

Abstract
In order to understand the function of genes expressed in specific region of the developing brain, including signaling molecules and axon guidance molecules, local gene transfer or knock- out is required. Gene targeting knock-in or knock-out into local regions is possible to perform with combination with a specific CRE line, which is laborious, costly, and time consuming. Therefore, a simple transfection method, an in utero electroporation technique, which can be performed with short time, will be handy to test the possible function of candidate genes prior to the generation of transgenic animals 1,2. In addition to this, in utero electroporation targets areas of the brain where no specific CRE line exists, and will limit embryonic lethality 3,4. Here, we present a method of in utero electroporation combining two different types of electrodes for simple and convenient gene transfer into target areas of the developing brain. First, a unique holding method of embryos using an optic fiber optic light cable will make small embryos (from E9.5) visible for targeted DNA solution injection into ventricles and needle type electrodes insertion to the targeted brain area 5,6. The patterning of the brain such as cortical area occur at early embryonic stage, therefore, these early electroporation from E9.5 make a big contribution to understand entire area patterning event. Second, the precise shape of a capillary prevents uterine damage by making holes by insertion of the capillary. Furthermore, the precise shape of the needle electrodes are created with tungsten and platinum wire and sharpened using sand paper and insulated with nail polish 7, a method which is described in great detail in this protocol. This unique technique allows transfection of plasmid DNA into restricted areas of the brain and will enable small embryos to be electroporated. This will help to, open a new window for many scientists who are working on cell differentiation, cell migration, axon guidance in very early embryonic stage. Moreover, this technique will allow scientists to transfect plasmid DNA into deep parts of the developing brain such as thalamus and hypothalamus, where not many region-specific CRE lines exist for gain of function (GOF) or loss of function (LOF) analyses.
Keywords: Neuroscience, Issue 54, In utero, electroporation, transfection, CNS, gene expression, gain of function, loss of function, neuron, axon
Protocol
1. Preparation of capillary and DNA solution for injection
Purify plasmid DNA with a Maxi-prep or equivalent method, and make a final concentration of 2-3 μg/μl. Aliquot 100 μg of DNA, add 10μl of fast green dye (1% stock) and TE (10mM Tris Base, 1mM EDTA Solution, pH 8.0) and adjust a volume to 100 μl to make the final DNA concentration for injection mix to 1μg/μl. The concentration of plasmid DNA can be changed depends on the size of plasmid, and also its transfection efficiency, which needs to be tested individually.
Spin the DNA solution for 5 minutes at 14,000 RPM at room temperature and collect supernatant to remove any residue.
Pull a glass capillary tubea glass capillary tubes using a micropipette puller. Pinch off the tip with forceps to make the tip has a 20–30 μm diameter. Measure 800 μm-1 mm from the tip and check the external diameter is less than 60 μm (Fig. 1A).
Connect the capillary to the micromanipulator, and fill with mineral oil. To fill the capillary with the DNA solution, submerge the tip into the solution and suck up the DNA solution.
2. Stick platinum electrodes
Here we used stick platinum electrodes (Nepe gene, CUY611P2-1) to electroporate into superficial region of brain, such as the cortex (Fig. 1F).
3. Preparation of micro-electrodes
Use micro-electrodes to electroporated into deep parts of the brain (i.e., thalamus and hypothalamus) (Fig. 2B-E).
One end of the tungsten (for negative electrodes) and platinum (for positive electrodes) wires is coiled on gold-plated pins and fixed with parafilm. Insert the wire to the stem of a cotton swab and wrap both ends with parafilm (Fig. 1B).
Evenly sharpen the tip of the wire with sandpaper until it reaches 20–30 μm external diameter and 60 μm from 800 μm-1 mm of the tip (Fig. 1C and D).
Apply a thin coat of nail polish to the wire for insulation. After the nail polish has dried, use an acetone-soaked cotton swab to remove it from the tip (about 200 μm from the tip). Important note: In order to prevent damage to the mouse’s uterus, the portion within 800 μm-1 mm of the tip should be no more than 70 μm in diameter (Fig. 1E).
4. Preparation of the surgery
Anesthetize a pregnant mouse (E9.5-E15.5). We demonstrate anesthetization with pharmaceutical grade sodium pentobarbital (Table II; 50 μg per gram body weight, inject intraperitoneally and wait for 5 min). Alternatives to sodium pentobarbital as an anesthetic include Ketamine/Xylazine injection or gas anesthetics (gas is preferable for short duration procedures). The injection should be done with attention to avoid any organs, otherwise it will cause to damage the uterus or kill animal. When successful IP is done, the survival rate is more than 95%. Also the surgery can be performed in open-air environment. Before started the operation, test for a lack of response by the animal to pinching their toe to confirm anesthetic.
Shave the hair from the abdomen using a razor blade and 50% EtOH. Prepare the skin with alternating scrubs of betadine and alcohol (70%).
Make an incision at the abdominal midline with fine scissors and pull out all uterine horns carefully onto a 37 °C pre-warmed phosphate-buffered saline (PBS)-moistened cotton gauze, which is placed around the wound. Keep the uterus moist with PBS all of the time.
Hold a flexible fiber optic cable between the index and middle fingers and place it under the uterine horn. Clean cable with 70% EtOH prior to use. No microscope or magnifier is required for visualization. Position uterus between the optic fiber light optic cable and the thumb, and squeeze gently to push up the embryo closer to the uterine wall.
5. Injection of DNA and electroporation
When embryo is positioned, insert glass capillary carefully into target ventricle and inject approx 1 μl of DNA solution (Fig. 2A).
For electroporation to superficial part of the brainIf using with stick platinum electrodes for electoporation, place the electrodes outside of the uterus and apply suggested square-wave current pulses according to manufacture’s instruction (Table I). If usingFor electroporation to deeper part of the brain, nmicro electroporation, eedle type electrodes are used. Iinsert a fine tungsten negative electrode into DNA injected ventricle and a platinum electrode into the uterus and place target region between two electrodes, then apply suggested square-wave current pulses (Table I) (Fig. 2B).
6. Post electroporation
Place the uterine horn back in its original location with and add 500 μL of 37 °C pre-warmed PBS.
Suture the inner layer with surgical suture and close outer layer with a 9 mm autoclipauto clip.
Place animals on a heating pad for 2 hours to allow recovery from anesthesia. Analgesics such as carprofen or buprenorphine can be administered for post operative pain.
7. Representative results:
Some examples of cortical electroporation of pCAG-EYFP construct using stick platinum electrodes at different time points are shown in figure Figure 32. Brains are electroporated at E13.5, E14.5 and E15.5, and harvested at postnatal day (P) 6. AThe ntibody staining of RORc reveals position of layer 4 and position of EYFP transfected cells are revealed by GFP antibody staining, and both images are super imposed (Fig. 3A and B). Labeled cortical cells are layer of cortical cells electroporated is clearly different in each experiment (Fig. 32), which suggests time- dependent electroporation makes cortical layer-specific GOF/LOF possible.
The viability of embryos and efficiency of cell transfection varies depending on the age of embryo and the location of electroporation. Samples are listed on Table 1, however, conditions should be optimized for each electroporation depending on the stage and part of the brain.
Electroporation into a deep tissue such as thalamus and hypothalamus with using stick platinum electrodes often give low efficiency (Table 1). This problem can be solved by using micro-electrodes whichelectrodes, which will reach to deep parts of the brain. Figure 3 4 shows example of pCAG-EYFP electroporation into the developing diencephalon. Plasmid DNA solution is injected into the 3rd ventricle at E11.5 and followed by micro electroporation (Fig. 3C4C). The electroporated area is restricted in the thalamus where most of the axons project to the cortex (Fig. 3B4B). Axon projection to layer 4 of the cortex can be also visualized by EYFP, which fillesfills up the entire neuron. (Fig. 3A4A).
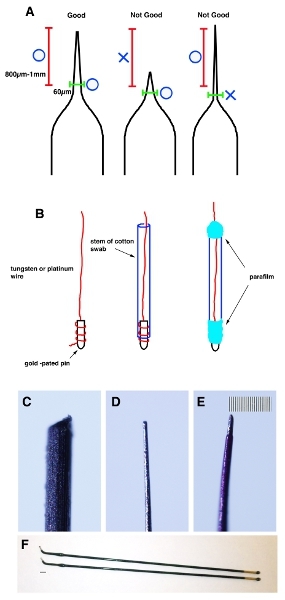 Figure 1. Cartoon schematic demonstrating the correct shape of the capillary. (A) A glass capillary tube is pulled using a micropipette puller to make certain shape. A tip is pinched off by forceps to make it 20–30 μm. Measure 800 μm-1mm from the tip (red bracket) and make sure that external diameter is less than 60 μm (green bracket). (B) One end of the tungsten or platinum wires is coiled on gold-plated pins and fixed with parafilm. Then use sand paper to make their tip to become 20-30 μm external diameters and 60 μm from 1 mm of the tip. (C) Image of the tungsten wire before shaping. (D) Image of the tungsten wire after shaping. (E) Image of the tungsten wire after applying thin coat of nail polish. Scale bar in E is 10 μm (smallest scale) for C-E. (F) Stick platinum electrodes (Nepa gene. CUY611P2-1). Scale bar in F is 2 mm.
Figure 1. Cartoon schematic demonstrating the correct shape of the capillary. (A) A glass capillary tube is pulled using a micropipette puller to make certain shape. A tip is pinched off by forceps to make it 20–30 μm. Measure 800 μm-1mm from the tip (red bracket) and make sure that external diameter is less than 60 μm (green bracket). (B) One end of the tungsten or platinum wires is coiled on gold-plated pins and fixed with parafilm. Then use sand paper to make their tip to become 20-30 μm external diameters and 60 μm from 1 mm of the tip. (C) Image of the tungsten wire before shaping. (D) Image of the tungsten wire after shaping. (E) Image of the tungsten wire after applying thin coat of nail polish. Scale bar in E is 10 μm (smallest scale) for C-E. (F) Stick platinum electrodes (Nepa gene. CUY611P2-1). Scale bar in F is 2 mm.
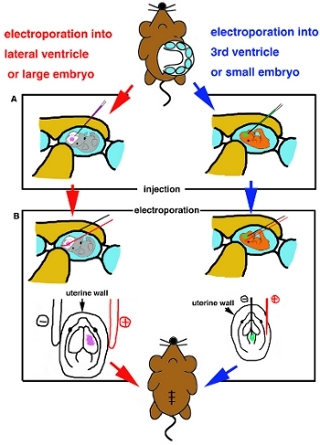 Figure 2. Cartoon schema of surgical procedure. (A) Diagram of injection into lateral ventricle or large embryos (left side) and into 3rd ventricle or small embryos (right side). (B) Diagram of electrporation by platinum stick electrode (left side) and by needle type electrode (right side).
Figure 2. Cartoon schema of surgical procedure. (A) Diagram of injection into lateral ventricle or large embryos (left side) and into 3rd ventricle or small embryos (right side). (B) Diagram of electrporation by platinum stick electrode (left side) and by needle type electrode (right side).
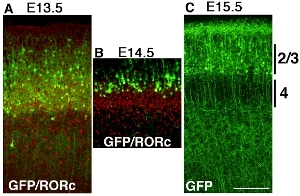 Figure 3. Developing mouse telencephalon was electroporated with pCAG-EYFP at E13.5 (A), E14.5 (B) and E15.5 (C) and harvested at P6. Brains were sectioned coronal and stained with GFP antibody or RORc antibody separately and images are merged. (A) Electroporation of pCAG-EYFP plasmid at E13.5 transfect many layer 4 neurons. which overlap with layer 4 markers RORc. (B) Electroporation of pCAG-EYFP plasmid at E14.5 transfect cells, which are more superficial than layer 4 neurons. (C) E15.5 electroporation label many superficial neurons such as layer 2/3. Scale bar 200 μm.
Figure 3. Developing mouse telencephalon was electroporated with pCAG-EYFP at E13.5 (A), E14.5 (B) and E15.5 (C) and harvested at P6. Brains were sectioned coronal and stained with GFP antibody or RORc antibody separately and images are merged. (A) Electroporation of pCAG-EYFP plasmid at E13.5 transfect many layer 4 neurons. which overlap with layer 4 markers RORc. (B) Electroporation of pCAG-EYFP plasmid at E14.5 transfect cells, which are more superficial than layer 4 neurons. (C) E15.5 electroporation label many superficial neurons such as layer 2/3. Scale bar 200 μm.
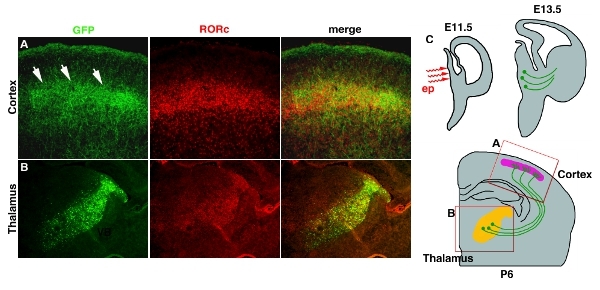 Figure 4. Transfection of EYFP into thalamo-cortical axons (TCA). (A) TCA terminus in cortical layer 4 is revealed by GFP antibody staining. RORc antibody staining is used to reveal a position of layer 4. Both images are taken from same section and merged. (B) Position of electroporation into the thalamus is also shown by GFP antibody staining and RORc staining, which expression is restricted in the thalamus 11. (C) Cartoon schematic for thalamic electroporation and TCA projection are shown. Scale bar 200 μm.
Figure 4. Transfection of EYFP into thalamo-cortical axons (TCA). (A) TCA terminus in cortical layer 4 is revealed by GFP antibody staining. RORc antibody staining is used to reveal a position of layer 4. Both images are taken from same section and merged. (B) Position of electroporation into the thalamus is also shown by GFP antibody staining and RORc staining, which expression is restricted in the thalamus 11. (C) Cartoon schematic for thalamic electroporation and TCA projection are shown. Scale bar 200 μm.
Discussion
In this protocol, we only described benefits of the technique of the pCAG-EYFP transfection into the restricted area of a small embryonic brain using different shaped electrodes. For comparison of transfection efficiency by different promoter is described previously, which shows cell type specificity 1. However, there are limitations for the technique, such as the transfection is only transient and varies depend on the plasmid, it does not have any cell specificity and it is hard to control numbers of transfected cells. ThereforeHowever, combining this protocol with other techniques will further provide possibilities for different manipulation methods. First, incorporating cell specific promoter into plasmid DNA will allow cell-type specific transfection, such as neurons, glial cells and astrocytes. Second, since the transfection is transient, it is not suitable for scientists who want to analyze the function of the gene in later life. Therefore, combination with a transposase to make plasmid DNA integrate into the genome will convert it into a stable transfection 8. Next, adoption of the tet-on or tet-off system will make it possible to manipulate the timing or cell type-specific gene expression in neural cells 8. Alternatively, one can combine this protocol with tamoxifen-inducible Cre-ER(T) recombinases 9 and reporter mice 10.
This broad accessibility of tissues will drastically change experimental designs used in neuroscience.
Disclosures
No conflicts of interest declared.
Acknowledgments
This work was funded by RIKEN Brain Science Institute (BSI), Human Frontier Science Program (HFSP) (awarded to T.S.) and RIKEN Junior Research Associate Program (JRA, awarded to M.K.)RIKEN Brain Science Institute (BSI), Human Frontier Science Program (HFSP) (awarded to T.S.) and RIKEN Junior Research Associate Program (JRA, awarded to M.K.) funded this work.
References
- Tabata H, Nakajima K. Efficient in utero gene transfer system to the developing mouse brain using electroporation: Visualization of neuronal migration in the developing cortex. Neuroscience. 2001;103:865–872. doi: 10.1016/s0306-4522(01)00016-1. [DOI] [PubMed] [Google Scholar]
- Saito T, Nakatsuji N. Efficient gene transfer into the embryonic mouse brain using in vivo electroporation. Developmental Biology. 2001;240:237–246. doi: 10.1006/dbio.2001.0439. [DOI] [PubMed] [Google Scholar]
- Fukuchi-Shimogori T, Grove EA. Neocortex patterning by the secreted signaling molecule FGF8. Science. 2001;294:1071–1074. doi: 10.1126/science.1064252. [DOI] [PubMed] [Google Scholar]
- Kataoka A, Shimogori T. Fgf8 controls regional identity in the developing thalamus. Development. 2008;135:2873–2881. doi: 10.1242/dev.021618. [DOI] [PubMed] [Google Scholar]
- Imayoshi I, Shimogori T, Ohtsuka T, Kageyama R. Hes genes and neurogenin regulate non-neural versus neural fate specification in the dorsal telencephalic midline. Development. 2008;135:2531–2541. doi: 10.1242/dev.021535. [DOI] [PubMed] [Google Scholar]
- Shimogori T, Banuchi V, Ng HY, Strauss JB, Grove EA. Embryonic signaling centers expressing BMP, WNT and FGF proteins interact to pattern the cerebral cortex. Development. 2004;131:5639–5647. doi: 10.1242/dev.01428. [DOI] [PubMed] [Google Scholar]
- Shimogori T, Ogawa M. Gene application with in utero electroporation in mouse embryonic brain. Development Growth & Differentiation. 2008;50:499–506. doi: 10.1111/j.1440-169X.2008.01045.x. [DOI] [PubMed] [Google Scholar]
- Takahashi Y, Watanabe T, Nakagawa S, Kawakami K, Sato Y. Methods in Cell Biology. 2nd Edition. Vol 87. Elsevier Academic Press Inc; 2008. Avian Embryology; pp. 271–271. [DOI] [PubMed] [Google Scholar]
- Feil R. Ligand-activated site-specific recombination in mice. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:10887–10890. doi: 10.1073/pnas.93.20.10887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Indra AK. Temporally-controlled site-specific mutagenesis in the basal layer of the epidermis: comparison of the recombinase activity of the tamoxifen-inducible Cre-ERT and Cre-ERT2 recombinases. Nucleic Acids Research. 1999;27:4324–4327. doi: 10.1093/nar/27.22.4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa Y, O'Leary DDM. Dynamic patterned expression of orphan nuclear receptor genes ROR alpha and ROR beta in developing mouse forebrain. Developmental Neuroscience. 2003;25:234–244. doi: 10.1159/000072271. [DOI] [PubMed] [Google Scholar]