

Abstract
Irbesartan (IRB) is an angiotensin II receptor blocker antihypertensive agent. The aim of the present investigation was to develop a self-nanoemulsifying drug delivery system (SNEDDS) to enhance the oral bioavailability of poorly water-soluble IRB. The solubility of IRB in various oils was determined to identify the oil phase of SNEDDS. Various surfactants and co-surfactants were screened for their ability to emulsify the selected oil. Pseudoternary phase diagrams were constructed to identify the efficient self-emulsifying region. The optimized SNEDDS formulation contained IRB (75 mg), Cremophor® EL (43.33%), Carbitol® (21.67%) and Capryol® 90 (32%). SNEDDS was further evaluated for its percentage transmittance, emulsification time, drug content, phase separation, dilution, droplet size and zeta potential. The optimized formulation of IRB-loaded SNEDDS exhibited complete in vitro drug release in 15 min as compared with the plain drug, which had a limited dissolution rate. It was also compared with the pure drug solution by oral administration in male Wister rats. The in vivo study exhibited a 7.5-fold increase in the oral bioavailability of IRB from SNEDDS compared with the pure drug solution. These results suggest the potential use of SNEDDS to improve dissolution and oral bioavailability of poorly water-soluble IRB.
Keywords: Bioavailability, irbesartan, poor water solubility, self-nanoemulsifying drug delivery system
INTRODUCTION
The oral route is the easiest and most convenient way of noninvasive administration. However, oral drug delivery may hamper drug molecules that exhibit a poor aqueous solubility. Approximately 40% of the new chemical entities exhibit poor aqueous solubility and present a major challenge to the modern drug delivery system, which leads to poor oral bioavailability, high intra- and inter-subject variability and lack of dose proportionality. These drugs are classified as class II drugs by the Biopharmaceutical Classification System, drugs with poor aqueous solubility and high permeability.[1] Different formulation approaches like solid dispersion and complexation with cyclodextrins have been already utilized to resolve the poor aqueous solubility of irbesartan (IRB).[2,3] Indeed, in some selected cases, these approaches have been successful, but they offer many other disadvantages like, in solid dispersion, the amount of carriers used is often large and, thus, if the dose of the active ingredient is high, the tablets or capsules formed will be large in volume and difficult to swallow. Moreover, because the carriers used are usually expensive and the freeze or spray-drying method requires particular facilities and processes, this leads to a high production cost. Although the traditional solvent method can be adopted instead, it is difficult to deal with co-precipitates with a high viscosity. Complexation with cyclodextrin techniques is not applicable for drug substances that are not soluble in both aqueous and organic solvents. Realization that the oral bioavailability of poor water-soluble drugs may be enhanced when co-administered with meal rich in fat has led to increasing the recent interest in the formulation of poorly water-soluble drugs in lipids. Lipid suspension, solutions and emulsions have all been used to enhance the oral bioavailability but, more recently, there has been increasing focus on the utility of self-nanoemulsifying drug delivery systems (SNEDDS). Being hydrophobic, i.e. more lipophilic, a lipid-based drug delivery system would ideally work for a poorly water-soluble drug. Lipid-based drug delivery systems have gained considerable interest after the commercial success of Sandimmune Neoral (Cyclosporine A), Fortovase (Saquinavir) and Norvir (Ritonavir).[3–6]
SNEDDS are defined as isotropic mixtures of natural or synthetic oils, solid or liquid surfactants or, alternatively, one or more hydrophilic solvents and co-solvents/surfactants that have the ability of forming fine oil-in-water (o/w) microemulsions upon mild agitation followed by dilution in aqueous media such as gastric fluids. SNEDDS spread readily in the gastrointestinal tract, and the digestive motility of the stomach and the intestine provide the agitation necessary for self-emulsification.[7]
IRB displace angiotensin II from the angiotensin I receptor and produce their blood pressure-lowering effects by antagonizing angiotensin II-induced vasoconstriction, aldosterone release, catecholamine release, arginine vasopressin release, water intake and hypertrophic response. IRB is practically insoluble in water (0.00884 mg/mL) and has a high hydrophobicity (log P 6), with 60–80% oral bioavailability. Hence, IRB was selected as a model drug for this study [Figure 1]. IRB is available in various doses (75, 150 and 300 mg), of which the 75 mg dose was selected as a working dose to limit the total formulation volume in the present investigation. Hence, the aim of this study was to develop a SNEDDS of a poorly water-soluble drug (IRB).[8,9]
Figure 1.
Structure of irbesartan
MATERIALS AND METHODS
Materials
IRB was obtained as a gift sample from Torrent Research Center, Bhat, Gandhinagar, India. The following materials were donated by Abitec Corporation, Columbus OH 43216-0569, USA and were used as received: Acconon® CC 400 (Polyoxyethylene 6 Capric esters), Acconon® Sorb 20 (Polyoxyethylene 20 Sorbitol), Acconon® E (Polyoxypropylene 15 Stearyl ether), Capmul® MCM (Glycerol mono-dicaprylate), Capmul® GMO (Glycerol mono/di-oleate), Capmul® MCM C8, Captex® 355 (Caprylic/Capric acid triglycerides) and Caprol® ET (Polyglycerol esters). Cremophor® RH 40 (Polyoxyl 40 hydrogenated castor oil) and Solutol® HS 15 (Macrogol 15 hydroxystearate) were also donated from BASF, Mumbai, India. Miglylol® 812 (Caprylic/Capric acid triglycerides) and Imwitor® 742 (Glycerol monocaprylocaprate) were generously gifted by Sasol, Germany. Sefsol® 218 (Propylene glycerol monocaprylate) was gifted by Nikko Chemicals, Tokyo, Japan. Labrafil® M 2125 CS (Linoleoyl macrogolglycerides), Plurol Oleique® (Polyglycerol oleate) and Capryol® 90 (Polypropylene glycol monocaprylate) were received as a gift sample from Gettefose, Mumbai, India. Acrysol® K 140 (Polyoxyl 40 hydrogenated castor oil) and Acrysol® EL 135 (Polyoxyl 35 castor oil) were also gifted by Corel Pharma, Gujarat, India. Cremophor® EL (Polyethoxylated castor oil) was purchased from Sigma Aldrich, Hyderabad, India. Other Chemicals like Span® 20 (sorbitan monolaurate), Span® 80 (Sorbitan monooleate), Tween® 20 (Polyoxyethylene sorbitan monolaurate), Tween® 80 (Polyoxyethylene sorbitan monooleate), Polyethylene glycol 400 (PEG 400), Polyethylene glycol 200 (PEG 200), Propylene glycol (PG), Carbitol® (Monoethyl ether of diethylene glycol), Glycerol, Castor oil, Olive oil, Cotton seed oil, Polaxomer 188 and Polaxomer 407 were bought from Merck, Mumbai, India and S.D. Fine Chem, Mumbai, India. Double-distilled water was used throughout the study. Acetonitrile and methanol used in the present study were of high-performance liquid chromatography (HPLC) grade. All other chemicals were reagent grade. Empty hard gelatin capsule shells were generously donated by Torrent Research Center.
Animals
Male Wister rats (weighing approximately 250±30 g) were used for the bioavailability studies. The animals were maintained at a temperature of 24–25°C and humidity of 60% and were supplied with food and water ad libitum. The animal requirement was approved by the Institute Animal Ethics Committee, and all experiments were conducted as per the norms of the Committee for the Purpose of Supervision of Experiments on Animals, India.
Selection of SNEDDS Components
Oil (solubility studies)
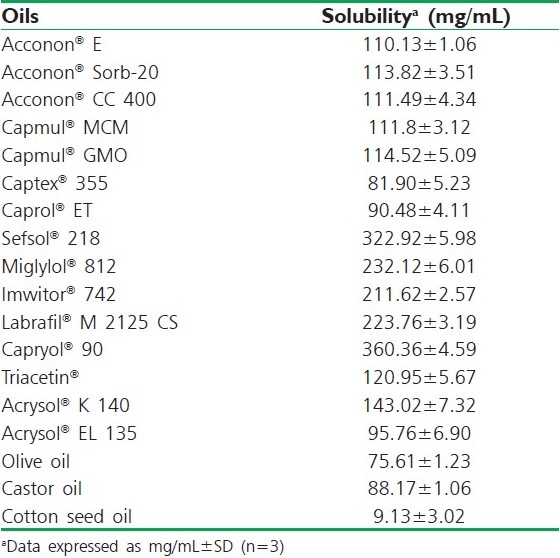
The solubility of IRB in various buffers, oils, surfactants and co-surfactants was measured using the shake flask method as suggested by Date and Nagarsenker, 2007.[10] An excess amount of IRB was introduced into 2 mL of each excipient and the mixture was kept in sealed vials. A vortex mixer (Remi, Mumbai, India) was used to facilitate the solubilization. Sealed vials were stirred in a water bath at 40°C for 24 h and allowed to reach equilibrium at 30°C for 72 h. Each vial was centrifuged at 15,000 rpm for 10 min using a centrifuge (Remi) followed by removal of the undissolved IRB by filtering with a membrane filter (0.45 μm). Samples were suitably diluted with methanol and drug concentration was obtained via a validated UV method at 244 nm using methanol as a blank, using a double-beam UV visible spectrophotometer (Shimadzu 1700, Shimadzu, Tokyo, Japan).[11] The experiment was repeated in triplicate and the results [Table 1] represent the mean value (mg/mL±SD).
Table 1.
Solubility study of IRB in various oils at 25°C
Surfactant (emulsification study)
Different surfactants (Cremophor® EL, Cremophor® RH 40, Solutol® HS 15, Span® 80, Tween® 20 and Tween® 80) were screened for emulsification ability of the selected oil phase. Surfactant selection was performed on the basis of %transparency and ease of emulsification. Briefly, 300 mg of the surfactants was added to 300 mg of the selected oily phase. The mixtures were gently heated at 50°C for homogenization of the components. Each mixture, 50 mg, was then diluted with distilled water to 50 mL in a stoppered conical flask. Ease of emulsification was judged by the number of flask inversions required to yield a homogenous emulsion.[10] The emulsions were allowed to stand for 2 h and their % transparency was evaluated at 650 nm by a double-beam UV spectrophotometer using distilled water as a blank. The emulsions were furthermore observed visually for any turbidity or phase separation.
Co-surfactant (emulsification study)
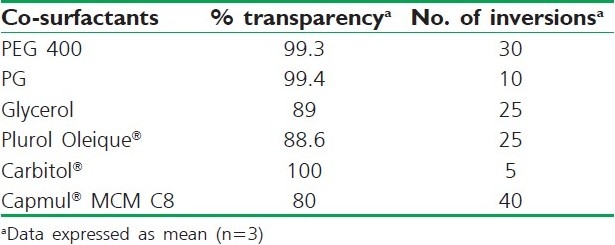
Six co-surfactants were screened for SNEDDS formulation, which included Carbitol®, PEG 400, PG, Capmul® MCM C8, Plurol oleique® and Glycerol. The screening of the co-surfactants was conducted on the basis of % transparency and ease of emulsification. Mixtures of 100 mg of the co-surfactant, 200 mg of the selected surfactant and 300 mg of the selected oil were prepared and evaluated in a similar fashion as described in the above section of surfactant selection.[10]
Construction of the Ternary Phase Diagram
On the basis of the solubility and emulsification study, Capryol® 90, Cremophor® EL and Carbitol® were selected as the oil, surfactant and co-surfactant, respectively. To determine the concentration of components for the existing range of SNEDDS, a pseudoternary phase diagram was constructed using the water titration method at ambient temperature (25°C).[12] The surfactant and co-surfactant were mixed in different volume ratios (1:1, 1:2, 1:3, 1:4, 4:1, 3:1 and 2:1). The oil and surfactant/co-surfactant mixtures (S/Co-S) were mixed thoroughly in different volume ratios (1:9, 1:8.5, 1:8, 1:7.5, 1:7, 1:6.5, 1:6, 1:5.5, 1:5, 1:4.5, 1:4, 1:3.5, 1:3, 1:2.5, 1:2, 1:1.5, 1:1, 1.5:1 and 2:1) and titrated with water by a drop-wise addition under gentle agitation. The proper ratio of one excipient to another in the SNEDDS formulation was analyzed and a pseudoternary plot was constructed using the TRIPLOT V14 software. All studies were repeated thrice, with similar observations being made between repeats. Moreover, to investigate the effects of IRB on the self-emulsifying performance of SNEDDS, the formulation amount of IRB was added to the boundary formulations of the self-emulsifying domain of the ternary phase diagrams. The self-emulsifying performance was visually assessed after infinite dilution using purified water.
Preparation of the Self-Nanoemulsified Formulations
IRB (75 mg) was added in accurately weighed amounts of oil into a screw-capped glass vial and melted in a water bath at 37°C. The surfactant and co-surfactant were added to the oily mix using a positive displacement pipette and stirred with a magnetic bar. The formulations were further sonicated (Frontline FS-4) for 15 min and stored at room temperature until their use in subsequent studies.[13]
Evaluation Parameters of IRB-Loaded SNEDDS
Emulsification time
The emulsification time (the time for a preconcentrate to form a homogeneous mixture upon dilution) was monitored by visually observing the disappearance of SNEDDS and the final appearance of the nanoemulsion in triplicate. A dissolution apparatus (Elactrolab Dissolution Tester USP, TDT-06 P, Electrolab, Mumbai, India) was employed with 500 mL water and with a paddle speed of 50 rpm at 37°C. The SNEDDS (1 mL) was added dropwise to the medium by dropping the pipette and time required for the disappearance of SNEDDS was recorded.[13]
Droplet size and zeta potential determination
Fifty milligrams of the optimized SNEDDS formulation was diluted with water to 1000 mL in a flask and gently mixed by hand. The droplet size distribution and zeta potential of the resultant emulsion was determined by laser diffraction analysis using a Particle size analyzer (Malvern Zetasizer, Malvern, Worcestershire WR14 1XZ, UK). The sizing of the emulsion was determined in a small volume module. The sample was directly placed into the module and the data were collected for 60s. Particle size was calculated from the volume size distribution. All studies were repeated in triplicate for good agreement being found between the measurements (P < 0.05).[14]
In vitro drug dissolution study
In vitro drug release of IRB from optimized SNEDDS was performed by a conventional method. A hard gelatin capsule size “0” filled with preconcentrate (equivalent to 75 mg IRB) and pure drug (75 mg) separately were put into each of the 900 mL water, pH 1.2 and pH 7.5 phosphate buffer at 37±0.5°C with 50 rpm rotating speed. Samples (5 mL) were withdrawn at regular time intervals (5, 10, 20, 30, 45, 60, 90 and 120 min) and filtered using a 0.45-μm filter. An equal volume of the respective dissolution medium was added to maintain the volume constant. The drug content of the samples was assayed using a UV visible spectrophotometric method. All measurements were performed in triplicate from three independent samples.[14]
Dilution studies/robustness on dilution
A dilution study was done to access the effect of dilution on the SNEDDS preconcentrate. In this study, the optimized formulation was subjected to various dilutions (i.e., 1:50, 1:100 and 1:500) with various diluents (i.e., water, 0.1N HCl, phosphate buffer pH 7.5) and the droplet size was recorded.[12]
Determination of drug content
IRB from an optimized SNEDDS formulation was extracted in methanol using the sonication technique. The methanolic extract was analyzed for IRB content spectrophotometrically at a wavelength of 244 nm after suitable dilution.[14]
In vivo studies
The rats were deprived of food but had free access to water 24 h before the day of the experiment. Two groups of rats were used for the experiments. Each group was either administered orally IRB aqueous suspension (control group) or IRB-loaded SNEDDS. The sample of IRB powder (75 mg) or IRB-SMEDDS (650 mg equivalent to IRB 75 mg) were accurately weighed and separately dispersed into distilled water (3 mL) by mixing homogeneously for 30 s prior to dosing. Each formulation was administered to rats by oral gavage using an animal feeding needle. Under ether anesthesia, blood samples (0.5 mL) were collected via the retro-orbital vein at 15, 30, 60, 120, 240, 360, 480 and 720 min after oral administration into heparinized microcentrifuge tubes. The samples were centrifuged at 15,000 rpm for 10 min at a temperature of 4°C. The plasma samples (100 μL) were separated and 1 mL of acetronitrile was added to each of the plasma samples to precipitate the protein. The samples were then centrifuged again at 15,000 rpm at 4°C for 5 min, and the supernatant (20 μL) was directly injected onto the HPLC (Shimadzu Corporation, LC-20AD, Shimadzu, Tokyo, Japan) with PDA detector for the estimation of IRB content by a validated chromatographic method (R2 = 0.9759, %error = 3.1, CV = 3.5%). Intra- and inter-day variations at the above two concentrations were lower than 10%. The limit of detection of IRB in this method was 10 ng/mL. The chromatographic column used was Luna C8 (150 cm and 4.6 mm i.d.) with a 5-μm particle size. Acetonitrile and methanol (55:45) were used as the mobile phase at a flow rate of 1.0 mL/min, with a total run time of 10 min. Data from these samples were used to plot curves for the IRB absorption with time.
Pharmacokinetic parameters
A noncompartmental model was employed to estimate the following pharmacokinetic parameters for individual rats in each group, peak plasma concentration (Cmax), the time to reach Cmax(Tmax) and area under the plasma concentration versus time curve from zero to the last sampling time (12 h) (AUC0→12h). Values are reported as mean±SD (n = 3) and the data were considered as statistically significant at P<0.05.
RESULTS AND DISCUSSIONS
Solubility Study (Screening of Oil)
Solubility studies were aimed at identifying a suitable oily phase for the development of IRB SNEDDS. Identifying the suitable oil having a maximal solubilizing potential for the drug under investigation is very important to achieve optimum drug loading.[6,7] Solubility of IRB in various oily phases, 10% (w/w) surfactant solutions and buffers is presented in Tables 1, 2 and 3, respectively. Among the various oily phases that were screened, Capryol® 90 could solubilize the target amount of IRB (75 mg) at a relatively small amount of 210 μL. The selection of the surfactant or co-surfactant in the further study was governed by the emulsification efficiency rather than the ability to solubilize IRB.
Table 2.
Solubility study of IRB in various surfactants at 25°C
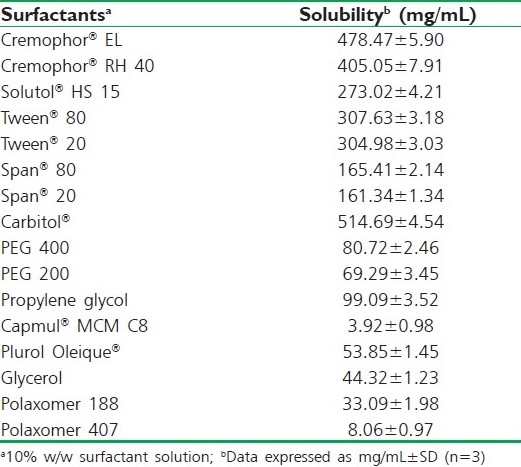
Table 3.
Solubility study of IRB in various buffers and media at 25°C

Preliminary screening of surfactants
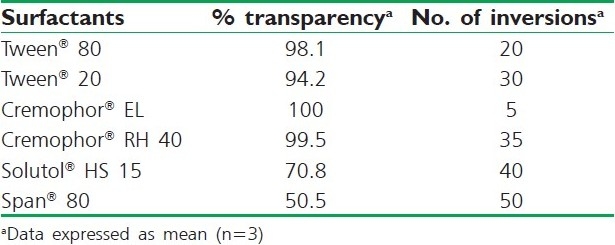
Nonionic surfactants are generally considered less toxic than ionic surfactants. They are usually accepted for oral ingestion. In this study, the six nonionic surfactants (Tween® 80, Tween® 20, Cremophor® EL, Cremophor® RH 40, Solutol® HS 15 and Span® 80) were selected, of which some are reported to have bioactive effects, like action on tight junction by Solutol® HS 15, lymphotropic characters by Tween® 80, Tween ® 20 and Span® 80 and inhibitory effect on p-gp and CYP enzymes such as Cremophor® RH40 and Cremophor® EL. These findings were confirmed by Zhang et al., 2003,[14] who demonstrated increased AUC and Cmax for orally administered digoxin in rats when co-administered with Cremophor®. It has been reported that well-formulated SNEDDS is dispersed within seconds under gentle stirring conditions, which ultimately depends on the emulsification ability of the surfactant. Results inferred that the oily phase Capryol® 90 exhibited the highest emulsification efficiency with Cremophor® EL [%transparency: 100, five flask inversions (5s)] for the homogenous emulsion formation. On the other hand, Capryol® 90 showed poor emulsification properties with other surfactants employed, requiring a higher number of flask inversions [Table 4]. The aforementioned results suggested the use of Capryol® 90 as an oily phase with Cremophor® EL as a surfactant for further study.
Table 4.
Emulsification efficiency of various surfactants
Preliminary screening of the co-surfactants
Addition of a co-surfactant to the surfactant-containing formulation was reported to improve dispersibility and drug absorption from the formulation.[6,7] In view of the current investigation, six co-surfactants, namely Carbitol®, PEG 400, PG, Capmul® MCM C8, Plurol oleique® and Glycerol, were compared. As depicted in Table 5, Capryol® 90 exhibited good emulsification with all co-surfactants, with Carbitol® showing maximum transmittance (100%) followed by PG (99.4%). Herein, solubility of the drug in different co-surfactants may judge the final selection. Results of the solubility study demonstrated in Table 1 inferred higher solubility in Carbitol®. It is worthy to note that all dispersions exhibited instantaneous emulsion formation with only five flask inversions [Table 5]. This could contend the importance of co-surfactant addition to the surfactant-containing dispersions.
Table 5.
Emulsification efficiency of various cosurfactants
Construction of Pseudo-Ternary Phase Diagrams
A series of SNEDDS were prepared and their self-emulsifying properties were observed visually. Pseudo-ternary phase diagrams were constructed in the absence of IRB to identify the self-emulsifying regions and to optimize the concentration of oil, surfactant and co-surfactant in the SNEDDS formulations. The ratio of surfactant to co-surfactant was very effective to a stable and efficient SNEDDS formation. The phase diagrams were constructed at the ratio of surfactant/co-surfactant 4:1, 2:1, 1:1, 1:2, 1:4 (w/w). The gel-like region was found to become large with the increasing concentration of Cremophor® EL, while the self-nanoemulsifying region expanded with increasing amounts of Carbitol®. The maximum self-nanoemulsifying region was to be at the ratio of 1:4 surfactant/co-surfactant. However, the drug precipitation was observed after several hours at the ratio of 1:2, 1:3 and 1:4. Co-surfactant will be beneficial to form nanoemulsion at a proper concentration range. However, excessive amount of co-surfactant will cause the system to become less stable for its intrinsic high-aqueous solubility and lead to the droplet size increasing as a result of the expanding interfacial film. Hence, the optimal ratio of surfactant to co-surfactant was selected to be 2:1 [Figure 2].
Figure 2.
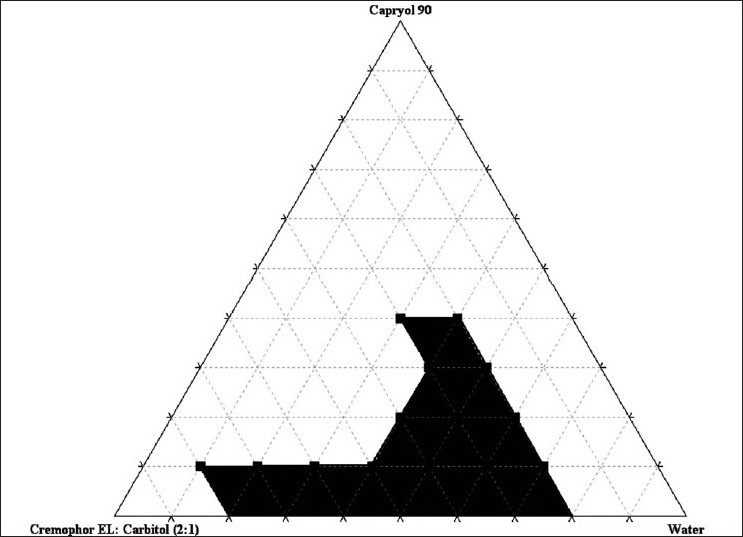
Pseudoternary phase diagram with surfactant/co-surfactant (Km) = 2:1
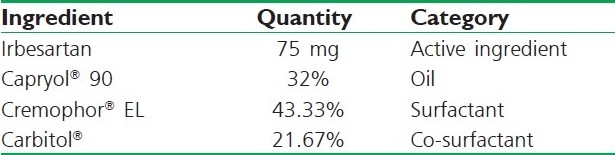
Based on the above results, a three-component SNEDDS formulation was established containing 32% Capryol® EL 135 as oil (on the basis of solubility study and required target amount of IRB-20 mg), 43.33% Cremophor® 20 as surfactant and 23.3% Carbitol® as co-surfactant (on basis of phase diagram) [Table 6]. It has been reported that the drug incorporated in the SNEDDS may have some effect on the self-emulsifying performance. In our study, no significant differences were found in self-emulsifying performance when compared with the corresponding formulations with IRB.
Table 6.
Composition of optimized SNEDDS formulation
Evaluation of Optimized SNEDDS
In the self-emulsifying systems, the free energy required to form an emulsion was very low (it was not practically measured), thereby allowing spontaneous formation of an interface between the oil droplets and water. Moreover, because the drug released will be in nano size, it will increase the effective surface area for dissolution and, ultimately, in vivo absorption.
Emulsification time
In SNEDDS, the primary means of self-emulsification assessment is visual estimation. The efficiency of self-emulsification could be estimated by primarily determining the rate of emulsification, which is an important index for the assessment of the efficiency of emulsification, i.e. the SNEDDS should disperse completely and quickly when subjected to aqueous dilution under mild agitation. The emulsification time study showed that the optimized formulation employed could emulsify within 25s, which suggests rapidity of the formulation.
Droplet size and zeta potential determination
The droplet size of the emulsion is a crucial factor in self-emulsification performance because it determines the rate and extent of drug release as well as absorption. We observed that the formulation composition ratio mentioned in Table 4 gave the smallest particle size (40±4.23 nm, mean±SD, n=3) than other SNEDDS formulations chosen for further studies. The charge of the oil droplets of SNEDDS is another property that should be assessed for increased absorption. The charge of oil droplets in SNEDDS was negative due to the presence of free fatty acids and the zeta potential of the optimized formulation was -23.9±0.42 (mean±SD, n=3). In general, a zeta potential value of±30 mV is sufficient for stability.[15] The optimized formulation posses a zeta potential of -23.9±0.42, which means that it does not comply with requirement of stability [Figure 3]. But, it has been found in some of the research articles that a zeta value in between -20 mV and -11 mV leads to threshold agglomeration. Hence, the optimized formulation of SNEDDS with a zeta potential -23.9 mV would not exhibit threshold agglomeration. Such kind of instability will only be observed on long-term storage, which is required to be further studied in the future.[16]
Figure 3.
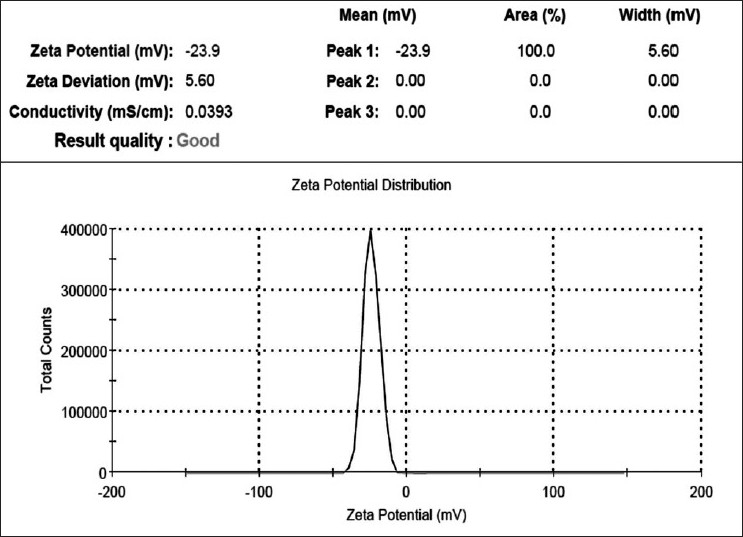
Zeta potential determinations of an optimized irbesartan-loaded self-nanoemulsifying drug delivery system
In vitro drug release
Dissolution studies were performed for the optimized SNEDDS formulation in water, pH 1.2 and pH 7.5, and the results were compared with pure drug. It was also seen that changes in the dissolution medium (buffer pH 1.2, 7.5 and water) had no effect on the drug release from either plain IRB or the SNEDDS formulation [Figure 4]. This observation can be explained by the fact that IRB has no ionizable group and thus its solubility and dissolution are pH-independent. As the emulsification time is below 25 s, the maximum percentage of the drug is released within 15 min; however, the dissolution studies are conducted for 2 h to observe the variation or occurrence of precipitation over time.
Figure 4.
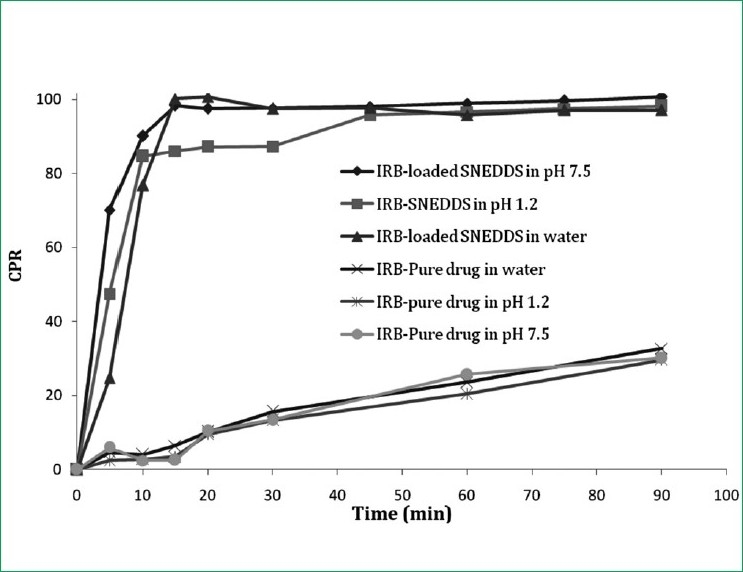
In vitro drug release of irbesartan from a self-nanoemul-sifying drug delivery system in water, pH 1.2 and pH 7.5 compared with pure drug
Effect of dilution studies/robustness on dilution
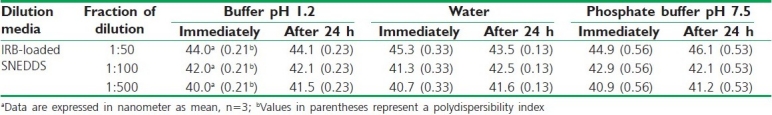
Distilled water was used as a dispersion medium in the present study. No significant difference is observed when the nanoemulsions prepared by nonionic surfactants were dispersed in water, phosphate buffer pH 7.5 or pH 1.2. Dilution studies of the optimized formulation have been shown in Table 7.
Table 7.
Dilution study of optimized IRB-loaded SNEDDS formulation
Drug content
Drug content of the optimized formulation was found to be 99.34±0.42% (mean±SD, n = 3).
In vivo studies
IRB determination in the rat blood was carried out using a validated HPLC technique that has been successfully developed in-house. The plasma concentrations vs. time profiles are shown in Figure 5, and the pharmacokinetic parameters are summarized in Table 6. Dosing the aqueous suspensions of IRB resulted in the lowest average IRB plasma concentrations. However, the AUC was 7.5-times greater when IRB was administered as SNEDDS compared with the AUC obtained for the aqueous IRB suspension. The SNEDDS gave mean values of Cmax 9.45 μg/mL, which was 16.87-fold higher than the Cmax obtained with the same dose of IRB administered as an aqueous suspension. The Tmax (100 min) after SNEDDS dosing was the same as the Tmax obtained within aqueous suspensions (100 min). These results reveal that the formulation of IRB as SNEDDS results in a significant increase in absorption compared with that from the aqueous suspensions [Table 8].
Figure 5.
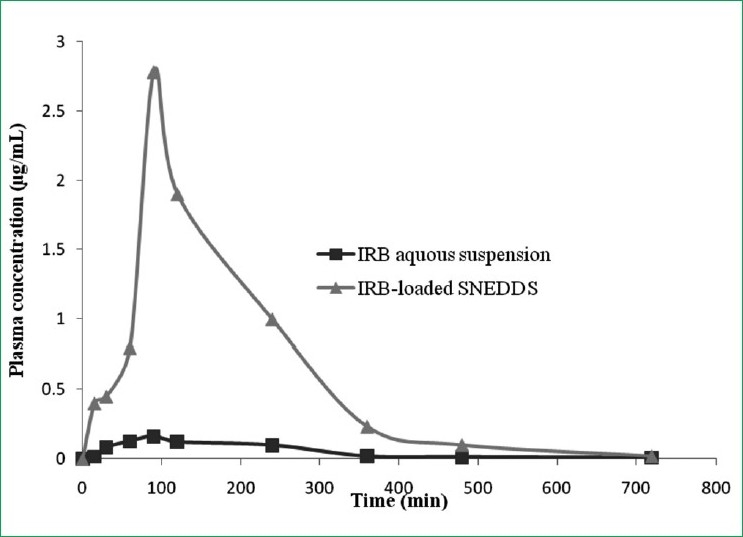
Plasma concentration time profile after oral administration of an irbesartan (IRB) - loaded self-nanoemulsifying drug delivery system, compared with IRB pharmacokinetics after dosing aqueous suspension
Table 8.
Pharmacokinetic parameter after oral administration of optimized formulation of IRBloaded SNEDDS

CONCLUSION
In this study, SNEDDS of IRB were prepared and evaluated for their in vitro and in vivo behavior. The optimized formulation consisting of Capryol® 90 (32%w/w), Cremophor® EL (43.33%w/w) and Carbitol (21.67%w/w) exhibited faster release profiles with a rapid rate of emulsification. The optimized SNEDDS formulation of IRB showed a significant increase in the dissolution rate and oral absorption compared with the aqueous drug suspension. Thus, SNEDDS can be regarded as a novel and commercially feasible alternative to the current IRB formulations. However, further studies in higher animals and human beings need to be performed before this formulation can be commercially exploited.
Footnotes
Source of Support: Nil
Conflict of Interest: Nil.
REFERENCES
- 1.Stegemanna S, Leveillerb F. When poor solubility becomes an issue: From early stage to proof of concept. Eur J Pharm Sci. 2007;31:249–61. doi: 10.1016/j.ejps.2007.05.110. [DOI] [PubMed] [Google Scholar]
- 2.Kane R, Naik S, Bumrela S, Kuchekar B. Preparation, physicochemical characterization, dissolution and formulation studies of irbesartan cyclodextrin inclusion complexes: Comparison beCremophor β-CD and HP-β-CD. J Pharm Res. 2009;2:1359–64. [Google Scholar]
- 3.Chavla G, Bansal AK. Improved dissolution of a poorly water soluble drug in solid dispersions with polymeric and non-polymeric hydrophilic additives. Acta Pharm. 2008;58:257–74. doi: 10.2478/v10007-008-0016-1. [DOI] [PubMed] [Google Scholar]
- 4.Hauss DJ. Oral lipid based formulations. Adv Drug Del Rev. 2007;59:667–76. doi: 10.1016/j.addr.2007.05.006. [DOI] [PubMed] [Google Scholar]
- 5.Pouton CW. Formulation of self-emulsifying drug delivery systems. Adv Drug Del Rev. 1997;25:47–58. [Google Scholar]
- 6.Pouton CW. Lipid formulations for oral administration of drugs: Non-emulsifying, self-emulsifying and ‘self-microemulsifying’ drug delivery systems. Eur J Pharm Sci. 2000;11:S93–8. doi: 10.1016/s0928-0987(00)00167-6. [DOI] [PubMed] [Google Scholar]
- 7.Pouton CW. Formulation of poorly water-soluble drugs for oral administration: Physicochemical and physiological issues and the lipid formulation classification system. Eur J Pharm Sci. 2006;29:278–87. doi: 10.1016/j.ejps.2006.04.016. [DOI] [PubMed] [Google Scholar]
- 8. [cited in 2010]. Available from: http://drugbank.wishartlab.com/drugs/DB01029 .
- 9. [cited in 2010]. Available from: http://en.wikipedia.org/wiki/Irbesartan .
- 10.Date AA, Nagarsenker MS. Design and evaluation of self nanoemulsified drug delivery systems (SNEDDS) for Cefpodoxime Proxetil. Int J Pharm. 2007;329:166–72. doi: 10.1016/j.ijpharm.2006.08.038. [DOI] [PubMed] [Google Scholar]
- 11.Hirlekar R, Kadam V. Preformulation Study of the Inclusion Complex Irbesartan-β-Cyclodextrin. AAPS Pharm Sci Tech. 2009;10:276–81. doi: 10.1208/s12249-009-9206-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Azeem A, Rizwan M. Nanoemulsion Components Screening and Selection: A Technical Note. AAPS Pharm Sci Tech. 2009;10:69–76. doi: 10.1208/s12249-008-9178-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Singh AK, Chaurasiya A, Singh M, Upadhyay SC, Mukherjee R, Khar RK. Exemestane Loaded Self-Microemulsifying Drug Delivery System (SMEDDS): Development and Optimization. AAPS Pharm Sci Tech. 2008;9:628–34. doi: 10.1208/s12249-008-9080-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang P, Liu Y, Feng N, Xu J. Preparation and evaluation of self microemulsifying drug delivery system of oridonin. Int J Pharm. 2008;355:269–76. doi: 10.1016/j.ijpharm.2007.12.026. [DOI] [PubMed] [Google Scholar]
- 15.Muller RH, Jacobs C, Kayser O. Nanosuspensions as particulate drug formulations in therapy rationale for development and what we can expect for the Future. Adv Drug Deli Rev. 2001;47:3–19. doi: 10.1016/s0169-409x(00)00118-6. [DOI] [PubMed] [Google Scholar]
- 16.Zang X, Xia Q, Gu N. Preparation of All-Trans Retinoic Acid Nanosuspensions Using a Modified Precipitation Method. Drug Dev Ind Pharm. 2006;32:857–63. doi: 10.1080/03639040500534184. [DOI] [PubMed] [Google Scholar]