualize axon projections, images in figure 1B and 1D had to be exposed at high levels such that the region of cell bodies were well over-exposed. A lower level of exposure (Fig. 1E) shows that cell bodies expressing GFP are indeed localized to the olfactory bulb (compare Fig. 1C and 1E; gray line marks the border of the olfactory bulb).
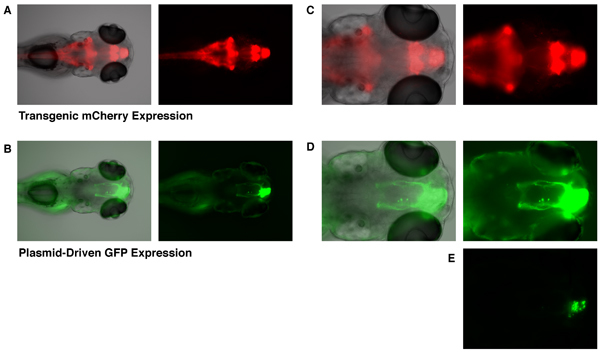 Figure 1. Targeted GFP expression in the olfactory bulb of a 5 dpf zebrafish embryo. The Et(zic4:Gal4TA4,UAS:mCherry)hzm5 transgenic enhancer trap line, displays constitutive expression of mCherry in the hindbrain, cerebellum, forebrain, and olfactory bulb (A and C, mCherry fluorescence shown in red). This expression of mCherry is mediated by transgenic Gal4-expression. Embryos that have been electroporated with a UAS-GFP expression vector at 28 hpf, display targeted GFP expression in the olfactory bulb at 5 days post-fertilization (B, D, and E, GFP fluorescence shown in green. Same embryo as in A and C). A lower exposure image at bottom (E) shows that GFP expression is limited to cell bodies of the olfactory bulb. The gray line indicates the border of the olfactory bulb. Bright field (gray scale), green fluorescence (green), and red fluorescence (red) images were all take with an Olympus BX60 fluorescence microscope.
Figure 1. Targeted GFP expression in the olfactory bulb of a 5 dpf zebrafish embryo. The Et(zic4:Gal4TA4,UAS:mCherry)hzm5 transgenic enhancer trap line, displays constitutive expression of mCherry in the hindbrain, cerebellum, forebrain, and olfactory bulb (A and C, mCherry fluorescence shown in red). This expression of mCherry is mediated by transgenic Gal4-expression. Embryos that have been electroporated with a UAS-GFP expression vector at 28 hpf, display targeted GFP expression in the olfactory bulb at 5 days post-fertilization (B, D, and E, GFP fluorescence shown in green. Same embryo as in A and C). A lower exposure image at bottom (E) shows that GFP expression is limited to cell bodies of the olfactory bulb. The gray line indicates the border of the olfactory bulb. Bright field (gray scale), green fluorescence (green), and red fluorescence (red) images were all take with an Olympus BX60 fluorescence microscope.
Discussion
Here we have described an in vivo electroporation method in zebrafish that utilizes an enhancer trap Gal4 transgenic line to target expression of the electroporated transgene to a specific population of cells in the developing olfactory bulb. This approach combines the excellent temporal resolution of in vivo electroporation 7 with the cell-type specific expression mediated by enhancer trap transgenic lines 8. Although here we have described the targeting of the olfactory bulb, in vivo electroporation can be used to target other regions of the developing nervous system 2-7, and enhancer trap lines are available with targeted expression of Gal4 in many different specific cell types or tissues 8, 11. Of course, this electroporation technique should also be suitable for other combinatioral genetic approaches such as the LexA or Tet systems 13, 14, 15. A major advantage of electroporation is that there is not need to make additional transgenic lines given that a suitable Gal4-line is available. This saves six months.
In vivo electroporation allows for the incorporation of oligonucleotides into neurons and their precursors at whatever specific stage of nervous systems development is of interest 7. This temporal resolution is particularly advantageous for loss-of-function analysis because it can circumvent problems with targeting genes that have essential functions at earlier developmental stages. The method we describe here can also be used to incorporate loss-of-function reagents including RNAi oligonucleotides or constructs and morpholino anti-sense oligonucleotides 4, 7. Plasmid-driven reagents such as dominant-negative protein expression plasmids or short-hairpin RNAi plasmids can also be placed under control of a Gal4 UAS, allowing for the precise cell type-specific expression we have shown here for expression of GFP.
Disclosures
No conflicts of interest declared.
Acknowledgments
This work was funded through an NIH R15 AREA grant to John Horne (NIMH; R15MH083221).
References
- Swartz M, Eberhart J, Mastick G, Krull CE. Sparking new frontiers: using in vivo electroporation for genetic manipulations. Dev Biol. 2001;233:13–21. doi: 10.1006/dbio.2001.0181. [DOI] [PubMed] [Google Scholar]
- Bhatt DH, Otto SJ, Depoister B, Fetcho JR. Cyclic AMP-induced repair of zebrafish spinal circuits. Science. 2004;305:254–258. doi: 10.1126/science.1098439. [DOI] [PubMed] [Google Scholar]
- Teh C, Parinov S, Korzh V. New ways to admire zebrafish: progress in functional genomics research methodology. Biotechniques. 2005;38:897–906. doi: 10.2144/05386RV01. [DOI] [PubMed] [Google Scholar]
- Cerda GA, Thomas JE, Allende ML, Karlstrom RO, Palma V. Electroporation of DNA, RNA, and morpholinos into zebrafish embryos. Methods. 2006;39:207–211. doi: 10.1016/j.ymeth.2005.12.009. [DOI] [PubMed] [Google Scholar]
- Hendricks M, Jesuthasan S. Electroporation-based methods for in vivo, whole mount and primary culture analysis of zebrafish brain development. Neural Development. 2007;15:2–6. doi: 10.1186/1749-8104-2-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tawk M, Bianco IH, Clarke JD. Focal electroporation in zebrafish embryos and larvae. Methods Mol Biol. 2009;546:145–151. doi: 10.1007/978-1-60327-977-2_10. [DOI] [PubMed] [Google Scholar]
- Kera SA, Agerwala SM, Horne JH. The temporal resolution of in vivo electroporation in zebrafish: a method for time-resolved loss-of-function. Zebrafish. 2010;7:97–108. doi: 10.1089/zeb.2009.0620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Distel M, Wullimann MF, Köster RW. Optimized Gal4 genetics for permanent gene expression mapping in zebrafish. Proc Natl Acad Sci U S A. 2009;106:13365–13370. doi: 10.1073/pnas.0903060106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoegler KJ, Horne JH. Targeting the zebrafish optic tectum using in vivo electroporation. Cold Spring Harb. Protoc. 2010. [DOI] [PMC free article] [PubMed]
- Nobuhiko M, Kozo M, Tatsuya T, Shin-ichi H, Hitoshi O, Yoshihiro Y. From the olfactory bulb to higher brain centers: genetic visualization of secondary olfactory pathways in zebrafish. J. Neurosci. 2009;29:4756–4766. doi: 10.1523/JNEUROSCI.0118-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davison JM, Akitake CM, Goll MG, Rhee JM, Gosse N, Baier H, Halpern ME, Leach SD, Parsons MJ. Transactivation from Gal4-VP16 transgenic insertions for tissue-specific cell labeling and ablation in zebrafish. Developmental Biology. 2007. pp. 304–811. [DOI] [PMC free article] [PubMed]
- Distel M, Kö ster, W R. in vivo time-lapse imaging of zebrafish embryonic development. CSH Protocols. 2007. [DOI] [PubMed]
- Emelyanov A, Parinov S. Mifepristone-inducible LexPR system to drive and control gene expression transgenic zebrafish. Developmental Biology. 2008;320:113–121. doi: 10.1016/j.ydbio.2008.04.042. [DOI] [PubMed] [Google Scholar]
- Knopf F, Schnabel K, Haase C, Pfeifer K, Anastassiasdis K, Weidinger G. Dually inducible TetON systems for tissue-specific conditional gene expression in zebrafish. PNAS. 1993. pp. 107–10. [DOI] [PMC free article] [PubMed]
- Zhu P, Narita Y, Bundschuh ST, Fajardo O, Schärer YP, Chattopadhyaya B, Bouldoires EA, Stepien AE, Deisseroth K, Arber S, Sprengel R, Rijli FM, Friedrich RW. Optogenetic Dissection of Neuronal Circuits in Zebrafish using Viral Gene Transfer and the Tet System. Front Neural Circuits. 2009;3:21–21. doi: 10.3389/neuro.04.021.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]