

Abstract
Viruses are obligate cellular parasites, and thus the study of their DNA requires isolating viral material away from host cell contaminants and DNA. Several downstream applications require large quantities of pure viral DNA, which is provided by this protocol. These applications include viral genome sequencing, where the removal of host DNA is crucial to optimize data output for viral sequences, and the production of new viral recombinant strains, where co-transfection of purified plasmid and linear viral DNA facilitates recombination.1,2,3
This procedure utilizes a combination of extractions and density-based centrifugation to isolate purified linear herpesvirus nucleocapsid DNA from infected cells.4,5 The initial purification steps aim to isolate purified viral capsids, which contain and protect the viral DNA during the extractions and centrifugation steps that remove cellular proteins and DNA. Lysis of nucleocapsids then releases viral DNA, and two final phenol-chloroform steps remove remaining proteins. The final DNA captured from solution is highly concentrated and pure, with an average OD260/280 of 1.90. Depending on the quantity of infected cells used, yields of viral DNA range from 150-800 μg or more. The purity of this DNA makes it stable during long-term storage at 4C. This DNA is thus ideally suited for high-throughput sequencing, high fidelity PCR reactions, and transfections.
Prior to beginning the protocol, it is important to know the average number of cells per dish (e.g. an average of 8 x 106 PK-15 cells in a confluent 15 cm dish), and the titer of the viral stock to be used (e.g. 1 x 108 plaque-forming units per ml). These are necessary to calculate the appropriate multiplicity of infection (MOI) for the protocol.6 For instance, to infect one 15 cm dish of PK-15 cells with the above viral stock, at an MOI of 5, you would use 400 μl of viral stock and dilute it with 3.6 ml of medium (total inoculation volume of 4 ml for one 15 cm plate).
Multiple viral DNA preparations can be prepared at the same time. The number of simultaneous preparations is limited only by the number of tubes held by the ultracentrifuge rotor (one per virus; see step 3.9 below). Here we describe the procedure as though being done for one virus.
Protocol
1. First Day: Viral Infection and Preparation of Buffers
Prepare 5-10 dishes (15 cm diameter) of tissue culture cells for infection, e.g. PK-15 cells for pseudorabies virus (PRV) or Vero cells for herpes simplex virus (HSV).
When cells are 95 - 100% confluent, infect them at a (MOI) of 5-10. To do this, inoculate each plate using virus stock in a total volume of 4 ml per plate, then incubate the plates for 1 hour at 37°C. Rock plates gently every 15 minutes to ensure that the cell monolayer remains fully coated by the virus inoculum. Meanwhile, warm the medium for the next step.
After one hour of infection, aspirate viral inoculum from the plates, add 15 ml of warm medium, and incubate at 37°C for 12-20 hours in a humidified incubator.
Prepare LCM buffers (see Table 1) and rock overnight at 4°C for thorough mixing. Prepare TNE for resuspension of viral DNA (see Table 2).
2. Second Day, Phase 1: Cell Lysis and Ultracentrifugation
Visually confirm that infection of cells has caused uniform cytopathic effect (CPE). Allow infection to progress until all cells have rounded up, but have not lifted off the plate.
Scrape the infected cells into the medium, and combine cells and medium into 50 ml conical tubes. The number of conical tubes needed for this step will depend on the number of dishes used for infection in Step 1.2 above, e.g. one 50 ml conical tube can be used to pellet a maximum of 3 dishes of 15 ml medium each. Optional: Rinse the plates with 10 ml PBS and combine with the cells and medium.
Centrifuge cells and medium for 10 minutes at 2000 relative centifugal force (rcf) at room temperature, then aspirate supernatant and resuspend each pellet in 10 ml of PBS. If the pellet from one viral infection was spread into multiple conical tubes in Step 2.2, these can be recombined into one conical tube at this step. Repeat centrifugation and aspiration. Optional: This pellet may be stored at -20°C and the protocol continued at a later time.
3. Second Day, Phase 2: Ultracentrifugation
Keep the cell pellet and LCM buffers on ice whenever possible during the following steps.
Add β-mercaptoethanol to 0% glycerol - LCM buffer. Begin cooling the ultracentrifuge (for step 3.9 below) by setting it to 4°C and turning on the vacuum.
Resuspend the cell pellet in 5 ml of 0% glycerol - LCM buffer until it is no longer clumpy. If necessary, vortex the tube to break up any undissolved cell pellet.
Extract by adding 1.5 ml Freon (1,1,2-trichloro-1,2,2-trifluoroethane) and vortexing vigorously. Immediately centrifuge for 10 min at 2000 rcf at a temperature of 4°C. Collect the top layer with a wide-bore pipette tip (avoiding the interface) and transfer to a new tube. Optional: If the pellet is a gelatinous solid, you can quickly pour the top layer into a new tube.
Repeat the Freon extraction and centrifuge again.
Collect the top layer (~5 ml) and transfer to a new polyallomer ultracentrifuge tube. Add β-mercaptoethanol to the two remaining glycerol - LCM buffers.
Prepare a gradient in the polyallomer centrifuge tube by underlaying the Freon extract (top layer) with 3 ml of 5% glycerol - LCM (middle layer), and then underlaying again with 2.5 ml of 45% glycerol - LCM (bottom layer). Use a thin pipette to avoid overflow of the tube contents.
If needed, prepare an equivalent balance tube using the same proportions of LCM buffers.
Transfer tubes into ultracentrifuge buckets. Balance buckets to within 0.1 g of each other, by gently adding extra 0% glycerol - LCM buffer to the top layer (~50-100 μl at a time).
Using a cold ultracentrifuge with a horizontal or swinging bucket rotor, spin the balanced samples for 1 hour at 77,000 rcf (e.g. 25,000 rpm for a SW41Ti rotor) at a temperature of 4°C.
During ultracentrifugation, make a glass hook to capture floating DNA at the ethanol precipitation step (4.6 below). Hold both ends of a glass Pasteur pipet and suspend the middle section over a flame. When a section of glass is almost molten, pull gently to stretch the glass, bend it gently to create a tightly angled hook (<90 degrees), and pull sharply to seal off the end. If the tip of the glass hook is open, hold the tip over the flame so that the glass melts enough to close it.
After the ultracentrifugation, you should see a thin opaque pellet with a dark spot in the middle. Carefully aspirate the liquid from the tube, including drippings along the sides, but avoiding the pellet at the bottom of the tube.
Add 0.5 ml TNE at room temperature.
Let the pellet sit for at least 10 minutes to allow hydration of the pellet. Optional: The pellet may also resuspend in TNE overnight, if kept at 4°C.
4. Third Day: "Ghost" DNA Precipitation
- Break up the pellet by pipetting with a small-bore pipette tip (e.g. a p200 pipette tip). Do not worry about shearing at this step, because the viral DNA is still contained in capsids at this point. Add the following reagents to the tube containing the viral pellet, at room temperature, to destroy capsids:
- 4.25 ml TNE
- 0.25 ml 10% SDS
- 2 mg Proteinase K
Beware of shearing from here on, e.g. use large-bore pipettes and do not vortex the viral material
Extract viral proteins by adding 5 ml of phenol-chloroform and immediately begin inverting or vortexing to maintain an emulsion. Mix for 10 seconds, then centrifuge the emulsion for 5 minutes at 2000 rcf at room temperature. Collect the top layer with a wide-bore pipette tip, avoiding the interface, and transfer to a new tube. Optional: Use phase lock gel (PLG) tubes in this step to avoid contamination from the interface.
Repeat the phenol-chloroform extraction and centrifuge again.
After the second phenol-chloroform extraction, collect the top layer into a 30 ml glass Corex tube and chill the tube at -20°C for 10-15 minutes, taking care that it does not freeze.
Add 10 ml of ice-cold ethanol, cover the tube (e.g. by stretching a piece of Parafilm M over the top), and invert to mix immediately. Watch for a DNA "ghost" to appear, which consists of a thin ropy precipitate. Invert the tube gently to mix further and cause the sticky DNA precipitates to cluster together.
Use a glass hook to capture the DNA ghost(s). Carefully blot the DNA to remove excess liquid, then place the hook (tip down) into a 1.5 ml tube and allow to dry. Add 0.25 - 0.5 ml of TE to dissolve the DNA ghost. Allow resuspension of DNA to proceed for at least one hour. Store DNA at 4°C. Optional: To maximize DNA yield, break off the glass hook into the 1.5 ml tube, so that resuspension off the glass fragments can continue over time.
5. Representative Results
With a sufficiently high MOI, cells should achieve full CPE within ~18 hours post infection (hpi) for wild-type PRV strains, and ~24 hpi for wild-type HSV-1 strains (Figure 1). Once the infected cell pellet is harvested, the remaining protocol steps can be complete in one long day or carried out over two days (Figure 2).
When preparing glass hooks to capture the DNA ghost, it is important to make the hook angle be less than 90 degrees, so that later it can fit into the base of an 1.5 ml tube (Figure 3). Sealing the tip of this hook prevents DNA loss inside the pipet.
DNA ghosts form during the precipitation step and will appear as white, ropy threads (Figure 4). These aggregate and stick to each other and/or the glass hook used to capture the DNA. Thus the same glass hook can be used to gather up multiple threads of DNA from within the precipitation solution.
| Choose column based on desired volume: | 10 ml | 15 ml | 20 ml | 40 ml |
| 1M KCL | 1.3 ml | 1.9 ml | 2.5 ml | 5 ml |
| 1M Tris (pH 7.4) | 300 μl | 450 μl | 600 μl | 1.2 ml |
| 1M MgCl2 | 50 μl | 75 μl | 100 μl | 200 μl |
| 0.5 EDTA | 10 μl | 15 μl | 20 μl | 40 μl |
| NP40/IGEPAL | 50 μl | 75 μl | 100 μl | 200 μl |
| β-mercaptoethanol (add immediately before use) | 4.3 μl | 6.5 μl | 8.6 μl | 17.2 μl |
| Use proportions for desired percent glycerol (0, 5, or 45%) | ||||
| 0% glycerol | None | None | None | None |
| water | 8.3 ml | 12.5 ml | 16.7 ml | 33.4 ml |
| 5% glycerol | 0.5 ml | 0.8 ml | 1.0 ml | 2.0 ml |
| water | 7.8 ml | 11.7 ml | 15.7 ml | 31.4 ml |
| 45% glycerol | 4.5 ml | 6.8 ml | 9.0 ml | 18.0 ml |
| water | 3.8 ml | 5.7 ml | 7.7 ml | 15.4 ml |
Table 1. Recipes for preparation of LCM buffers
| For 50 ml volume |
| 5 ml 1M NaCl |
| 2.5 ml 1M Tris, pH 7.5 |
| 1 ml 0.5 EDTA |
| 41.5 ml H2O |
Table 2. Preparation of TNE (0.1 M NaCl; 50 mM Tris, pH 7.5; 10 mM EDTA)
| For 1 L volume |
| 0.2 g KCl |
| 0.2 g KH2PO4 |
| 8 g NaCl |
| 1.15g Na2HPO4 |
| H2O to 1L |
Table 3. Preparation of PBS (2.7 mM KCl; 1.5 mM KH2PO4; 137 mM NaCl; 8.1 mM Na2HPO4; pH 7.0)
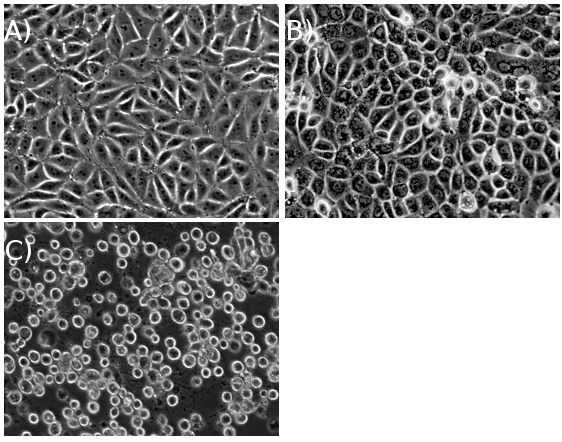 Figure 1. PK15 cells (A) prior to infection, (B) midway through infection, and (C) at cytopathic effect (CPE), after a high multiplicity of infection.
Figure 1. PK15 cells (A) prior to infection, (B) midway through infection, and (C) at cytopathic effect (CPE), after a high multiplicity of infection.
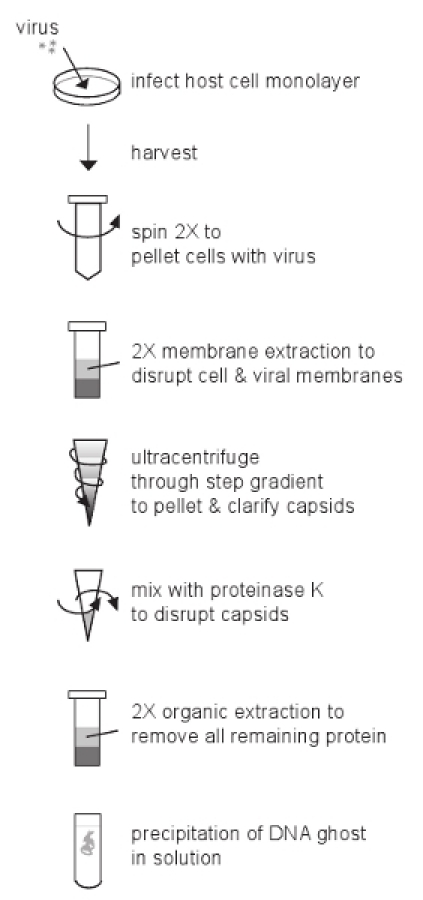 Figure 2. Overview of the protocol.
Figure 2. Overview of the protocol.
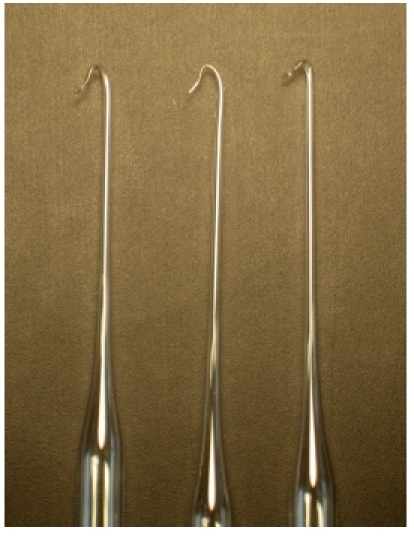 Figure 3. Three representative examples of glass hooks used to collect the viral DNA ghosts.
Figure 3. Three representative examples of glass hooks used to collect the viral DNA ghosts.
 Figure 4. Representative example of a well-formed and abundant DNA "ghost".
Figure 4. Representative example of a well-formed and abundant DNA "ghost".
Discussion
Portions of this protocol were originally developed for viral DNA isolation in BSL4 conditions, but it adapts equally well to non-BSL conditions.4 We commonly use this protocol to isolate DNA from the alpha-herpesviruses PRV and HSV-1, which have DNA genomes enclosed in a proteinaceous capsid and surrounded by a lipid envelope.7,8 However it is likely directly adaptable to other large DNA viruses, including beta- and gamma-herpesviruses and adenoviruses. Similar extractions are commonly used for RNA viruses as well.9
The robustness of this DNA preparation likely results from the multiple methods of purification that are included. The initial Freon extractions denature lipid membranes, separating cell components and also releasing the lipid envelope and outer tegument proteins that surround herpesvirus capsids. This is followed by ultracentrifugation, where the heavy viral nucleocapsids pellet through two gradient cushions, effectively separating them from most other cellular components. These capsids are resuspended, and the viral nucleocapsid DNA is released by rupturing the capsids with proteinase K and detergent. Two phenol-chloroform extractions thoroughly remove nucleocapsid components and any remaining cellular proteins. Finally, precipitation of the large viral DNA genomes in solution reduces carryover of salts or particulate contaminants.
This procedure to isolate viral nucleocapsid DNA provides abundant, high purity material for downstream applications. The combination of multiple extraction and centrifugation steps distinguishes this protocol from simpler single-extraction protocols that leave more cellular debris or degraded protein products with the DNA10. Protein contaminants can decrease the storage stability of viral DNA, and reduce transfection efficiency into cells. Cellular DNA contaminants negatively impact PCR reactions and high-throughput sequencing protocols.1 The DNA yield of this protocol is also high, making it well suited to restriction fragment length polymorphism (RFLP) analysis and Southern blotting.5,11 One nucleocapsid preparation provides sufficient material for all of these analyses.
The quantity of cells used for infection impacts the eventual yield of viral DNA. For PRV strains with a wild-type growth rate, input of 5-10 dishes of PK-15 cells (one 15 cm diameter dish holds approximately 8 x 106 cells) typically yields 250-500 μg of viral DNA. For viral mutants with a decreased yield of infectious virions, the input material should be scaled upward accordingly. Doubling the input number of dishes will approximately double the yield, and can be processed with the same quantities of reagents described here. To increase the input beyond this point, we recommend separating the input material into two parallel handling streams (e.g. two cell pellets and two extraction tubes) through step 3.13. Two ultracentrifugation pellets of the same viral strain can be combined into one tube at this resuspension step. If the DNA fails to cleanly form a ghost at the end of the procedure, the primary troubleshooting approach is to increase the input quantity of cells, which will increase the DNA yield and improve its precipitation.
Other conditions can also affect the success of ghost formation. For instance, it is important to time the harvest of infections at a point when viral nucleocapsids are abundant, but are mostly intracellular or cell-associated. Virions released to the medium will not be captured by this procedure, and therefore harvesting cells too late in infection (e.g. when CPE has progressed to the point where the cells detach from the dish) will reduce potential DNA yield and ghost formation. It is also important to completely dissolve the harvested cell pellet in LCM buffer (Step 3.2) to take full advantage of the following extraction step; this resuspension requires more time and care if the cell pellet has been frozen at the preceding step. Minor procedural details affect the DNA yield as well. While Steps 3 and 4 can be carried out without using chilled solutions and keeping all tubes on ice, the success rate of DNA ghosting is much higher when the reagents are kept cold. Similarly, the stability of β-mercaptoethanol in solution declines over time, and therefore adding it to the LCM solutions immediately before use optimizes its reducing capacity, improving protein disruption and increasing DNA yield and ghost formation. Careful attention to these details will improve the output of high quality viral DNA.
Because viral DNA is viscous, the resulting solution can be heterogeneous. Allowing more time for resuspension from the glass hook increases the useful DNA yield and improves solution homogeneity. Standard spectrophotometry can be used to quantitate DNA yield and purity. DNA fluorimetry provides even more precise measures of DNA yield (e.g. Invitrogen PicoGreen dsDNA stain).
Disclosures
No conflicts of interest declared.
Acknowledgments
The authors appreciate the contributions of Greg Smith, Lisa Pomeranz, Matt Lyman, Marlies Eldridge, Halina Staniszewska Goraczniak, and members of the Enquist lab in fine-tuning this protocol.
References
- Szpara ML, Parsons L, Enquist LW. Sequence variability in clinical and laboratory isolates of herpes simplex virus 1 reveals new mutations. J Virol. 2010;84:5303–5313. doi: 10.1128/JVI.00312-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banfield BW, Kaufman JD, Randall JA, Pickard GE. Development of pseudorabies virus strains expressing red fluorescent proteins: new tools for multisynaptic labeling applications. J Virol. 2003;77:10106–10112. doi: 10.1128/JVI.77.18.10106-10112.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobiler O, Lipman Y, Therkelsen K, Daubechies I, Enquist LW. Herpesviruses carrying a Brainbow cassette reveal replication and expression of limited numbers of incoming genomes. Nat Commun. 2010;1:146–146. doi: 10.1038/ncomms1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enquist LW, Madden MJ, Schiop-Stanley P, Vande Woude GF. Cloning of herpes simplex type 1 DNA fragments in a bacteriophage lambda vector. Science. 1979;203:541–544. doi: 10.1126/science.216076. [DOI] [PubMed] [Google Scholar]
- Smith GA, Enquist LW. Construction and transposon mutagenesis in Escherichia coli of a full-length infectious clone of pseudorabies virus, an alphaherpesvirus. J Virol. 1999;73:6405–6414. doi: 10.1128/jvi.73.8.6405-6414.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flint SJ, Enquist LW, Racaniello VR, Skalka AM. Principles of virology. 3rd edn. ASM Press; 2008. [Google Scholar]
- Pomeranz LE, Reynolds AE, Hengartner CJ. Molecular biology of pseudorabies virus: impact on neurovirology and veterinary medicine. Microbiol Mol Biol Rev. 2005;69:462–500. doi: 10.1128/MMBR.69.3.462-500.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roizman B, Pellett PE. In: Fields Virology. 2 ed. Knipe DM, Howley PM, editors. Lippincott Williams & Wilkins; 2001. pp. 2381–2397. [Google Scholar]
- Mendez II, Hermann LL, Hazelton PR, Coombs KM. A comparative analysis of freon substitutes in the purification of reovirus and calicivirus. J Virol Methods. 2000;90:59–67. doi: 10.1016/s0166-0934(00)00217-2. [DOI] [PubMed] [Google Scholar]
- Gharabaghi F, Aymard M, Trotemann P, Gerdil C. A rapid and simplified micromethod for subtyping varicella-zoster virus. J Med Virol. 1990;31:129–134. doi: 10.1002/jmv.1890310210. [DOI] [PubMed] [Google Scholar]
- Granstedt AE, Szpara ML, Kuhn B, Wang SS, Enquist LW. Fluorescence-based monitoring of in vivo neural activity using a circuit-tracing pseudorabies virus. PLoS One. 2009;4:e6923–e6923. doi: 10.1371/journal.pone.0006923. [DOI] [PMC free article] [PubMed] [Google Scholar]