

Abstract
The electronic structure of terpyrimidinethiols is investigated by means of density-functional theory calculations for isolated molecules and monolayers. In the transition from molecule to self-assembled monolayer (SAM), we observe that the band gap is substantially reduced, frontier states increasingly localize on opposite sides of the SAM, and this polarization in several instances is in the direction opposite to the polarization of the overall charge density. This behavior can be analyzed by analogy to inorganic semiconductor quantum-wells, which, as the SAMs studied here, can be regarded as semiperiodic systems. There, similar observations are made under the influence of a, typically external, electric field and are known as the quantum-confined Stark effect. Without any external perturbation, in oligopyrimidine SAMs one encounters an energy gradient that is generated by the dipole moments of the pyrimidine repeat units. It is particularly strong, reaching values of about 1.6 eV/nm, which corresponds to a substantial electric field of 1.6 × 107 V/cm. Close-lying σ- and π-states turn out to be a particular complication for a reliable description of the present systems, as their order is influenced not only by the docking groups and bonding to the metal, but also by the chosen computational approach. In the latter context we demonstrate that deliberately picking a hybrid functional allows avoiding pitfalls due to the infamous self-interaction error. Our results show that when aiming to build a monolayer with a specific electronic structure one can not only resort to the traditional technique of modifying the molecular structure of the constituents, but also try to exploit collective electronic effects.
Self-assembled monolayers (SAMs) of organic molecules on noble metals have raised enormous interest over the past decades1−6 and have become important for a number of applications.7−10 SAMs, and even individual molecules, are subject to research also as the active elements in molecular electronic devices.8,11−24 There, especially, the alignment of the states in the SAM relative to the metal Fermi-level as well the degree of “delocalization” of the transport channels between the electrodes are of uttermost importance.25−29
When functional molecules are assembled into SAMs as the active region of electronic devices, control of the electronic structure can be achieved by tuning the properties of the isolated molecule via targeted chemical substitution. Relating molecular and SAM properties, is, however, far from trivial, and the peculiarities of SAM electrostatics have been pointed out repeatedly.30−35,6,36−40 They are essentially related to the fact that well-ordered SAMs of polar molecules correspond to quasi-infinite dipolar layers splitting space into two regions with different vacuum energies with the magnitude of this difference depending on the molecular dipole moments.(38) The latter can be modified by attaching polar donor or acceptor substituents (so-called tail groups) to otherwise apolar molecules, which in the individual molecules significantly modifies the positions of the eigenstates such as the highest occupied molecular orbital (HOMO) relative to the common vacuum level. Surprisingly, however, the level alignment between the SAM states and the Fermi-level of the substrate is typically tail-group independent.33,41 This finding has been rationalized by an electrostatic decoupling of the attached polar groups from the molecular backbones in combination with depolarization effects within the SAM, thus underlining the impact of collective interaction of molecules on the properties of monolayers.42,43
Another approach to adjust molecular dipole moments is to use polar building blocks such as pyrimidines to form molecular backbones, which then can be bonded to metal surfaces by suitable docking groups, e.g., thiolates.(44) By attaching this docking group to one or the other “end” of the oligopyrimidine molecule, two variants with opposite dipole orientation can be realized (cf., “3Nup” and “3Ndown” in Figure 1a).(44) Monolayers formed from these molecules display a number of intriguing and a priori unexpected features, whose discussion and explanation provide interesting new insights into effects that can be exploited to alter the electronic structure of SAMs, especially when they are made of oligomers with polar repeat units. These are briefly outlined in the following:
Figure 1.
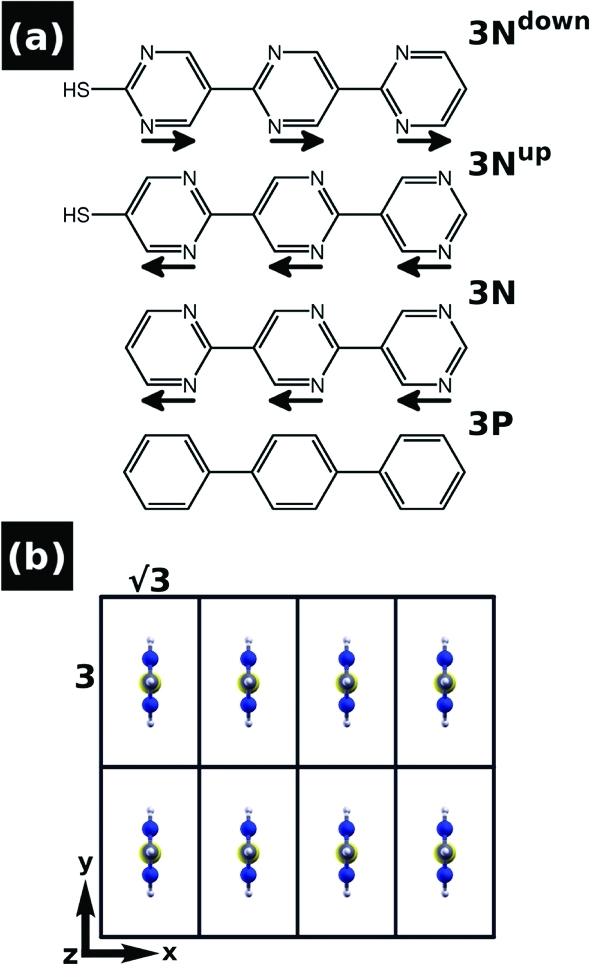
(a) Molecular structures of the terpyrimidinethiols ([2,5′:2′,5″-terpyrimidine]-2″-thiol, 3Ndown, and [2,5′:2′,5″-terpyrimidine]-5-thiol, 3Nup), unsubstituted terpyrimidine (3N), and terphenyl (3P). The permanent dipole moments of the pyrimidines are indicated by black arrows (direction defined as pointing from negative to positive charge). (b) Top view of the investigated monolayers in the (√3 × 3) surface unit-cell. The Cartesian axes are indicated.
-
I.
As mentioned above, tail-group substitutions in many cases33,41 change the molecular eigenenergies but do not impact the level alignment at a metal/organic interface. We find that the exact opposite is true for 3Nup and 3Ndown gold/monolayer systems: The frontier molecular orbital energies for 3Nup and 3Ndownmolecules are essentially identical, whereas for 3Nup and 3Ndownmonolayers the alignment of the electronic states with the gold Fermi-level is totally different.(44)
-
II
. External fields have been shown to modify orbital (band) energies, the frontier orbital (band) gap, and the energetic ordering of the electronic states in molecules and monolayers;45−54 related mechanisms have recently been suggested also for inorganic cluster assemblies.(55) The field distorts and localizes eigenstates,53,54 and recent studies imply that such processes are relevant also when the field originates from dipoles embedded within the layer,(34) resulting in a shift in the optical absorption spectrum.45,46 Internally generated electric fields can be expected to be at work also in pyrimidine-based SAMs, as every pyrimidine unit carries an electric dipole moment. In contrast to tail-group substituted SAMs, where the resulting potential gradient is confined essentially to the substituent region,56,42,33 the distributed dipoles in an oligopyromidine SAM give rise to a potential gradient throughout the entire monolayer. The associated effective internal fields are comparably large (in the range of 107 V/cm), which makes oligopyrimidines an ideal test-bed for studying their impact on the electronic structure of a SAM. This aspect, to the best of our knowledge, has not yet been studied systematically.
-
III.
Upon increasing the SAM packing density and thereby the electric field, the molecular dipoles depolarize each other. Such (de)polarization effects in SAMs arising from internal31,56,57 as well as external58,59,56 electric fields have been the topic of several studies. Here we explain why, in spite of the depolarization of the overall charge density, the collectively generated field polarizes rather than depolarizes the electron densities associated with the frontier orbitals.
In addressing the above aspects, we establish a microscopic understanding of the relevant physicochemical processes in SAMs of molecules consisting of repeated dipolar building blocks. In particular, we describe collective effects that strongly modify SAM properties and, thus, need to be considered in “molecular design” approaches and their optimization. Prior to discussing the electronic structure of the oligopyrimidine layers, it is, however, necessary to provide some details on the chosen model system and also to clarify an important methodological issue that arises from the use of density-functional theory (DFT). The latter is essential, as DFT is the only approach allowing for a realistic, fully quantum-mechanical treatment of two-dimensional periodic arrangements of the size explored here.
Results and Discussion
System
The focus of the present contribution is on processes within the molecular monolayer. Therefore, as a first step we avoid effects involving the interaction with a metallic substrate by excluding the metal from most of our calculations. This is insofar justified as numerous studies have found that the bonding-induced charge rearrangements are confined to the very vicinity of the docking groups,60,61 unless so-called Fermi-level pinning occurs.(62) Such charge rearrangements should, thus, have only a minor effect on the field distribution within the SAM, which is crucial for the effects discussed in this paper. Moreover, in densely packed monolayers the collectively induced electric field acting within the monolayer virtually does not extend onto the metal for electrostatic reasons, as its decay length amounts to only roughly one-sixth of the interdipole distance.(42) The docking chemistry and the interaction with the metal, however, directly affect the order of σ- and π-states. This is addressed later in the paper.
In ref (44), where certain properties of the bonded monolayer are discussed, a herringbone arrangement of the oligopyrimidinethiols in the (√3 × 3) Au(111) surface unit-cell has been assumed. This is a plausible choice, as it has been reported that the related biphenylthiols arrange in this way on Au(111).(63) In fact, oligophenylenes generally tend to crystallize in the herringbone pattern,(64) and this might hold true also for longer oligopyrimidinethiols. At least for monopyrimidinethiols, however, this appears not to be the case: For them, a mixed structure of flat lying and upright standing molecules in larger unit cells has been reported.65,66 The situation is further complicated by the observation that for substituted monopyrimidinethiols the molecules were found to arrange in parallel rows at an even less dense packing.(67) As we are not aware of a work in which assemblies of unsubstituted oligopyrimidinethiols of the type shown in Figure 1 have been studied, it is a plausible compromise to consider such parallel orientations, but in a denser packing of one molecule per (√3 × 3) cell (shown in Figure 1b). Also from a practical point of view the chosen surface unit-cell is convenient as it allows an easy comparison of the various systems and between the electronic structure of isolated and assembled molecules. For consistency with previous work,44,57 where each (√3 × 3) cell contained two molecules in a herringbone pattern (vide supra), we denote the present full coverage as θ = 1/2. Finally, we note that in test calculations on the herringbone-packed SAM at the coverage used in ref (44) we qualitatively recover the results reported below.
Determining a Suitable Computational Approach
Before discussing the electronic structure of the above-discussed SAMs, it is imperative to critically assess which of the available computational approaches is most suitable for reliably predicting the quantities of interest. We expect that to be of distinct relevance also for future studies of related systems.
As we aim at describing monolayers, i.e., systems that are infinitely extended in two dimensions, periodic boundary conditions are required. Considering the size of the investigated unit cells, any approach other than DFT is computationally not affordable. In spite of its great success in quantum-chemistry and physics, (semi)local DFT, however, also suffers from a number of shortcomings.
In the present context especially the infamous self-interaction error (SIE) in (semi)local DFT, i.e., the erroneous interaction of each orbital with itself,68,69 can become particularly relevant (cf., extended discussion in the Supporting Information). This is because in pyrimidines there are close-lying σ- and π-orbitals in the vicinity of the HOMO/LUMO gap,70−72 intertwined with significant variations in orbital localization, a scenario in which drastic failures of a (semi)local DFT-based description have been reported.73,74 In oligopyrimidines this could very well result in an incorrect prediction of the actual nature (σ vs π) of the frontier states.
To estimate the possible impact of the SIE, we follow the strategy presented in ref (74), where a simple-to-evaluate predictor for the presence of a strongly orbital-dependent SIE has been proposed. For every Kohn–Sham orbital, it calculates the amount of Coulomb self-repulsion that is not canceled by exchange-correlation (xc) self-attraction. To evaluate the importance of self-interaction in the present systems, we performed this test for the isolated 3Nup molecule. The orbital SIEs, ei, for the HOMO–10 to HOMO (i.e., valence orbitals 36 to 46) relative to that of the HOMO, Δei:= (ei – e46), are shown as solid orange circles in Figure 2a.(76) The displayed values have been calculated using the local density approximation (LDA), but essentially equivalent results are obtained when using gradient-corrected (GGA) functionals.
Figure 2.

Self-interaction error and electronic structure of the isolated 3Nup molecule. (a) The self-interaction error of the LDA orbitals is shown (orange) together with the GKLI SIC (green) and the HSE corrections (black). All quantities are given relative to the value for the HOMO (orbital 46). (b) LDA (left) and HSE (right) orbital energies, aligned to the respective HOMO energies. The arrows link associated orbitals. (c) Absolute HSE-computed eigenenergies of 3Ndown (left panel) and 3Nup molecule (right panel), broadened by a Gaussian with σ = 0.1 eV. The insets show orbital charge-densities of the frontier states; the HOMO–LUMO gap is indicated.
Indeed, strongly varying values of Δei are found for the high-lying orbitals, which are most important in applications. An inspection of the real-space representations of the orbital charge-densities (contained in the Supporting Information) shows that the magnitude of the SIE predictor indeed depends on the visual impression of orbital localization (while, apparently, it is not significantly affected by whether an orbital displays σ- or π-character). These results clearly emphasize the importance of using a description that is free from self-interaction.
To achieve this goal, we performed a self-interaction correction (SIC) employing the generalized optimized effective potential (GOEP) method(77) using the Krieger–Li–Iafrate approximation (GKLI). The latter has been shown to accurately reproduce the results of numerically far more demanding full GOEP calculations.(77) The obtained relative orbital energy shifts, ΔεiGKLI = (εi – εiLDA) – (εHOMO – εHOMOLDA), are depicted in Figure 2a as solid green squares. Besides opposite signs, we find systematic differences between Δei and Δεi. This is insofar expected, as there is only an approximate correlation between the SIE and its correction.(78) Different from Δei, the values ΔεiGKLI appear to be primarily sensitive to the character of an orbital; i.e., there is a common correction for all σ-states and a different common one for all π-states irrespective of the intuitive impression of localization (see Supporting Information). Most importantly, the correction shifts the HOMO–2 up in energy by 0.59 eV, to above the LDA-HOMO, demonstrating that the SIE severely distorts the electronic structure of terpyrimidinethiols.
An alternative approach to obtain improved orbital energies is the use of hybrid functionals. These, however, only partially correct for self-interaction, as they include only a fraction of exact exchange. Still (as rationalized in the Supporting Information), often good comparability with experiment and SIC or GW calculations is found for the low binding energy states. A functional which is readily applicable to both molecular and periodic systems is the Heyd–Scuseria–Ernzerhof screened-exchange hybrid functional, HSE06,79−81 where the long-range part of the Fock exchange is absent by construction. As discussed in ref (82), the absence of the long-range Fock exchange is not detrimental to the molecular electronic structure, as it hardly influences the highly localized orbitals which exhibit a large SIE. The open black squares in Figure 2a depict the correction to the LDA orbital energies as calculated with HSE, again relative to the shift of the LDA-HOMO, ΔεiHSE:= (εi – εiLDA) – (εHOMO – εHOMOLDA).(76) Clearly, HSE and GKLI corrections compare very well, and both result in a strong modification of the order of the occupied frontier orbitals compared to the LDA (and also GGA) calculations (cf. Figure 2b). This way the two approaches mutually support each other, and we conclude that HSE is well-suited for describing the systems at hand.
Certain shortcomings, however, remain: Hybrid functionals do not account for the narrowing of the fundamental gap by dielectric screening in the SAM, as this requires nonlocal correlation.83−85 Such effects should, however, have no qualitative impact on the SAM’s electronic structure, although they will need to be revisited in a later chapter, when the interaction of the SAM with the metal is discussed.
Another potential pitfall linked to DFT is the reliable calculation of molecular polarizabilities. For example, Champagne et al.(86) have argued that typical exchange-correlation functionals tend to overestimate polarizabilities. Some of us have shown that this is not always the case87,88 and that it depends on the particular electronic structure of the system in question.(89) An (at least partial) remedy is typically the inclusion of exact exchange. As molecular dipole moments and polarizabilities are absolutely crucial parameters for the present paper, we have performed extensive test calculations employing a number of different DFT functionals, Hartree–Fock (HF) and second-order Møller–Plesset perturbation theory (MP2). These tests (which are discussed in considerable detail in the Supporting Information) show that for the present system all methods give consistent results for the polarizabilities as well as for the dipole moments; the hybrid functional calculations yield a HOMO–LUMO gap that is somewhat larger than the optical gap calculated by time-dependent density-functional theory (in contrast to the LDA and GGA functionals, which severely underestimate the gap). This further confirms the suitability of HSE for describing the problems at hand.
Molecule-to-Monolayer Transition
With the appropriate tools in hand, we can now turn to first calculating the molecular and then the monolayer properties. Figure 2c shows the density of states (DOS) of 3Nup and 3Ndown molecules as calculated with HSE. This plot illustrates that the position of the thiol docking group has only minor impact on the molecular electronic structure. The ordering of the frontier orbitals is the same with all frontier orbitals having π-character (see insets), and also the eigenvalues are comparable; the gap differs by only 0.2 eV.
Assembling the polar molecules into a monolayer creates a two-dimensional array of dipoles, μ, which give rise to a change in the electron electrostatic energy between the docking- and the tail-group sides of the SAM. The net effect is described via the Helmholtz solution to the Poisson equation (ΔEvac = −eμ/ε0A) and is proportional to the areal dipole density.90,91 In densely packed SAMs, in which ΔEvac originates from polar tail-group substituents (the commonly studied situation), the corresponding shift in the potential landscape is strongly confined to the region of the tail-group.(33) This can be explained by purely electrostatic arguments, as it can be shown that for a square two-dimensional array of point dipoles the decay-length of the field is equivalent to the interdipole distance divided by 2π.(42) In SAMs consisting of polarizable molecules, this confinement is further enhanced by depolarization effects.92,93,31,94,30,59,56,57,41,43
The situation is markedly different for oligopyrimidines as shown for a 3Nup SAM at the highest investigated packing density, θ = 1/2, in Figure 3a. As the total molecular dipole originates from each of the dipolar pyrimidine rings, ΔEvac is established across the SAM in a more or less continuous way.(44) Even though the plane-averaged electrostatic energy displayed in Figure 3a oscillates due to the nuclei, one can clearly see that it is superimposed with an approximately linear potential gradient, which is due to the combination of the fields generated by the pyrimidine dipoles. This is best seen when averaged over the length scale of interatomic distances (red curve).95,96 The collectively generated potential-energy gradient exists only within the SAM and can be associated with an effective internal field. The magnitude of that “collectively-induced” field can be estimated to be 1.6 eV/nm from the overall potential energy increase and the thickness of the monolayer. Such simple model considerations have been found to be of great explanatory power in previous studies of inorganic systems,97−99 and in the following sections we describe how they are applicable also to oligomeric SAMs made of molecules that consist of polar repeat units. In passing, we mention that one should be able to observe this potential gradient, for example, by high-resolution X-ray photoelectron spectroscopy experiments,100,101 as it results in the core electrons of carbon and nitrogen atoms in the backbone being located at different relative electrostatic energies.
Figure 3.
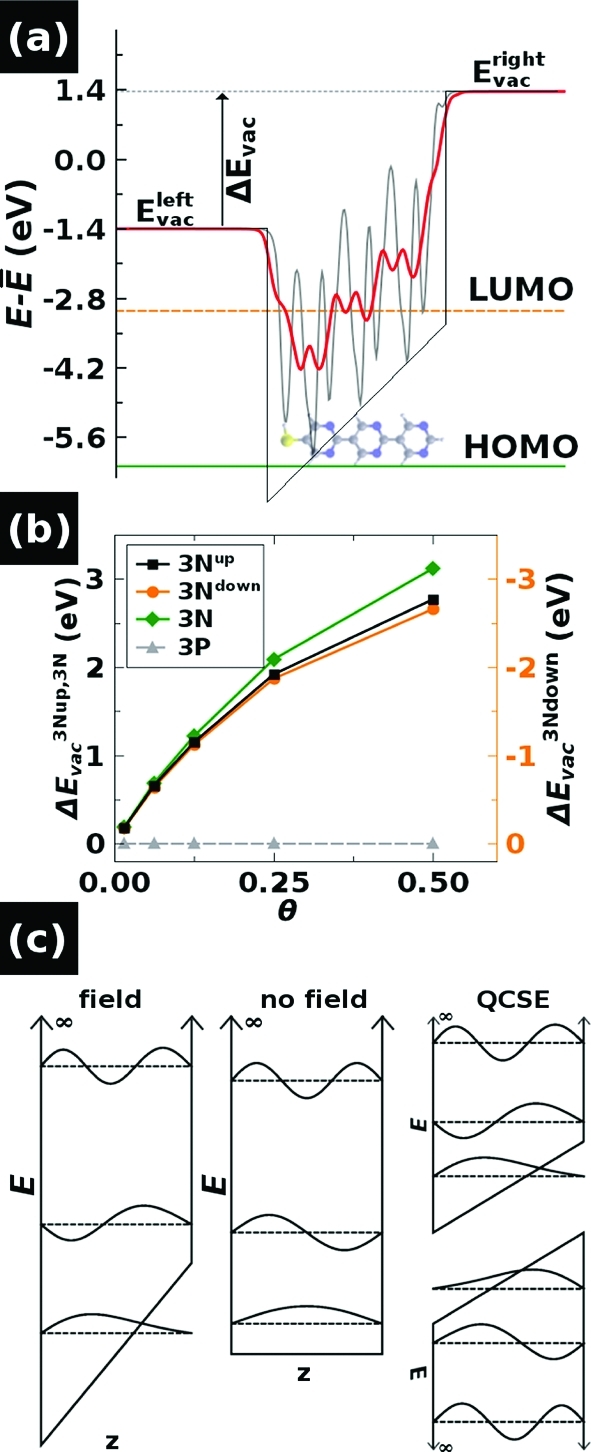
(a) (x, y)-averaged electrostatic energy E of an electron across a hypothetical free-standing 3Nup-SAM at θ = 1/2 (gray curve; for details on the further averaged red curve, see text). The molecule in the background and the “box-potential” serve as guide to the eye. The zero of the energy axis is set to the average of the left and the right vacuum energy, E̅ = (Evacleft + Evac)/2 . (b) Step in the electrostatic energy across the SAM, ΔEvac = Evacright – Evac, as a function of the SAM packing density θ for 3Nup (black squares), 3Ndown (orange disks), 3N (green diamonds). The flat evolution for the apolar 3P SAM (gray triangles) is shown for comparison (see Figure 1b for the molecular structures). Note that the energy scale is reversed for 3Ndown. (c) Sketches of the potential and the lowest eigenstates of 1D potential wells with infinitely high barriers for a particle in a box exposed to an electric field (left panel) and without electric field (middle panel). The rightmost panel sketches the situation encountered for the envelope functions in the valence and conduction bands that are responsible for the quantum-confined Stark effect (QCSE) in a semiconductor quantum-well structure.
Also in oligopyrimidine SAMs, the overall magnitude of ΔEvac is reduced by the above-mentioned depolarization effects, as can be inferred from its sublinear increase with packing density shown by the green diamonds in Figure 3b. A comparison between the trend for 3N (green diamonds) and 3Nup (black squares) shows that the thiol docking group does not qualitatively influence the overall electrostatic situation. In 3Ndown, the pyrimidines are oriented differently (see Figure 1a), which reverses the sign of ΔEvac, keeping its magnitude, however, largely unchanged (orange disks; note the reversed scale).
The overall situation of the free-standing SAM in Figure 3a can be viewed in analogy to the well-understood case of an (inorganic) semiconductor quantum-well under the influence of an electric field. In semiconductor quantum-wells the field is typically externally applied, although the role of internal fields is well acknowledged (for instance, see ref (102)). Here we are dealing exclusively with the latter: a packing-density dependent collectively induced field that originates from the distributed molecular dipoles within the system. In both, semiconductor quantum-wells and oligopyrimidine SAMs, one has to deal with infinitely extended periodic systems in the x- and y-direction, while there are a finite number of repeat units in z-direction. As a consequence, the wave functions of the quantum-well retain their Bloch-type character in x- and y-direction (i.e., they can be described as lattice-periodically modulated plane waves), while in z-direction the so-called envelope-function approximation comes into play.(103) There, the plane-wave parts of the Bloch-type states, which require infinite periodicity, are replaced by the eigenfunctions of the quantum-well (reflecting the well properties); the lattice-periodic parts of the wave functions still determine which band the states are associated with. Describing the orbitals of oligomers (like terpyrimidine) in such a quasiband structure picture is a well established concept(104) which is consistent with Hückel theory(105) and has been confirmed experimentally by inelastic electron scattering106−108 and photoelectron spectroscopy.109−112
The envelope functions play a defining role for the properties of the orbitals and are key to understanding the electronic structure of the SAMs discussed here. When, for the sake of simplicity, infinitely high barriers are assumed, the well-known eigenfunctions of such a quantum-well in the presence of a constant potential gradient (i.e., a homogeneous electric field) are linear combinations of Airy functions.(113) The few lowest are depicted in the left panel of Figure 3c. In contrast to the field-free situation (middle panel of Figure 3c), they are polarized by the electric field. The lowest state shows pronounced localization on the low-energy (left) side of the well and for moderate fields the probability density for higher states increases in the high-energy (right) side of the potential well.113−115 This phenomenon of qualitatively different localization for different states has been denoted as anomalous polarization in the literature,114,115 and is relevant for the discussion below.
For describing both the valence and the conduction band, one has to keep in mind that holes carry a positive charge, which reverses the corresponding potential well leading to the situation depicted in the right panel of Figure 3c. The latter is characteristic of the quantum-confined Stark effect (QCSE). It is exploited, for example, in absorptive modulators, where the confinement imposed by the well-structure is necessary to prevent dissociation of the excitons.116,103 The simple picture in Figure 3c implies that (i) the gap between the occupied and unoccupied states should decrease upon increasing the field strength (in the oligopyrimidines this means increasing the packing density); (ii) electrons and holes are localized at opposite edges of the quantum structure; (iii) the degree of this localization should increase with the field.
Collectively Induced QCSE: Band Gap and Localization
We start our discussion with terpyrimidine, where no thiol group is attached (3N in Figure 1a). Comparing the DOS of this molecule with that of the corresponding SAM (θ = 1/2) in the upper panel of Figure 4, it is found that the HOMO–LUMO gap is reduced by no less than 0.8 eV. In contrast, the gap of the nonpolar terphenyl (3P in Figure 1a) remains essentially unchanged upon monolayer formation (Figure 4, lower panel). Consequently, it is the continuous electric field induced within the ensemble of 3N molecules which is responsible for the observed reduction of the band gap in the spirit of the above-described QCSE. This is consistent with the results of others for molecules45−50,52 and SAMs45,46,53 in electric fields. It also implies that the calculated gap-reduction is not a consequence of dielectric screening by neighboring molecules, an effect that is not captured by hybrid calculations (cf., Determining a Suitable Computational Approach section).
Figure 4.
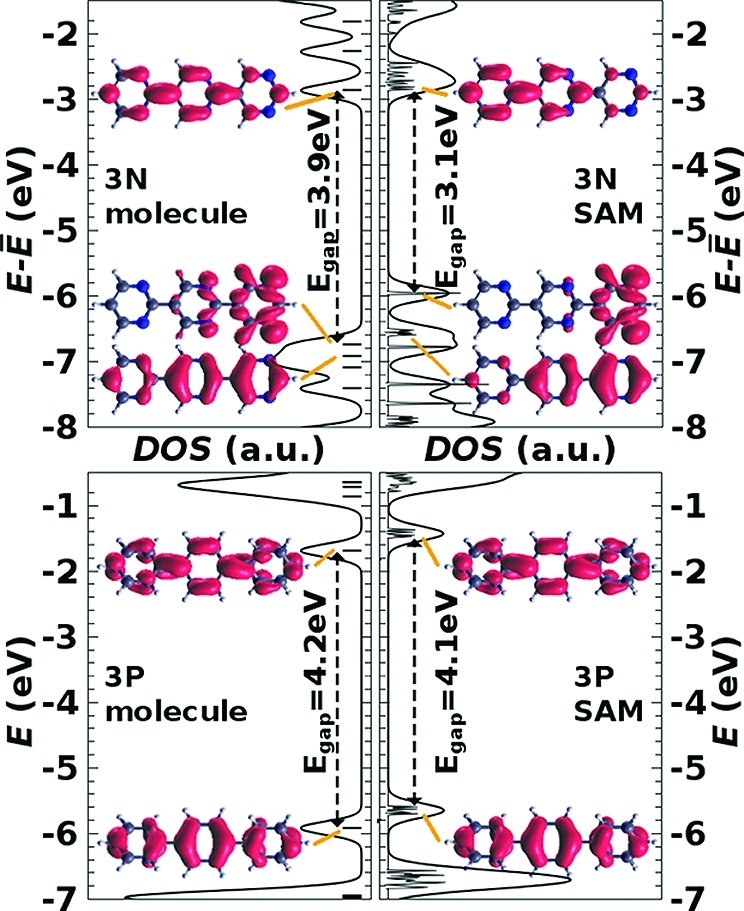
Density of states (DOS) for the 3N (upper panels) and 3P (lower panels) molecules (left panels) and high-coverage SAMs (θ = 1/2, right panels), aligned at the average electrostatic energy across the SAM, at E̅ = (Evacleft + Evac)/2 (cf. Figure 3a). The thick black curves are Gaussian-broadened (σ = 0.1 eV) convolutions of the results of the calculation. The insets show orbital/band charge densities of the frontier states, and the band gap is indicated. It is determined from the onsets of the respective nonbroadened DOS peaks.
The real-space representations in Figure 4 can also be associated with the collectively induced QCSE: In the case of terphenyl (lower panel), both HOMO and LUMO are delocalized over the molecule, and this does not change upon monolayer formation. In contrast, for terpyrimidine the HOMO and LUMO are to some degree localized on opposite ends of the molecule already in the isolated system (molecular effect). This localization is further enhanced in the monolayer (collective effect). Because the electrostatic energy increases across the SAM (cf., Figure 3a), this corresponds to a localization of the HOMO level at the high-energy side of the SAM as in the QCSE model in Figure 3c (right panel). In other words, the corresponding electron density is shifted along the electric field. Note that this is counterintuitive from the perspective of depolarization effects (Figure 3a), as the total electron density is (naturally) shifted opposite to the electric field direction as a consequence of the negative charge of electrons.
To better quantify this effect, we calculated orbital charge-densities of the frontier states integrated over the (x, y)-plane using the definition given in the Methods section. They are shown for the isolated 3N molecule as orange areas in the upper panels of Figure 5. The black curves show the corresponding charge densities for the bands derived from these orbitals in the SAM (θ = 1/2). As inferred already from the contour plots in Figure 4, HOMO contributions on the central ring are shifted to the rightmost pyrimidine and “virtual” charge density corresponding to the molecule’s LUMO accumulates on the leftmost ring at the expense of the rightmost ring. This trend is even more clearly resolved when plotting the respective charge-density differences, Δρi(z) = ρiSAM(z) – ρi(z), in the central panel of Figure 5, where red (blue) areas show accumulation (depletion) of electron density. In this context it is also useful to plot the cumulative charge rearrangement, Qi(z), defined as Qi(z) = ∫0z Δρi(z′) dz′ (see bottom panel of Figure 5). Qi(z) > 0 gives the number of electrons that have been transferred from the region right of z to its left. For Qi(z)< 0, the direction of the charge transfer is reversed. The latter quantity shows that the effect of self-localization is sizable: for the HOMO, 0.3 electrons are accumulated on the rightmost ring. The reorganization of the LUMO is of the same magnitude, but opposite in sign.
Figure 5.
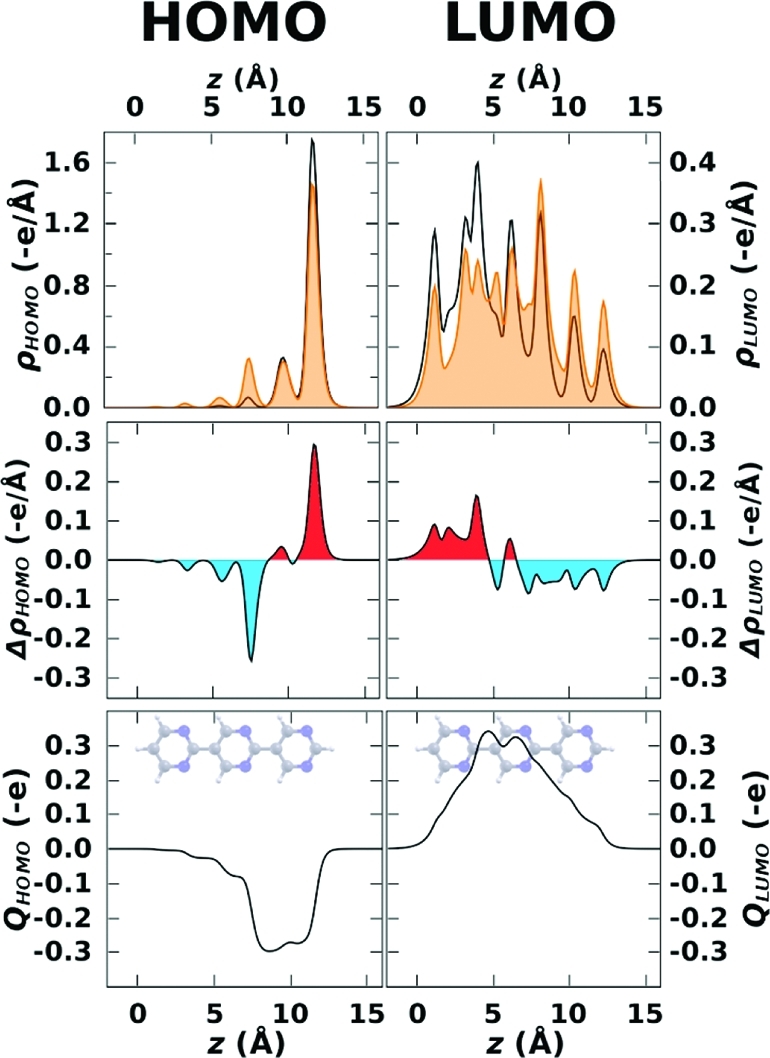
Upper panels: (x, y)-plane-integrated orbital charge-densities for the HOMO and LUMO of 3N, to quantify orbital localization in the direction of the long molecular axis (orange areas). The black curves show the corresponding data for the band derived from those orbitals in the SAM at θ = 1/2. Middle panels: Charge rearrangements Δρi(z) = ρiSAM(z) – ρi(z) for the HOMO (left) and LUMO (right) upon monolayer formation. Red (blue) areas show accumulation (depletion) of electron density. Lowest panels: Cumulative charge rearrangements Qi(z) for these states upon monolayer formation. The molecules in the background serve as guide to the eye.
Collectively Induced QCSE: Eigenenergies
The above findings lead to the question of how the collectively induced electric field influences the electronic structure of the SAM beyond orbital localization. The simplified potential wells in Figure 6a sketch the electrostatics across the SAM at low (gray) and high (black) packing density. The orange markings illustrate how the energy is changed differently at different positions in the well. As the orbitals are differently localized along the long molecular axis already in the isolated molecule and even more so in the SAM (vide supra), it can be expected that their energies are differently affected by increasing the packing density (and, thus, increasing the internal field). To illustrate that such shifts are actually observed, Figure 6b shows the evolution of the HOMO, HOMO–1, and LUMO derived states of 3N as a function of the packing density relative to E̅ = (Evacleft + Evac)/2 (cf., Figure 3a). The HOMO shifts up by up to 0.9 eV, while the LUMO is destabilized by only 0.1 eV, consistent with the above-discussed gap-reduction by 0.8 eV. The observation that the LUMO is destabilized instead of stabilized (as one might expect from its localization) is most likely due to the somewhat arbitrary choice of the average vacuum level as energy reference, but could also be an indication that, while the model in Figure 6a provides a consistent qualitative picture of the situation, it does not reproduce all quantitative details.
Figure 6.
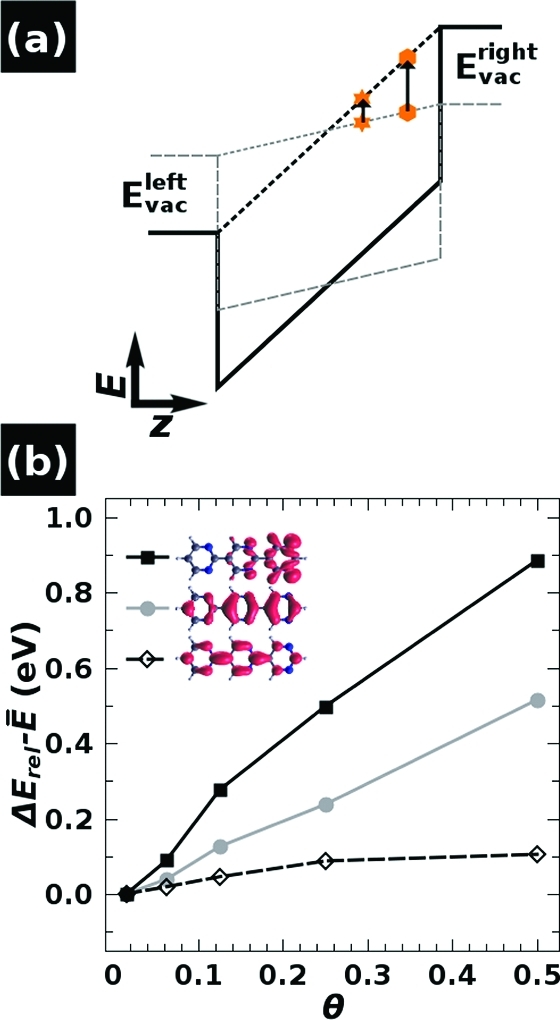
(a) Simplified sketch of the electron electrostatic energy across a SAM of 3N molecules at low (dashed gray) and high (solid black) packing density. The orange symbols illustrate for two positions how the energy is changed differently at different positions in the well upon increasing the SAM packing density. (b) Energetic shifts of the HOMO (black squares), HOMO–1 (gray circles), and LUMO (black diamonds) derived bands in a SAM of 3N molecules upon increasing the SAM packing density θ, aligned at the average electrostatic energy E̅ = (Evacleft + Evac)/2 (cf. Figure 3a). The energies are determined from the onsets of the respective peaks in the DOS.
Furthermore, the energetic distance between HOMO and HOMO–1 is increased by nearly 0.4 eV (cf., Figure 4) because of the very different localization of those states. As a consequence, the π–π* gap (i.e., the gap between the HOMO–1 and the LUMO) is reduced by only 0.4 eV. An interesting observation concerns the significant shifts already at very low packing densities. The energy difference between HOMO and HOMO–1, for example, is increased by 0.2 eV already between θ = 1/64 (considered as the isolated molecule) and θ = 1/8, and the gap is reduced even more. This clearly shows that the observed orbital-energy shifts are primarily of electrostatic origin, fully consistent with the occurrence of a collectively induced QCSE.
QCSE and Depolarization
Finally, it needs to be understood how the fact that the HOMO is shifted toward the “right” side of the SAM upon increasing the packing density (which corresponds to increasing the molecular dipole) can be reconciled with the generally observed depolarization effects in polar SAMs. That the latter occur also in oligopyrimidine SAMs can be inferred, for example, from the sublinear increase of ΔEvac with coverage in Figure 3b. This can only be explained by the total charge density reacting in a qualitatively different manner to the electric field than the charge density associated with the HOMO-derived band.
To explain that, it is useful to identify “related” molecular orbitals. This can be done by applying the quasiband picture outlined earlier, in which each orbital of an oligomer is interpreted as the product of a quantum-well eigenfunction (the envelope function) and a function derived from an orbital of a single repeat unit (e.g., the HOMO or LUMO of pyrimidine).103−112 The latter then determines which quasiband a given orbital of the oligomer belongs to, with the number of states in a quasiband equaling the number of repeat units. Figure 7a shows the orbitals related in this way to the HOMOs and LUMOs of 3N and 3P. As terphenyl is an apolar molecule, sine functions are suitable envelope functions (cf., middle panel of Figure 3c).(110) In contrast, significant asymmetries are found in the orbitals of terpyrimidine, which is due to the envelope functions in this polar molecule being better described by linear combinations of Airy functions (cf., left panel of Figure 3c). Note that, although this very simple model does not quantitatively reproduce the localization of each orbital, its value is evident as it perfectly reproduces the changes of sign of the wave functions along the backbone.
Figure 7.

(a) Isodensity plots for related occupied (lower plots) and unoccupied (upper plots) molecular orbitals of 3N (left) and 3P (right).(117) Also shown are schematic standing-wave envelope functions. (b) Upper panels: Charge rearrangements Δρi(z) = ρiSAM(z) –ρi(z) for the HOMO–2 (left) and HOMO–3 (right) upon monolayer formation. Red (blue) areas show accumulation (depletion) of electron density. For the equivalent plot for the HOMO, see Figure 5. Lower panels: Cumulative charge rearrangements upon SAM formation. Left panel: Qi(z) for the three related states HOMO (black), HOMO–2 (crossed gray), and HOMO–3 (light gray) of 3N. Right panel: Sum over the three Qi-curves shown in the left panel (black color) and sum over the Qi of all occupied states (gray color).
Bearing in mind that the QCSE “picture” in the rightmost panel of Figure 3c is derived from the envelope-function approximation, it is useful to consider all orbitals belonging to a quasiband “simultaneously”. The corresponding Δρi(z)- and Qi(z)-plots for the HOMO-related states (the HOMO–2 and the HOMO–3) in 3N are, therefore, included in Figure 7b. For the lowest-lying orbital (HOMO–3) the opposite trend than for the HOMO is observed (cf., Figure 5); i.e., electron density is shifted to the left part of the molecule. The HOMO–2, on the other hand, experiences a shift of charge density from the perimeters to the center of the molecule, almost perfectly canceling the combined effect of HOMO and HOMO–3. Indeed, the sum of all three Qi(z) (Figure 7b, lower right panel, black curve) shows that the net polarization due to the HOMO-related quasiband essentially vanishes. This simultaneous occurrence of polarization and anomalous polarization within a quasiband is fully consistent with previous findings for the eigenfunctions of quantum-wells (cf., Figure 3c).114,115,113 Whether a small polarization or depolarization remains within a given quasiband then depends on the details of the involved orbitals. The overall response obtained from summing over all orbitals must, of course, reflect a depolarization (cf., Figure 7b, lower right panel, gray curve). Interestingly, the maximum of this net cumulative transfer amounts to <0.1 electrons, which is only about 1/4 of what is observed for the HOMO alone.
Bonding the SAM to the Metal Substrate: Experimentally Accessible Quantities
Attaching the monolayer to the metal substrate can conceptually be viewed as a two-step process: First a docking group needs to be attached to the terpyrimidine SAM, which then binds the layer to the metal. The attachment of the docking group has two consequences: (i) It determines in which orientation the terpyrimidines are bonded to the metal and whether they induce a work-function decrease (3Ndown) or a work-function increase (3Nup) (cf., ΔEvac in Figure 3b and ref (44)). (ii) Typically, docking group orbitals will hybridize with the states of the terpyrimidines. For thiols, the latter is evident already in Figure 2c, where for the isolated molecules we find the HOMO to display π-character, as the corresponding 3N orbital (the HOMO–1) has been destabilized due to the contribution of the sulfur to the molecular π-system. As a consequence of the orbital localization in terpyrimidine (cf., Figure 4), the effect is more pronounced in 3Ndown. Consistently, there the degree of localization of the highest occupied π-state is increased by the thiol, whereas it is decreased for 3Nup.
In 3Ndown, when forming a monolayer from the isolated molecules, the collectively induced field is oriented such that it enhances the localization of the HOMO (and HOMO–1) and strongly destabilizes it relative to the average vacuum level. This results in the particularly small band gap of 2.8 eV (see left plot of Figure 8a). In the 3Nup case the highest occupied σ-type orbital experiences a similar shift as in 3Ndown, while the energetic position of the highest occupied π-state relative to E̅ is hardly affected by the collectively induced field. This results in the gap of the 3Nup SAM being of σ–π* character as in the terpyrimidine SAM (cf., Figure 8a). A similar situation is encountered also when considering isocyanide as docking group. Also, there the π-electrons of the −NC group hybridize with the π-system of 3N, modifying orbital localization and the packing-density dependent shifts of the eigenenergies. A more detailed discussion is contained in the Supporting Information.
Figure 8.

(a) DOS of 3Ndown and 3Nup SAMs at θ = 1/2, aligned at the average electrostatic energy across the SAM, E̅ (see Figure 3a). The thick black curves are Gaussian-convolutions (σ = 0.1 eV) of the results of the calculation. The insets show band charge-densities of the frontier states, and the band gap is indicated. It is determined from the onsets of the respective nonbroadened DOS peaks. (b) Energetic shifts of the HOMO, HOMO–1, and LUMO derived bands in a SAM of 3Ndown (left) and 3Nup (right) molecules relative to the molecular eigenvalues upon increasing the SAM packing density θ. The energies are determined from the onsets of the respective peaks in the DOS. They are aligned at the left vacuum energy Evacleft; the absolute molecular eigenvalues (approximated by θ = 1/64) are −6.52 eV (−6.57 eV), −7.02 eV (−6.74 eV), and −3.02 eV (−2.90 eV) for the π-HOMO, σ-HOMO, and LUMO of 3Ndown (3Nup). (c) DOS of the full metal/SAM system projected onto the molecular region (PDOS) for the 3Ndown and 3Nup SAMs, with broadening and insets as in part a.(118) Only a small fraction of metal atoms is shown. The Fermi energy is indicated as gray horizontal line.
To understand qualitatively how the collectively induced QCSE affects the alignment between the molecular states and the Fermi-level, i.e., addressing question I raised in the Introduction, it is useful to analyze the position of the electronic states relative to the vacuum level at the docking-group end of the SAM (Evacleft in Figure 3a) as detailed in refs (119, 61, 91, and 38).
The alignment of the states relative to Evacleft for 3Ndown and 3Nup shown in Figure 8b can be understood in a straightforward manner from the schematic sketch in Figure 6a and the localization of the orbitals in the isolated molecules (Figure 2c) and the monolayers (Figure 8a). In 3Ndown, the potential energy gradient is opposite to that sketched in Figure 6a. This is, compared to Evac all states are shifted down in energy upon increasing the field (i.e., increasing the packing density). The effect is relatively weak for the HOMO and the HOMO–1 as a result of their localization close to the docking-group end of the SAM and amounts to only ca. 0.5 eV. For the 3Nup SAM, the energy gradient is opposite to that in 3Ndown, resulting in an upward shift of all states for increasing coverage. The net shift between the isolated molecule and the θ = 1/2 SAM amounts to ca. 1.3 eV for the (SAM) HOMO–1 and the LUMO and becomes as large as 2.0 eV for the (SAM) HOMO, which is localized on the ring furthest away from the docking group end of the SAM. This already strongly suggests that the level alignment of the 3Nup and 3Ndown SAMs bonded to the metal will differ by several electron volts as a result of the collectively induced QCSE. This is a spectacular effect, considering that the molecular ionization potentials of 3Nup and 3Ndown are essentially the same.
To obtain a more complete picture, one needs to explicitly consider the bonding to the metal. This is a computationally formidable task when using hybrid functionals. We also note that it is still not fully quantitative: In the case of molecules adsorbed on a metallic substrate,(83) describing the narrowing of the gap due to polarization of the metal will generally require both long-range exchange and long-range correlation, neither of which is present with HSE.85,120
Figure 8c shows the density of states projected onto the SAM region (PDOS) and the local density of states plotted for certain energy ranges(118) for the 3Ndown and 3Nup metal-adsorbed SAMs. Also, in the presence of the metal the frontier states are localized on opposite “ends” of the monolayer in the 3Nup and 3Ndown SAMs, and indeed a completely different level alignment is found in the two systems. While for 3Ndown the unoccupied states are closest to the Fermi-level (see Figure 8c), it is the occupied states for 3Nup.44,121 However, the thiolate–Au bond also modifies the SAM electronic structure. First, strongly dispersing metal-induced intragap states appear around 1.7 eV below the Fermi-level (overlapping with sharper peaks for 3Nup). They are strongly localized on the S atom (see inset for 3Ndown). This changes its hybridization with the π-electrons of the backbone upon adsorption, and the π-states no longer have any weight on the S-atom unlike in the freestanding SAMs shown in Figure 8a. The highest occupied π-states are strongly stabilized and for both SAMs shift down to partly overlap with the next σ-states. This has next to no influence on the band gap of 3Nup (see Figure 8c). However, adsorption changes the symmetry of the 3Ndown HOMO, and the gap is again opened by 0.5 eV. Interestingly, this way a value close to the HSE-computed gas phase gap is reached (although with different character of the HOMO).
Having analyzed the electronic structure of the SAM bonded to the metal substrate one can now wonder how the effects described in the paper would impact experimentally accessible physical observables. Beyond the potential of directly imaging the collectively induced electric field in pyrimidine SAMs by high resolution XPS mentioned earlier, especially the QCSE-induced shifts of the eigenenergies should have drastic effects. The different positions of the molecular states relative to the Fermi-level will strongly impact charge-carrier injection into oligopyrmidine SAMs, for example, in scanning tunneling microscopy and spectroscopy experiments. For a given tunnel bias, it will also result in drastically different tunneling microscopy images. In fact, the results in Figure 8c imply that for imaging 3Ndown SAMs a negative tip bias inducing the tunneling of electrons might be preferable, while the opposite (i.e., a preferred tunneling of holes) applies to 3Nup SAMs. The different level alignment for the two terpyrimidine orientations should also be observable in UV photoelectron spectroscopy and will modify the effective injection barrier when using SAMs to modify the work function of metal electrodes in organic devices. Charge transport through oligopyrimidine layers will be affected also by orbital localization effects, as they tend to deteriorate the transmittance of the transport channels. Moreover, excitation energies in oligopyrimidine layers will be reduced and two-photon photoemission provides a technique to study the relevant excited states.(133) Whether or not certain experimental observations are indeed due to the collectively induced QCSE can then be checked in an (at least conceptually) straightforward manner, as they should all display a distinct dependence on the molecular packing density that might, for example, be varied by forming mixed monolayers with “inert” molecules.
Relevance of the QCSE beyond Oligopyrimidines
As stressed repeatedly, all above effects are essentially caused by the collectively generated electric fields. Therefore, they should by no means be limited to oligopyrimidines, but occur in any SAMs made of molecules with a distributed dipole character, i.e., molecules consisting of a series of polar building blocks. A few examples for alternative (mostly heterocyclic) molecules are shown in Figure 9. A quick theoretical screening shows that most of them have appreciable dipole moments, which for the methineimines is even larger than in the oligopyrimidines; i.e., while the above-described effects are expected to be weaker for the displayed oxazines, thiazoles, imidazoles, and oxazoles, they should be significantly stronger in the methineimines. While the latter have been treated theoretically,(122) their synthesis will pose a considerable challenge.
Figure 9.

Alternative molecules (names given in the Supporting Information) consisting of polar building blocks with equivalent numbers of heteroatoms along the backbone. The HSE06/6-311++G(d,p) calculated total molecular dipole moments are also listed, and terpyrimidine is included for the sake of comparison.
Summary and Conclusions
In summary, using a carefully selected theoretical approach that accounts for the infamous self-interaction error of density-functional theory, we have shown that the electronic structure of oligopyrimidine SAMs is strongly influenced by the electric field that results from a collective action of the intrinsic molecular dipole moments: Upon gradual transition from the isolated molecule to the densely packed monolayers, we observe (i) an increasing localization (and anomalous polarization) of the molecular orbitals (respectively the real space representations of the associated bands), (ii) a significant decrease of the computed band gap by up to 0.8 eV, and (iii) concomitantly, a strong impact of the collectively induced electric field on the level alignment with the metal Fermi-level. These trends can be rationalized combining the quasiband/envelope-function picture with the Quantum-Confined Stark effect typically observed in semiconductor quantum-wells under the influence of strong external fields. The peculiarity of the present systems is that here the potential energy gradient within the layer is collectively induced by the interacting polar molecular building blocks. For the sake of clarity, these effects are primarily discussed for the hypothetical free-standing monolayers, but essentially prevail also when the SAMs are bonded to a metal substrate.
The above considerations underline that periodically assembled molecules can display properties that hugely differ from those they possess in gas phase. Therefore, when designing molecular building blocks for novel self-assembled materials and hybrid systems, collective effects have to be carefully considered. This is particularly relevant when working with molecules containing polar building blocks, where electrostatic effects can have a huge impact on the electronic properties. Apart from complicating the bottom-up design of new systems, the presented results, however, also provide tools that can be exploited as additional handles for tuning the properties of also more complex systems than the ones discussed here. As a long-term vision, one could imagine exploiting collective electrostatic effects that occur as a result of monolayer formation to shift the electronic levels “by design” in various parts of extended molecules giving molecular electronics a “collectively-controlled” twist.
Methods
All molecular geometries were obtained by optimizations with the PBE functional using the Gaussian 03(123) package and the aug-cc-pVTZ basis set. Subsequent LDA and GKLI calculations were done with the PARSEC real-space code124,125,77 and norm-conserving pseudopotentials(126) and HSE06 calculations with the VASP code(127) with a plane-wave basis set (kinetic energy cutoff of approximately 20 Ry) and the projector augmented-wave (PAW) method.128,129 For LDA and PBE, the comparability of the eigenvalues resulting from the different codes was carefully tested. For details on the GKLI methodology the reader is referred to the original publication.(77)
In the periodic calculations, a Methfessel–Paxton occupation scheme with the temperature set to 100 K (0.008 62 eV) was used, and Monkhorst-Pack(130)k-point grids of 8 × 5 × 1, 4 × 5 × 1, 2 × 5 × 1, 2 × 2 × 1 and 1 × 1 × 1 were chosen for the SAM packing densities of 1/2, 1/4, 1/8, 1/16, and 1/64, respectively. The corresponding unit cells were obtained by doubling the short lattice vector (see Figure 1b) once (θ = 1/4) and twice (θ = 1/8); θ = 1/16 was obtained by doubling the shorter lattice vector of the cell at θ = 1/8, and θ = 1/64 by again doubling both lattice vectors. The latter was used as approximation for the isolated molecule. Even at such low packing density, ΔEvac does not vanish exactly. We have found values ΔEvac < 0.2 eV, and account for this in Figures 4, 6b, and 8b by using the reference energy E̅ (and Evacleft in Figure 8b) also in the case of the isolated molecule. The molecular geometries were not reoptimized in the periodic calculations, and the molecules were oriented parallel to the z-axis of the unit cell (see Figure 1); this procedure facilitates an easy comparison of molecular orbitals and orbital-derived bands. To exclude spurious interaction between subsequent slabs in the z-direction, a vacuum gap of >20 Å was introduced together with a dipole layer within the vacuum to compensate for the net dipole moment of the slab.
Orbital charge-densities ρi(z) were defined as
in units of −e/Å, with a and b being the lattice constants of the surface unit-cell and e the (positive) elementary charge. The sum is taken over all reciprocal-space vectors k⃗ included in the calculation. Note that the unit cell size (i.e., a and b) and the included vectors k⃗ differ for “isolated” and periodic systems as detailed above. ρi(z) comprises the charge of a non-spin-polarized band; i.e., the area below a curve amounts to 2 electrons.
Five layers of the nonrelaxed Au(111) surface in the above-used p(√3 × 3) unit cell were used for the calculations on the full metal/SAM systems. No geometry relaxation of the upright-standing molecules was performed, and the position of the bonded sulfur atom was determined from previous work.(44)
Representations of the systems were generated with XCrysDen.(131) 3D orbital/band charge density plots were generated at the isovalue of 0.02 −e/Å3. HSE06 calculations for the orbital representations in Figure 7a and dipole moments in Figure 9 were done with Gaussian 09(132) using the 6-311++G(d,p) basis set. An isovalue of 0.04 a.u. was used.
Acknowledgments
The authors thank A.M. Kelterer, O.T. Hofmann, C. Slugovc, Z.Y. Ma, and L.J. Wang for fruitful discussions, and Z.Y. Ma from the Institute of Chemistry of the Chinese Academy of Sciences also for support with the Gaussian HSE06 calculations. We thank the ZID of the TU Graz for providing computational resources. The research was largely funded by the Austrian Science Fund (FWF): P20972–N20. D.A.E. is recipient of a DOC-fellowship of the Austrian Academy of Sciences. T.K. acknowledges financial support by the Alexander-von-Humboldt-Foundation. S.K. acknowledges financial support by GRK 1640 and the German-Israeli-Foundation. L.K. acknowledges financial support by the Israel Science Foundation, the German-Israeli-Foundation, and the Lise Meitner Minerva Center for Computational Chemistry.
Author Present Address
¶ Department of Physical Electronics, Tel-Aviv University, 69978 Tel-Aviv, Israel
Supporting Information Available
Detailed discussion of the role of self-interaction, assessment of the accuracy of DFT for molecular polarizabilities, dipole moments and energy gaps, additional information on the role of the docking group; full name, geometry and total energy of optimized molecular structures, complete references (11), (123), and (132). This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
∥ These authors contributed equally to this work.
Supplementary Material
References
- Ulman A. Chem. Rev. 1996, 96, 1533–1554. [DOI] [PubMed] [Google Scholar]
- Schreiber F. J. Phys.: Condens. Matter 2004, 16, R881–R900. [Google Scholar]
- Kind M.; Wöll C. Prog. Surf. Sci. 2009, 84, 230–278. [Google Scholar]
- Love J. C.; Estroff L. A.; Kriebel J. K.; Nuzzo R. G.; Whitesides G. M. Chem. Rev. 2005, 105, 1103–1170. [DOI] [PubMed] [Google Scholar]
- Gooding J. J.; Mearns F.; Yang W.; Liu J. Electroanalysis 2003, 15, 81–96. [Google Scholar]
- Naaman R.; Vager Z. MRS Bull. 2010, 35, 429–434. [Google Scholar]
- Fontaine P.; Goguenheim D.; Deresmes D.; Vuillaume D.; Garet M.; Rondelez F. Appl. Phys. Lett. 1993, 62, 2256–2258. [Google Scholar]
- Guo X.; Myers M.; Xiao S.; Lefenfeld M.; Steiner R.; Tulevski G. S.; Tang J.; Baumert J.; Leibfarth F.; Yardley J. T.; Steigerwald M. L.; Kim P.; Nuckolls C. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 11452–11456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bock C.; Pham D. V.; Kunze U.; Käfer D.; Witte G.; Wöll C. J. Appl. Phys. 2006, 100, 114517. [Google Scholar]
- Pacher P.; Lex A.; Proschek V.; Etschmaier H.; Tchernychova E.; Sezen M.; Scherf U.; Grogger W.; Trimmel G.; Slugovc C.; Zojer E. Adv. Mater. 2008, 20, 3143–3148. [Google Scholar]
- Smits E. C. P.; et al. Nature 2008, 455, 956–959. [Google Scholar]
- Chen J.; Reed M. A.; Rawlett. A. M.; Tour J. M. Science 1999, 286, 1550–1552. [DOI] [PubMed] [Google Scholar]
- Kushmerick J.; Holt D.; Yang J.; Naciri J.; Moore M.; Shashidhar R. Phys. Rev. Lett. 2002, 89, 086802. [DOI] [PubMed] [Google Scholar]
- Akkerman H. B.; Blom P. W. M.; de Leeuw D. M.; de Boer B. Nature 2006, 441, 69–72. [DOI] [PubMed] [Google Scholar]
- Pace G.; Ferri V.; Grave C.; Elbing M.; von Hanisch C.; Zharnikov M.; Mayor M.; Rampi M. A.; Samori P. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 9937–9942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bumm L. A.; Arnold J. J.; Cygan M. T.; Dunbar T. D.; Burgin T. P.; Jones L.; Allara D. L.; Tour J. M.; Weiss P. S. Science 1996, 271, 1705–1707. [Google Scholar]
- Reed M. A.; Zhou C.; Muller C. J.; Burgin T. P.; Tour J. M. Science 1997, 278, 252–254. [Google Scholar]
- Donhauser Z. J.; Mantooth B. A.; Kelly K. F.; Bumm L. A.; Monnell J. D.; Stapleton J. J.; Price D. W. Jr.; Rawlett A. M.; Allara D. L.; Tour J. M.; Weiss P. S. Science 2001, 292, 2303–2307. [DOI] [PubMed] [Google Scholar]
- Park J.; Pasupathy A. N.; Goldsmith J. I.; Chang C.; Yaish Y.; Petta J. R.; Rinkoski M.; Sethna J. P.; Abruña H. D.; McEuen P. L.; Ralph D. C. Nature 2002, 417, 722–725. [DOI] [PubMed] [Google Scholar]
- Nitzan A.; Ratner M. A. Science 2003, 300, 1384–1389. [DOI] [PubMed] [Google Scholar]
- Xu B.; Tao N. J. Science 2003, 301, 1221–1223. [DOI] [PubMed] [Google Scholar]
- Lörtscher E.; Ciszek J. W.; Tour J.; Riel H. Small 2006, 2, 973–977. [DOI] [PubMed] [Google Scholar]
- Venkataraman L.; Klare J. E.; Nuckolls C.; Hybertsen M. S.; Steigerwald M. L. Nature 2006, 442, 904–907. [DOI] [PubMed] [Google Scholar]
- Selzer Y.; Salomon A.; Ghabboun J.; Cahen D. Angew. Chem., Int. Ed. 2002, 41, 827–830. [DOI] [PubMed] [Google Scholar]
- Ishii H.; Sugiyama K.; Ito E.; Seki K. Adv. Mater. 1999, 11, 605–625. [Google Scholar]
- Koch N. J. Phys.: Condens. Matter 2008, 20, 184008. [Google Scholar]
- Koch N. ChemPhysChem 2007, 8, 1438–1455. [DOI] [PubMed] [Google Scholar]
- Hwang J.; Wan A.; Kahn A. Mater. Sci. Eng., R 2009, 64, 1–31. [Google Scholar]
- Kamenetska M.; Quek S. Y.; Whalley A. C.; Steigerwald M. L.; Choi H. J.; Louie S. G.; Nuckolls C.; Hybertsen M. S.; Neaton J. B.; Venkataraman L. J. Am. Chem. Soc. 2010, 132, 6817–6821. [DOI] [PubMed] [Google Scholar]
- Sushko M. L.; Shluger A. L. Adv. Funct. Mater. 2008, 18, 2228–2236. [Google Scholar]
- Natan A.; Zidon Y.; Shapira Y.; Kronik L. Phys. Rev. B 2006, 73, 193310. [Google Scholar]
- Deutsch D.; Natan A.; Shapira Y.; Kronik L. J. Am. Chem. Soc. 2007, 129, 2989–2997. [DOI] [PubMed] [Google Scholar]
- Heimel G.; Romaner L.; Brédas J. L.; Zojer E. Phys. Rev. Lett. 2006, 96, 196806. [DOI] [PubMed] [Google Scholar]
- Monti O. L. A.; Steele M. P. Phys. Chem. Chem. Phys. 2010, 12, 12390–12400. [DOI] [PubMed] [Google Scholar]; Steele M. P.; Kelly L. L.; Ilyas N.; Monti O. L. A. J. Chem. Phys. 2011, 135, 124702. [DOI] [PubMed] [Google Scholar]
- Cahen D.; Naaman R.; Vager Z. Adv. Funct. Mater. 2005, 15, 1571–1578. [Google Scholar]
- Piacenza M.; D’Agostino S.; Fabiano E.; Della Sala F. Phys. Rev. B 2009, 80, 153101. [DOI] [PubMed] [Google Scholar]
- De Renzi V. Surf. Sci. 2009, 603, 1518–1525. [Google Scholar]
- Heimel G.; Rissner F.; Zojer E. Adv. Mater. 2010, 22, 2494–2513. [DOI] [PubMed] [Google Scholar]
- Kimura S. Org. Biomol. Chem. 2008, 6, 1143–1148. [DOI] [PubMed] [Google Scholar]
- Vilan A.; Cahen D. Trends Biotechnol. 2002, 20, 22–29. [DOI] [PubMed] [Google Scholar]
- Wang L.; Rangger G. M.; Romaner L.; Heimel G.; Bučko T.; Ma Z.; Li Q.; Shuai Z.; Zojer E. Adv. Funct. Mater. 2009, 19, 3766–3775. [Google Scholar]
- Natan A.; Kronik L.; Haick H.; Tung R. T. Adv. Mater. 2007, 19, 4103–4117. [Google Scholar]
- Rissner F.; Egger D. A.; Romaner L.; Heimel G.; Zojer E. ACS Nano 2010, 4, 6735–6746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egger D. A.; Rissner F.; Rangger G. M.; Hofmann O. T.; Wittwer L.; Heimel G.; Zojer E. Phys. Chem. Chem. Phys. 2010, 12, 4291–4294. [DOI] [PubMed] [Google Scholar]
- Nakayama H.; Morita T.; Kimura S. Phys. Chem. Chem. Phys. 2009, 11, 3967–3976. [DOI] [PubMed] [Google Scholar]
- Nakayama H.; Morita T.; Kimura S. J. Phys. Chem. C 2010, 114, 4669–4674. [Google Scholar]
- Staykov A.; Nozaki D.; Yoshizawa K. J. Phys. Chem. C 2007, 111, 11699–11705. [Google Scholar]
- Li Y.; Zhao J.; Yin X.; Yin G. J. Phys. Chem. A 2006, 110, 11130–11135. [DOI] [PubMed] [Google Scholar]
- Yin X.; Li Y.; Zhang Y.; Li P.; Zhao J. Chem. Phys. Lett. 2006, 422, 111–116. [Google Scholar]
- Choi Y. C.; Kim W. Y.; Park K.-S.; Tarakeshwar P.; Kim K. S.; Kim T.-S.; Lee J. Y. J. Chem. Phys. 2005, 122, 094706. [DOI] [PubMed] [Google Scholar]
- Kim W. Y.; Kim K. S. Acc. Chem. Res. 2010, 43, 111–120. [DOI] [PubMed] [Google Scholar]
- Rai D.; Joshi H.; Kulkarni A. D.; Gejji S. P.; Pathak R. K. J. Phys. Chem. A 2007, 111, 9111–9121. [DOI] [PubMed] [Google Scholar]
- Tóbik J.; Dal Corso A.; Scandolo S.; Tosatti E. Surf. Sci. 2004, 566–568, 644–649. [Google Scholar]
- Arnold A.; Weigend F.; Evers F. J. Chem. Phys. 2007, 126, 174101. [DOI] [PubMed] [Google Scholar]
- Chaki N. K.; Mandal S.; Reber A. C.; Qian M.; Saavedra H. M.; Weiss P. S.; Khanna S. N.; Sen A. ACS Nano 2010, 4, 5813–5818. [DOI] [PubMed] [Google Scholar]
- Romaner L.; Heimel G.; Ambrosch-Draxl C.; Zojer E. Adv. Funct. Mater. 2008, 18, 3999–4006. [Google Scholar]
- Romaner L.; Heimel G.; Zojer E. Phys. Rev. B 2008, 77, 045113. [Google Scholar]
- Tóbik J.; Dal Corso A. J. Chem. Phys. 2004, 120, 9934–9941. [DOI] [PubMed] [Google Scholar]
- Natan A.; Kuritz N.; Kronik L. Adv. Funct. Mater. 2010, 20, 2077–2084. [Google Scholar]
- Wang J. G.; Prodan E.; Car R.; Selloni A. Phys. Rev. B 2008, 77. [Google Scholar]
- Heimel G.; Romaner L.; Zojer E.; Brédas J. L. Nano Lett. 2007, 7, 932–940. [DOI] [PubMed] [Google Scholar]
- Ma Z.; Rissner F.; Wang L.; Heimel G.; Li Q.; Shuai Z.; Zojer E. Phys. Chem. Chem. Phys. 2011, 13, 9747–9760. [DOI] [PubMed] [Google Scholar]
- Azzam W.; Fuxen C.; Birkner A.; Rong H.-T.; Buck M.; Wöll C. Langmuir 2003, 19, 4958–4968. [Google Scholar]
- Baker K.; Fratini A.; Resch T.; Knachel H.; Adams W.; Socci E.; Farmer B. Polymer 1993, 34, 1571–1587. [Google Scholar]
- Yoshimoto S. Bull. Chem. Soc. Jpn. 2006, 79, 1167–1190. [Google Scholar]
- Taniguchi I.; Yoshimoto S.; Sunatsuki Y.; Nishiyama K. Electrochemistry 1999, 67, 1197–1199. [Google Scholar]
- Sek S. Langmuir 2009, 25, 13488–13492. [DOI] [PubMed] [Google Scholar]
- Perdew J. P.; Zunger A. Phys. Rev. B 1981, 23, 5048–5079. [Google Scholar]
- Kümmel S.; Kronik L. Rev. Mod. Phys. 2008, 80, 3–60. [Google Scholar]
- Potts A. W.; Holland D. M. P.; Trofimov A. B.; Schirmer J.; Karlsson L.; Siegbahn K. J. Phys. B: At. Mol. Opt. Phys. 2003, 36, 3129–3143. [Google Scholar]
- Lottermoser U.; Rademacher P.; Mazik M.; Kowski K. Eur. J. Org. Chem. 2005, 2005, 522–531. [Google Scholar]
- van den Ham D. M. W.; van der Meer D.; Feil D. J. Electron Spectrosc. Relat. Phenom. 1974, 3, 479–487. [Google Scholar]
- Dori N.; Menon M.; Kilian L.; Sokolowski M.; Kronik L.; Umbach E. Phys. Rev. B 2006, 73, 195208. [Google Scholar]
- Körzdörfer T.; Kümmel S.; Marom N.; Kronik L. Phys. Rev. B 2009, 79, 201205. [Google Scholar]; Körzdörfer T.; Kümmel S.; Marom N.; Kronik L. Phys. Rev. B 2010, 82, 129903. [Google Scholar]
- When comparing gas-phase calculations with experimental spectra, a constant, orbital-independent SIE is only of minor importance as it can be accounted for by a rigid shift of the spectrum. For the alignment of the electronic states in a SAM with the metallic Fermi-level of the substrate this is not sufficient, as will be discussed in a later section.
- Körzdörfer T.; Kümmel S.; Mundt M. J. Chem. Phys. 2008, 129, 014110. [DOI] [PubMed] [Google Scholar]
- Körzdörfer T. J. Chem. Phys. 2011, 134, 094111. [DOI] [PubMed] [Google Scholar]
- Heyd J.; Scuseria G. E.; Ernzerhof M. J. Chem. Phys. 2003, 118, 8207–8215. [Google Scholar]
- Heyd J.; Scuseria G. E. J. Chem. Phys. 2004, 121, 1187–1192. [DOI] [PubMed] [Google Scholar]
- Heyd J.; Scuseria G. E.; Ernzerhof M. J. Chem. Phys. 2006, 124, 219906. [Google Scholar]
- Marom N.; Hod O.; Scuseria G. E.; Kronik L. J. Chem. Phys. 2008, 128, 164107. [DOI] [PubMed] [Google Scholar]
- Neaton J.; Hybertsen M.; Louie S. Phys. Rev. Lett. 2006, 97, 216405. [DOI] [PubMed] [Google Scholar]
- Garcia-Lastra J. M.; Rostgaard C.; Rubio A.; Thygesen K. S. Phys. Rev. B 2009, 80, 245427. [Google Scholar]
- Biller A.; Tamblyn I.; Neaton J. B.; Kronik L.. J. Chem. Phys., in press. [DOI] [PubMed]
- Champagne B.; Perpète E. A.; van Gisbergen S. J. A.; Baerends E.-J.; Snijders J. G.; Soubra-Ghaoui C.; Robins K. A.; Kirtman B. J. Chem. Phys. 1998, 109, 10489. [Google Scholar]
- Kronik L.; Vasiliev I.; Jain M.; Chelikowsky J. R. J. Chem. Phys. 2001, 115, 4322. [Google Scholar]
- Kümmel S.; Akola J.; Manninen M. Phys. Rev. Lett. 2000, 84, 3827–3830. [DOI] [PubMed] [Google Scholar]
- Kümmel S.; Kronik L.; Perdew J. Phys. Rev. Lett. 2004, 93, 213002. [DOI] [PubMed] [Google Scholar]
- Adam N. K.; Danielli J. F.; Harding J. B. Proc. R. Soc. London, Ser. A 1934, 147, 491–499. [Google Scholar]
- Heimel G.; Romaner L.; Zojer E.; Brédas J. L. Acc. Chem. Res. 2008, 41, 721–729. [DOI] [PubMed] [Google Scholar]
- Gershevitz O.; Sukenik C. N.; Ghabboun J.; Cahen D. J. Am. Chem. Soc. 2003, 125, 4730–4731. [DOI] [PubMed] [Google Scholar]
- Fukagawa H.; Yamane H.; Kera S.; Okudaira K.; Ueno N. Phys. Rev. B 2006, 73, 041302(R). [Google Scholar]
- Cornil D.; Olivier Y.; Geskin V.; Cornil J. Adv. Funct. Mater. 2007, 17, 1143–1148. [Google Scholar]
- We average E over the length scale of interatomic distances, a = 1.5 Å, using the equation Ê(z) = 1/(2a) ∫z-az+aE(z′) dz′ to reduce the oscillations due to the ionic potential wells.
- Baldereschi A.; Baroni S.; Resta R. Phys. Rev. Lett. 1988, 61, 734–737. [DOI] [PubMed] [Google Scholar]
- Zhao X.; Wei C. M.; Yang L.; Chou M. Y. Phys. Rev. Lett. 2004, 92, 236805. [DOI] [PubMed] [Google Scholar]
- Popovic Z.; Satpathy S. Phys. Rev. Lett. 2005, 94, 176805. [DOI] [PubMed] [Google Scholar]
- Wu Z.; Neaton J. B.; Grossman J. C. Phys. Rev. Lett. 2008, 100, 246804. [DOI] [PubMed] [Google Scholar]
- Zharnikov M. J. Electron Spectrosc. Relat. Phenom. 2010, 178–179, 380–393. [Google Scholar]
- Doron-Mor I.; Hatzor A.; Vaskevich A.; van der Boom-Moav T.; Shanzer A.; Rubinstein I.; Cohen H. Nature 2000, 406, 382–385. [DOI] [PubMed] [Google Scholar]
- Leroux M.; Grandjean N.; Laügt M.; Massies J.; Gil B.; Lefebvre P.; Bigenwald P. Phys. Rev. B 1998, 58, R13371–R13374. [Google Scholar]
- Miller D.; Chemla D.; Schmitt-Rink S. Phys. Rev. B 1986, 33, 6976–6982. [DOI] [PubMed] [Google Scholar]
- Heilbronner E.; Bock H.. Das HMO-Modell und seine Anwendung Bd. 1. Grundlagen und Handhabung; Verlag Chemie: Weinheim, 1968. [Google Scholar]
- Hoffman R.Solids and Surfaces: A Chemist‘s View of Bonding in Extended Structures; Wiley-VCH: New York, 1988. [Google Scholar]
- Pellegrin E.; Fink J.; Drechsler S. Phys. Rev. Lett. 1991, 66, 2022–2025. [DOI] [PubMed] [Google Scholar]
- Zojer E.; Knupfer M.; Resel R.; Meghdadi F.; Leising G.; Fink J. Phys. Rev. B 1997, 56, 10138–10144. [Google Scholar]
- Zojer E.; Shuai Z.; Leising G.; Brédas J. L. J. Chem. Phys. 1999, 111, 1668–1675. [Google Scholar]
- Seki K.; Karlsson U. O.; Engelhardt R.; Koch E.-E.; Schmidt W. Chem. Phys. 1984, 91, 459–470. [Google Scholar]
- Narioka S.; Ishii H.; Edamatsu K.; Kamiya K.; Hasegawa S.; Ohta T.; Ueno N.; Seki K. Phys. Rev. B 1995, 52, 2362–2373. [DOI] [PubMed] [Google Scholar]
- Koller G.; Berkebile S.; Oehzelt M.; Puschnig P.; Ambrosch-Draxl C.; Netzer F. P.; Ramsey M. G. Science 2007, 317, 351–355. [DOI] [PubMed] [Google Scholar]
- Häming M.; Ziroff J.; Salomon E.; Seitz O.; Cahen D.; Kahn A.; Schöll A.; Reinert F.; Umbach E. Phys. Rev. B 2009, 79, 155418. [Google Scholar]
- Miller D.Quantum Mechanics for Scientists and Engineers; Cambridge University Press: New York, 2009. [Google Scholar]
- Nguyen D.; Odagaki T. Am. J. Phys. 1987, 55, 466–469. [Google Scholar]
- Puri A.; Sun J.; Odagaki T. Solid State Commun. 1989, 70, 907–910. [Google Scholar]
- Miller D.; Chemla D.; Damen T.; Gossard A.; Wiegmann W.; Wood T.; Burrus C. Phys. Rev. Lett. 1984, 53, 2173–2176. [Google Scholar]
- Orbital plots (instead of charge density plots) are chosen to illustrate the correspondence between the sign of a wave function and the associated envelope function.
- Used energy windows for the local DOS insets: 0.2 eV around the peak maxima for the occupied states, 0.3 eV above the onsets of the lowest unoccupied states, and 0.8 eV for the broad intragap states.
- This is useful as it has been shown that this quantity together with the metal work-function and the potential energy shift due to the bonding-induced charge redistributions determines the level-alignment. The bonding-induced energy shift, however, is only weakly affected by the nature of the molecular backbone as long as one does not encounter Fermi-level pinning.
- The situation is complicated by the fact that the actual screening will depend on the distance between the orbital and the metal substrate; i.e., differently localized orbitals can be expected to be differently affected by the screening.
- In the GGA calculations of ref (44), pinning of the HOMO-derived band at EF was found. These states are shifted to well below EF due to the significantly increased band gap when using HSE.
- Blumenfeld M. L.; Steele M. P.; Monti O. L. A. J. Phys. Chem. Lett. 2010, 1, 145. [Google Scholar]
- Jacquemin D.; Adamo C. J. Chem. Theory Comput. 2011, 7, 369–376. [DOI] [PubMed] [Google Scholar]
- Frisch M. J.;et al. Gaussian 03, Revision C.02; Gaussian Inc.: Wallingford CT, 2004.
- Chelikowsky J.; Troullier N.; Saad Y. Phys. Rev. Lett. 1994, 72, 1240–1243. [DOI] [PubMed] [Google Scholar]
- Kronik L.; Makmal A.; Tiago M. L.; Alemany M. M. G.; Jain M.; Huang X.; Saad Y.; Chelikowsky J. R. Phys. Status Solidi B 2006, 243, 1063–1079. [Google Scholar]
- Troullier N.; Martins J. L. Phys. Rev. B 1991, 43, 1993–2006. [DOI] [PubMed] [Google Scholar]
- Kresse G.; Furthmüller J. Phys. Rev. B 1996, 54, 11169–11186. [DOI] [PubMed] [Google Scholar]
- Blöchl P. Phys. Rev. B 1994, 50, 17953–17979. [DOI] [PubMed] [Google Scholar]
- Kresse G.; Joubert D. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar]
- Monkhorst H.; Pack J. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar]
- Kokalj A. Comput. Mater. Sci. 2003, 28, 155–168. [Google Scholar]
- Frisch M. J.;et al. Gaussian 09, Revision A.02; Gaussian Inc.: Wallingford CT, 2009.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.