

Abstract
Nuclear estrogen receptor α (ERα) regulates target gene expression in response to ligands through two distinct mechanisms: direct binding to DNA and indirect tethering through other DNA-bound transcription factors, such as AP-1. In the studies described herein, we examined the molecular mechanisms underlying the activation of ERα in the AP-1 tethering pathway by the selective estrogen receptor modulator (SERM) raloxifene (Ral). Our results with the MMP1 and PRUNE genes indicate that the c-Fos component of the AP-1 tethering factor and the c-Jun N-terminal kinase 1 (JNK1) are constitutively bound at the promoter regions prior to Ral exposure. Ral then promotes the binding of ERα at the promoter in a c-Fos-dependent manner. Interestingly, we found that JNK1 enzymatic activity is required for Ral-dependent gene activation through ERα. Our results suggest that one role for Ral-dependent recruitment of ERα to the AP-1 binding site is to stimulate JNK1 enzymatic activity. Alternatively, Ral-occupied ERα might recruit protein substrates to promoter-bound JNK1 without any change in JNK1 activity. Collectively, our studies have revealed a new role for JNK1 in determining gene regulatory outcomes by ERα.
Keywords: Raloxifene, Estradiol, Activating Protein-1 (AP-1), c-Fos, c-Jun N-Terminal Kinase (JNK1), Estrogen Receptor, Transcriptional Activation, Promoter, Chromatin Immunopreciptation
1. Introduction
Estrogens control physiological and pathophysiological processes in a wide variety of tissues, exerting their effects through two distinct estrogen receptor proteins, ERα and ERβ (Heldring et al., 2007; Warner et al., 1999). ERs acts as ligand-regulated, DNA-binding transcription factors that regulate distinct subsets of genes across the genome (Cheung and Kraus, 2010; Heldring et al., 2007; Kininis and Kraus, 2008; Warner et al., 1999). ERs can bind directly to genomic DNA as dimers through specific sequences called estrogen response elements (EREs). ERs can also be recruited to genomic DNA by indirect tethering through other DNA-bound transcription factors, including members of the activating protein-1 (AP-1) family of transcription factors (e.g., heterodimers of c-Jun and c-Fos) (Heldring et al., 2007; Heldring et al., 2011; Kushner et al., 2000). In either case, chromatin associated ERs nucleate the recruitment of a variety of coregulator proteins (e.g., coactivators and corepressors) to activate or repress gene transcription (Acevedo and Kraus, 2004; Glass and Rosenfeld, 2000). By now, hundreds of potential coregulators with diverse functions – from histone modification and chromatin remodeling to RNA polymerase II recruitment and mRNA splicing – have been identified (Lonard and O’Malley, 2006). The specific requirements for and functions of these coregulators, and their modulation by different ER ligands, have not been fully defined.
A wide variety of both natural and synthetic ER ligands have been identified and characterized. Selective estrogen receptor modulators (SERMs), such as tamoxifen (Tam) and raloxifene (Ral), are a class of ER ligands that exhibit context-specific agonist and anatagonist activities (Kuiper et al., 1999; McDonnell et al., 2001). For example, Tam and Ral act predominantly as ER antagonists in the mammary glands, but exhibit pronounced agonistic effects in other tissues, including bone (Tam and Ral) and uterus (Tam only) (Kuiper et al., 1999; McDonnell et al., 2001; Shang and Brown, 2002). The ER/AP-1 tethering pathway is activated in response to both estrogens and SERMs in a cell-specific and ER isoform-specific manner (Kushner et al., 2000; Webb et al., 1995). In some cellular contexts, 17β-estradiol (E2), the predominant naturally occurring estrogen, activates ERα, but not ERβ, in the tethering pathway, while Tam and Ral may activate both ER isoforms in the tethering pathway (Paech et al., 1997). The different ligand responses observed with different cellular contexts, different modes of ER recruitment to genomic DNA (i.e., ERE versus tethering), and different ER isoforms (i.e., ERα versus ERβ) are likely due to differential use of coregulator proteins (Cheung et al., 2005; Kushner et al., 2000; Webb et al., 2003).
c-Jun N-terminal kinase 1 (JNK1) is a mitogen-activated protein kinase that interacts with and phosphorylates c-Jun in response to cellular signaling pathways (Barr and Bogoyevitch, 2001; Dunn et al., 2002). Although the traditional view has been that MAPK-mediated phosphorylation events (e.g., phosphorylation of transcription factors) do not occur at the downstream target genes that they ultimately regulate, the terminal kinases of various signaling pathways are found in the nucleus under activating conditions (Edmunds and Mahadevan, 2004; Turjanski et al., 2007). In addition, gene-specific and genomic analyses in cells from yeast (Pascual-Ahuir et al., 2006; Pokholok et al., 2006), Drosophila (Suganuma et al., 2010), and mammals (Bruna et al., 2003; Bungard et al., 2010; Dawson et al., 2009; Edmunds and Mahadevan, 2004; Hu et al., 2009; Madak-Erdogan et al., 2011; Vicent et al., 2006) have shown that some signaling kinases bind to the promoters of genes whose expression they regulate. In the studies described herein, we used cell-based reporter assays, gene-specific mRNA analyses, and chromatin immunoprecipitation (ChIP) assays to explore SERM-dependent activation of ERα in the AP-1 tethering pathway and characterize the role of JNK1 in this pathway. Our results indicate that JNK1 enzymatic activity plays a key role in supporting Ral agonistic actions in the tethering pathway for some genes.
2. Materials and Methods
2.1. Antibodies
The JNK1/3 antibody was from Santa Cruz Biotechnology (sc-474). The custom rabbit polyclonal antisera against c-Fos and ERα were produced by Pocono Rabbit Farms and Laboratory.
2.2. Plasmid DNA constructs
The following luciferase reporter plasmids were used in these studies: (1) MMP1-Luc, which was constructed by cloning the −73 to +63 fragment of the human collagenase/matrix metalloproteinase-1 (MMP-1) gene (Angel et al., 1987) upstream of the luciferase reporter gene in the pXP1 reporter plasmid (Nordeen, 1988) (provided by Dr. Steve Nordeen, University of Colorado Health Sciences Center), (2) pGL3-2ERE-pS2-Luc (referred to herein as 2ERE-TFF1-Luc), which contains two estrogen response elements (EREs) and a fragment of the pS2/trefoil factor 1 promoter upstream of a luciferase reporter gene (Kim et al., 2006), (3) PRUNE-Luc, which contains the −461 bp to +55 bp fragment of the PRUNE promoter upstream of a luciferase reporter gene (kindly provided by Dr. Miltos Kininis from the Kraus laboratory), and (4) UGT2B15-Luc, which contains the −449 bp to +114 bp fragment of the UGT2B15 promoter upstream of the luciferase reporter (Kininis et al., 2007). The MMP1, PRUNE, and UGT2B15 promoter fragments lack identifiable EREs, but contain an AP-1 binding site, as illustrated in Fig. 1A.
Fig. 1. Schematic diagrams of the promoters used for the luciferase, gene expression, and ChIP assays and verification of the experimental system.

(A) The structures of the MMP1, PRUNE, and UGT2B15 promoters from −700 bp to +200 bp relative to the transcription start site (TSS) are shown. The locations of the known AP-1 binding sites (boxes labeled “AP-1”), promoter DNA fragments used in the luciferase reporters (horizontal brackets), and locations of the primers used for ChIP-qPCR are indicated. The bent arrow denotes the TSS for each gene. (B) E2 and TPA activate their cognate reporter genes in HeLa cells. An E2-responsive luciferase reporter vector (2ERE-TFF1-Luc) or a TPA-responsive reporter vector (MMP1-Luc) was transfected into HeLa cells with or without an expression vector for ERα. The cells were then treated with E2 (100 nM) or TPA (ng/mL TPA), as indicated, before determination of luciferase activity. Each bar represents the mean ± SEM for n ≥ 4 separate experiments. Bars marked with an asterisk are significantly different from the corresponding controls (Student’s t-test, p < 0.05).
The following expression vectors were used: (1) pRSV, an “empty” expression vector containing the Rous sarcoma virus promoter, (2) RSV-hERα, which expresses human estrogen receptor alpha (provided by Dr. Benita Katzenellenbogen, University of Illinois, Urbana-Champaign) (Reese and Katzenellenbogen, 1991), (3) pCMV, an “empty” expression vector containing the cytomegalovirus promoter, (4) pCMV-A-Fos, which expresses a dominant-negative version of c-Fos (provided by Dr. Charles Vinson, NCI, and Nina Heldring from the Kraus laboratory) (Heldring et al., 2011; Olive et al., 1997), and (5) pCMVβ, a constitutive β-galactosidase expression vector used for transfection normalization (Clontech).
2.3. Cell growth and maintenance
HeLa cells were purchased from ATCC. HeLa-ERα cells were kindly provided by Dr. David Shapiro (University of Illinois, Urbana-Champaign) (Zhang et al., 1999). Both cell types were maintained in DME/F12 (Sigma, D2906) supplemented with 10% charcoal-dextran stripped calf serum (CDCS), 100 units/ml penicillin, and 100 μg/ml streptomycin. The HeLa-ERα cell medium also contained 100 μg/ml G418.
2.4. Transient transfection reporter gene assays
Luciferase assays were performed as described, with modifications (Heldring et al., 2011; Kim et al., 2006). HeLa cells were plated in 6-well dishes in DME/F12 containing 10% CDCS 24 hrs prior to transfection, grown to about 80% confluence, and transfected using Gene Juice transfection reagent (EMD Biosciences). For luciferase reporter experiments, each well received a total of 1 μg of plasmid DNA including combinations of the following as noted in the main text and figures legends: (1) 400 ng of one of the luciferase reporter plasmids described above, (2) 50 ng of pRSV-ERα or an empty pRSV control vector, and (3) 100 ng of pCMV-A-Fos or an empty pCMV control vector, and carrier plasmid DNA (pET15b or pET28c) to balance the total amount of plasmid DNA in the transfection and to achieve a proper ratio of DNA to Gene Juice according to the manufacturer’s specifications. Twenty-four hrs after transfection, the cells were treated with ethanol (vehicle control), Raloxifene (Ral; 100 nM), or 17β-estradiol (E2; 100 nM) for 18 hr. For experiments with the JNK inhibitor SP600125 (SP; Enzo Life Sciences, EI-305) (Bennett et al., 2001), the cells were treated with 20 μM of the drug for 1 hr before treatment with Ral and E2. The cells were washed with once with PBS, collected in 1x lysis buffer (Promega Corp.), and centrifuged to eliminate cell debris. The supernatant was then used to measure luciferase (Promega Corp. Luciferase Assay Kit) in a 96-well plate assay using a Beckman Coulter LD400 luminometer. Luciferase activity was normalized to total protein content using a Bradford assay (BioRad) or to β-galactosidase activity using a standard β-gal assay. Each experiment was performed a minimum of three times to ensure reproducibility.
2.5. Gene expression analyses by reverse transcription-qPCR
Expression of the endogenous genes, MMP1 and PRUNE, was determined by RT-qPCR. HeLa-ERα cells were plated in 6-well dishes in DME/F12 containing 2% CDCS for 48 hrs; the medium was then changed to DME/F12 containing 10% CDCS. The cells were treated with ethanol, Ral (100 nM), or E2 (100 nM) for 3 hrs. For experiments with SP, the cells were treated with 20 μM of the drug for 30 min before treatment with Ral and E2. Total RNA was isolated with Trizol reagent (Invitrogen) according to the manufacturers specifications and reverse transcribed using 4 μg of total RNA, 4 μg oligo(dT), and 600 units of MMLV reverse transcriptase (Promega). The resulting cDNA from each sample was treated with 3 units of RNAse H (Ambion) for 30 min at 37°C and then diluted 1:5 with water. Samples were further diluted 1:10 and analyzed by qPCR with gene-specific primers using a 96-well DNA Engine Opticon (MJ Research) or a 384-well LightCycler 480 (Roche) real-time PCR thermocycler for 45 cycles (95°C for 15 sec, 60°C for 1 min) following an initial 10 min incubation at 95°C. The sequences of the primers used for qPCR were: (1) MMP1 forward 5′-GCCATATATGGACGTT-3′, (2) MMP1 reverse 5′-CACTTCTCCCCGAATCGT-3′, (3) PRUNE forward 5′-ACATACACATTTGTTTACCGAACGA-3′, and (4) PRUNE reverse 5′-TCCGCAATGTCCCTAGCAA-3′. The fold change in expression for each condition was calculated using a standard curve of diluted cDNA from untreated samples (1:1, 1:10, 1:100) normalized to β-actin transcript as an internal standard. Independent experiments were scaled in relation to the no inhibitor/ethanol condition. Each experiment was performed a minimum of three times to ensure reproducibility.
2.6. Chromatin immunoprecipitation (ChIP)
ChIP assays were performed as described previously (Kininis et al., 2007) with minor modifications. HeLa-ERα cells were plated in 10 cm diameter dishes in DME/F12 containing 2% CDCS for 48 hrs; the medium was then changed to DME/F12 containing 10% CDCS. The cells were grown to ~80% confluence, then treated with ethanol, Ral (100 nM), or E2 (100 nM) for 3 hrs. The cells were then crosslinked with 10 mM dimethyl suberimidate•HCl (DMS; Pierce, 20700) for 10 min at room temperature, followed by 1% formaldehyde for 10 min at 37°C, with subsequent quenching by 125 mM glycine for 5 min. The crosslinked cells were collected by centrifugation and processed as described previously (Kininis et al., 2007). The ChIP DNA was analyzed by qPCR with gene-specific primers using a 96-well DNA Engine Opticon (MJ Research) or a 384-well LightCycler 480 (Roche) real-time PCR thermocycler for 40 cycles (95°C for 15 sec, 60°C for 1 min) following an initial 10 min incubation at 95°C. The primers used for ChIP-qPCR were: (1) MMP1 forward 5′-TCTCCTTCGCACACATCTTG-3′, (2) MMP1 reverse 5′-TAGAGTCCTTGCCCTTCCAG-3′, (3) PRUNE forward 5′-ACATACACATTTGTTTACCGAACGA-3′, and (4) PRUNE reverse 5′-TCCGCAATGTCCCTAGCAA-3′. Standard curves were produced based on percent input from each treatment (ethanol, Ral or E2) and IP percent inputs were plotted relative to this curve. At least three independent ChIP experiments were conducted to ensure reproducibility.
3. Results
3.1. Characterization of the experimental system
To investigate the molecular mechanisms of Ral-dependent ERα gene activation through the AP-1 tethering mechanism, we performed a series of luciferase-based reporter assays. In these assays, we used reporter transgenes containing promoter fragments from native E2-regulated genes (e.g., MMP1, PRUNE, and UGT2B15), which contain an AP-1 response element, but lack a discernible ERE (Fig. 1A). The assays were performed in HeLa human cervical epithelial cancer cells, which were cotransfected with the reporter genes and an ERα expression vector or an empty control vector. In our initial assays, we demonstrated E2-, ERα-, and AP-1-dependent reporter gene activity in these cells (Fig. 1B). As expected, a reporter construct containing two EREs upstream of the TFF1 promoter was activated in the presence of ERα and 100 nM E2 (Fig. 1B). A reporter construct containing the MMP1 promoter, which has a well-characterized AP-1 binding site (Angel et al., 1987), was activated by 12-O-tetradecanoylphorbol-13-acetate (TPA; a stimulator of AP-1-dependent signaling pathways; (Karin, 1995)) in the presence or absence of ERα, although addition of ERα enhanced the response by two-fold (Fig. 1B). These results indicate that ERα and AP-1 pathways are active under these experimental conditions in HeLa cells.
3.2. Ral activates AP-1 site-containing promoters in an ERα-dependent manner in HeLa cells
Previous studies have indicated that SERMs can act as ERα agonists on AP-1 targeted promoters in a cell type-specific manner (Kushner et al., 2000; Paech et al., 1997; Webb et al., 1995). To determine if the Ral can act as an agonist for ERα in the AP-1 tethering pathway in HeLa cells, we cotransfected the MMP1-luc, PRUNE-luc, or UGT2B15-luc reporter vectors with an ERα expression vector or an empty control vector. The transfected cells were then treated with 100 nM Ral or 100 nM E2. The three reporter constructs showed a 1.7 to 3-fold increase in response to Ral, but not E2, and this activation was dependent on the presence of ERα (Fig. 2). These results demonstrate Ral- and ERα-dependent activation of the tethering pathway in HeLa cells.
Fig. 2. Ral activates AP-1 site-containing promoters in an ERα-dependent manner in HeLa cells.

HeLa cells were transfected with MMP1, PRUNE, or UGT2B15 luciferase reporter vectors with or without an expression vector for ERα, as indicated. The cells were then treated with Ral (100 nM) or E2 (100 nM), as indicated, before determination of luciferase activity. Each bar represents the mean ± SEM for n = 3 separate experiments. Bars marked with an asterisk are significantly different from the corresponding controls (Student’s t-test, p < 0.05).
3.3. Inhibition of AP-1 blocks Ral-dependent activation of the ERα-dependent tethering pathway in HeLa cells
As noted above, AP-1 is a heterodimeric transcription factor composed of c-Jun and c-Fos (Hess et al., 2004; Karin et al., 1997). We used a dominant negative mutant of c-Fos, called A-Fos (Olive et al., 1997), to investigate whether c-Fos or its dimerization partners are involved in mediating the effects of Ral-activated ERα on AP-1 dependent promoters. The A-Fos dominant negative mutant prevents wild-type c-Fos and its dimerization partners from binding to DNA (Heldring et al., 2011; Olive et al., 1997). We cotransfected HeLa cells with the MMP1-luc, PRUNE-luc, or UGT2B15-luc reporter vectors, an ERα expression vector, and an A-Fos expression vector or an empty control vector. The transfected cells were then treated with Ral or E2. As shown above, each reporter construct was stimulated by Ral, but the response was abrogated in the presence of A-Fos (Fig. 3). E2 was a much less effective stimulator of reporter gene activity; nonetheless, the modest response observed in this assay was abrogated in the presence of A-Fos (Fig. 3). These experiments suggest that Ral-activated ERα requires c-Fos (or related Fos proteins) and its heterodimerization partners (e.g., c-Jun) to mediate its stimulatory effects on AP-1 dependent promoters.
Fig. 3. Inhibition of AP-1 by a dominant-negative c-Fos blocks Ral-dependent activation of the ERα-dependent tethering pathway in HeLa cells.

HeLa cells were transfected with MMP1, PRUNE, or UGT2B15 luciferase reporter vectors and an expression vector for ERα, with or without an expression vector for A-Fos. The cells were then treated with Ral (100 nM) or E2 (100 nM), as indicated, before determination of luciferase activity. Each bar represents the mean ± SEM for n ≥ 3 separate experiments. Bars marked with an asterisk are significantly different from the corresponding controls (Student’s t-test, p < 0.05).
3.4. JNK kinase activity is required for Ral-dependent activation of the MMP1 and PRUNE promoters in reporter gene assays and with native genes
JNK1 is a mitogen-activated protein kinase that interacts with and phosphorylates c-Jun in response to cellular signaling pathways (Barr and Bogoyevitch, 2001; Dunn et al., 2002). Given the potential role of c-Jun, acting with c-Fos as a component of AP-1, in Ral-mediated, ERα-dependent activation in the tethering pathway (Fig. 3 and (Paech et al., 1997)), we considered a possible role for JNK1. As a starting point to address this issue, we cotransfected HeLa cells with an ERα expression vector and one of the three reporter vectors (i.e., MMP1-luc, PRUNE-luc, or UGT2B15-luc), then treated the cells with the JNK inhibitor, SP600125 (SP; 20 μM) (Bennett et al., 2001) for 1 hour before treatment with Ral and E2. SP inhibited the Ral-mediated responses with the MMP1 and PRUNE promoter constructs, but had little to no effect with the UGT2B15 promoter construct (Fig. 4). Again, E2 was not an effective stimulator of reporter gene activity, but SP reduced the reporter activity observed with the MMP1 and PRUNE promoter constructs in the presence of E2 by about 50 percent (Fig. 4). This result suggests that JNK kinase activity is required for Ral-dependent stimulation of the MMP1 and PRUNE promoters, but not the UGT2B15 promoter, which may require other proteins instead. Although possible that SP might cause some inhibition of other cellular kinases, we used the drug under conditions that should allow maximal efficacy and excellent specificity (Bennett et al., 2001).
Fig. 4. JNK kinase activity is required for Ral-dependent activation of the MMP1 and PRUNE promoters in reporter gene assays.

HeLa cells were transfected with MMP1 or PRUNE luciferase reporter vectors and an expression vector for ERα. The cells were then treated with the JNK inhibitor SP (20 μM) for 1 hour prior to treatment with Ral (100 nM) or E2 (100 nM), as indicated, before determination of luciferase activity. Each bar represents the mean ± SEM for n ≥ 3 separate experiments. Bars marked with an asterisk are significantly different from the corresponding controls (Student’s t-test, p < 0.05).
To explore the role of JNK1 kinase activity in more detail, we examined the effects of SP on the Ral-dependent stimulation of the expression of the native MMP1 and PRUNE genes. For these experiments, we used HeLa cells stably expressing ERα (HeLa-ERα) and assessed MMP1 and PRUNE mRNA expression following Ral or E2 treatment with or without JNK inhibition. Similar to the results from the transient transfection assays shown in Fig. 4, the expression of both MMP1 and PRUNE was induced by Ral, and the Ral-dependent activation was reduced to basal levels when the cells were pretreated with SP (Fig. 5). These results confirm that JNK kinase activity is required for Ral-dependent stimulation of the MMP1 and PRUNE promoters. The expression of the native MMP1 gene was largely unaffected by E2 in the absence or presence of SP (Fig. 5, left panel). Surprisingly, however, E2 induced the expression of PRUNE to the same extent as Ral (Fig. 5, right panel), in contrast to our results from the reporter gene assays (Fig. 6). These results suggest additional aspects of the regulation of the native PRUNE gene that are not recapitulated with the 517 bp promoter fragment used in the reporter gene assays (e.g., missing promoter element or native chromatin structure).
Fig. 5. JNK kinase activity is required for Ral-dependent activation of the native MMP1 and PRUNE genes.

HeLa-ERα cells were treated with SP for 1 hour prior to a 3 hour treatment with Ral (100 nM) or E2 (100 nM), as indicated. The cells were collected and subjected to RT-qPCR expression analyses, and the results were standardized to β-actin. Each bar represents the mean ± SEM for n ≥ 3 separate experiments. White bars marked with a single asterisk are significantly different from the corresponding “Veh” controls (Student’s t-test, p < 0.05). Black bars marked with two asterisks are significantly different from the corresponding “−SP” controls for that particular treatment (Student’s t-test, p < 0.05).
Fig. 6. Ral promotes the activation of constitutively bound JNK1 at the native MMP1 and PRUNE gene promoters.

HeLa-ERα cells were treated with Ral (100 nM) or E2 (100 nM) for 3 hours, as indicated. The cells were crosslinked with formaldehyde, collected, and subjected to ChIP-qPCR analyses for (A) ERα, (B) c-Fos, and (C) JNK1. Each bar represents the mean ± SEM for n ≥ 3 separate experiments. Filled bars (i.e., the specific antibody condition in each panel; “+Ab”) marked with an asterisk are significantly different from the corresponding “+Ab/Veh” controls (Student’s t-test, p < 0.05). All “+Ab” conditions, except the E2 conditions in panel C, are significantly different from the corresponding “No Ab” conditions, indicating a positively enriched ChIP signal.
3.5. Ral promotes the activation of constitutively bound JNK1 at the native MMP1 and PRUNE gene promoters
A number of recent studies have indicated that terminal kinases in cellular signaling pathways may localize to the nucleus, be recruited to gene promoters, and function as coregulators (Bruna et al., 2003; Bungard et al., 2010; Dawson et al., 2009; Edmunds and Mahadevan, 2004; Hu et al., 2009; Madak-Erdogan et al., 2011; Vicent et al., 2006). To determine if ERα and AP-1 might function together to recruit JNK1 to target gene promoters in response to Ral or E2, we performed ChIP-qPCR assays for ERα, c-Fos, and JNK1 in HeLa-ERα cells. ERα showed a moderate level of occupancy at the MMP1 and PRUNE promoters in the absence of ligand treatment (~5- to 6-fold over background; Fig. 6A). The occupancy of ERα at the MMP1 promoter was stimulated about 3-fold by Ral (Fig. 6A, left panel), whereas the recruitment of ERα to the PRUNE promoter was stimulated about 2-fold by both Ral and E2 (Fig. 6A, right panel). These results correspond with the cognate gene expression results presented in Fig. 5, which show that PRUNE expression, but not MMP1 expression, was stimulated by E2.
c-Fos and JNK1 constitutively occupied both the MMP1 and PRUNE promoters in the basal (untreated) condition (~3- to 5-fold over background; Fig. 6, B and C). The occupancy of c-Fos and JNK1 was not affected by Ral on either the MMP1 or PRUNE promoters (Fig. 6, B and C), outcomes that corresponded with enhanced Ral-dependent gene expression (Fig. 5). In contrast, E2 reduced the levels of c-Fos and JNK1 on the MMP1 promoter (Fig. 6, B and C), an outcome that corresponded with an absence of E2-dependent gene expression (Fig. 5). E2 had no statistically significant effect on the levels of c-Fos on the PRUNE promoter (Fig. 6B), but reduced the levels of JNK1 (Fig. 6C); this outcome corresponded with enhanced E2-dependent gene expression (Fig. 5). The results from the JNK1 ChIP experiments, in conjunction with the experiments using SP shown in Fig. 5, suggest that Ral-dependent expression of MMP1 and PRUNE requires the recruitment of ERα through an AP-1-mediated tethering mechanism, as well as the enzymatic activity of JNK1. The effects of E2 are less consistent with respect to the expression of MMP1 and PRUNE, but suggest that the basal occupancy of JNK1 at the promoters is blocked by E2.
4. Discussion
In this paper, we used cell-based assays with reporter genes and native genes to explore the molecular mechanisms underlying Ral-dependent activation of ERα in the AP-1 tethering pathway. Our results with the MMP1 and PRUNE genes indicate the following: (1) JNK1 and the c-Fos component of the AP-1 tethering factor are constitutively bound at the promoter region prior to Ral exposure (Fig. 6, A and B), (2) Ral promotes the binding of ERα at the promoter (Fig. 6A), likely in a c-Fos-dependent manner (Fig. 3 and (Heldring et al., 2011)), and (3) JNK1 enzymatic activity is required for Ral-dependent gene activation through ERα (Figs 4 and 5). These results are summarized schematically in Fig. 7. Our results also indicate that not all promoters activated by Ral through an ERα tethering pathway require JNK1 enzymatic activity (e.g., UGT2B15) (Fig. 4). The effects of E2 in our assays, which we examined for comparison, were less consistent with respect to the effects on target gene expression, ERα recruitment, and c-Fos occupancy. Nonetheless, our results did show a clear inhibition of JNK1 occupancy at the promoters by E2, suggesting that E2-liganded ERα can impact the activity or function of JNK1 in this pathway.
Fig. 7. Model for Ral-, ERα-, and JNK1-dependent gene regulation at an AP-1 binding site.
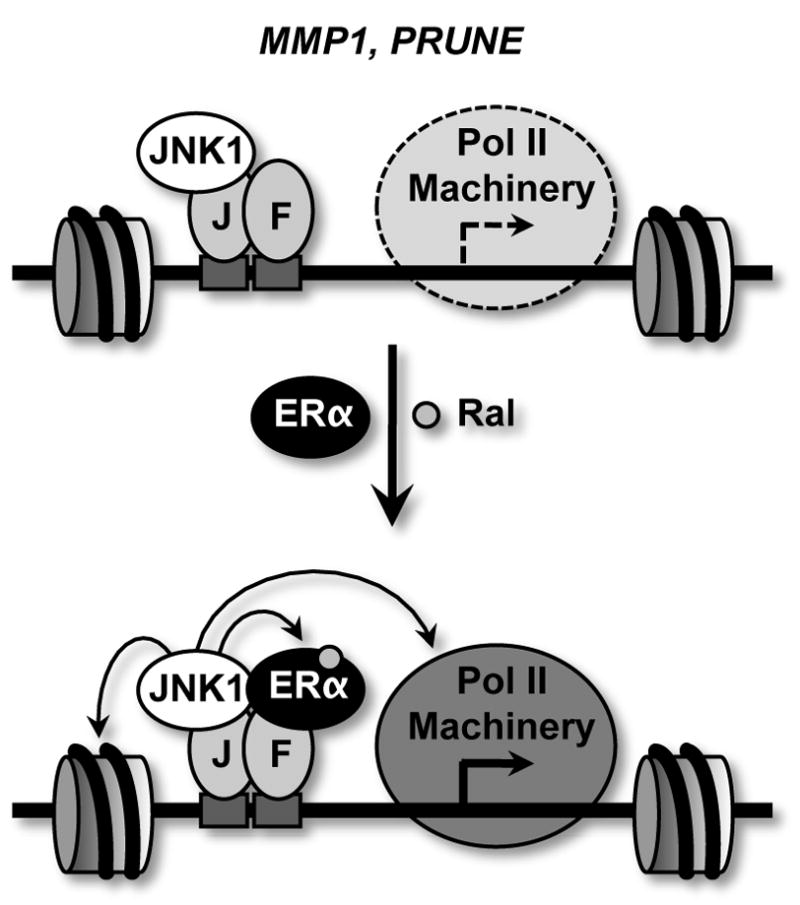
See the text for a detailed explanation. Prior to Ral treatment, the MMP1 and PRUNE genes show modest levels of expression, as indicated by a reduced presence of the RNA polymerase II machinery (“Pol II Machinery”) relative to the condition with Ral. In this case, JNK1 and AP-1 are constitutively bound. Upon treatment with Ral, the tethering of ERα through DNA-bound AP-1 and JNK1 enzymatic activity are both stimulated. JNK1 may phosphorylate histones, ERα, coregulators (not shown) or components of the Pol II machinery. J = c-Jun; F = c-Fos.
The importance of the ERα tethering pathway for cellular responses to estrogens and SERMs has been accumulating support in the literature over the past two decades (Kushner et al., 2000; Paech et al., 1997; Webb et al., 1995; Webb et al., 1999). A key aspect of this pathway is that classical ERα antagonists may function as agonists, giving rise to altered pharmacological responses that can affect the overall biological responses. In addition, the ERα tethering pathway likely plays a key role in determining cell type-specific responses to SERMs (Kushner et al., 2000; Paech et al., 1997; Webb et al., 1995). Recent studies have focused on two fundamental aspects of this pathway: (1) the nature of the tethering factor and (2) the role of coregulators in determining gene regulatory outcomes. With respect to the former, previous studies have shown that factors other than AP-1 may also function as ERα tethering factors, including Runx1 and CREB (Heldring et al., 2011; Lalmansingh and Uht, 2008; Stender et al., 2010). With respect to the latter, previous studies have shown that altered recruitment of coregulators in response to different ligands in the tethering pathway can play a key role in determining ligand-specific responses (Cheung et al., 2005; Webb et al., 1999; Webb et al., 2003).
Our results presented herein suggest that JNK1 can act as a coregulator and play a key role in determining ligand-specific responses by ERα in the AP-1 tethering pathway. In this regard, we observed that JNK1 is bound at the promoters of ERα/AP-1-regulated genes and that JNK1 catalytic activity is required for the Ral-dependent activation of these genes. JNK1 is a kinase with numerous potential targets in the nucleus. Recent studies examining the activity of other cellular kinases in the nucleus, including JAK and AMPK, have shown that histones are targets for phosphorylation, which plays a role in determining gene expression outcomes (Bungard et al., 2010; Dawson et al., 2009; Vicent et al., 2006). Other targets, however, are also possible, including ERα, coregulators, and components of the transcription machinery (Fig. 7). The nuclear targets that are actually phosphorylated by JNK1 in response to Ral signaling in the tethering pathway will be determined in future studies.
Previous models of ligand-dependent gene regulation by ERα have suggested a key role for the receptor in nucleating the formation of complexes on DNA that contain the receptor and its associated regulatory proteins (Acevedo and Kraus, 2004; Kushner et al., 2000). Interestingly, our results with the MMP1 and PRUNE promoters indicate that Ral-activated ERα has little effect on the recruitment of c-Fos and JNK1. Rather, we observed that these proteins were pre-bound at the AP-1 binding site in the promoter region prior to treatment with Ral (Fig. 6). This result is in contrast to our previous results with different target genes in a different cell line showing that ERα ligands can drive the assembly of protein complexes at ERα tethering sites (Heldring et al., 2011). Thus, different types of ERα-dependent regulatory mechanisms are likely to exist in the tethering pathway. Given the requirement for JNK1 enzymatic activity, as indicated by the experiments with SP (Figs. 5 and 6), and the constitutive binding of JNK1 (Fig. 6), our results suggest that one role for Ral-dependent recruitment of ERα to the AP-1 binding site might be to stimulate JNK1 enzymatic activity. Alternatively, these results may suggest a scenario in which Ral-occupied ERα recruits protein substrates to promoter-bound JNK1 without any change in JNK1 activity.
Collectively, our studies have revealed a new role for JNK1 in determining gene regulatory outcomes by ERα. The generality of these responses across the set of all ERα target genes will be examined in future genomic studies.
Highlights.
JNK1 and the c-Fos (of AP-1) are constitutively bound at Raloxifene (Ral)-regulated promoters prior to Ral exposure.
Ral promotes the binding of ERα at the promoter in a c-Fos-dependent manner.
JNK1 enzymatic activity is required for Ral-dependent gene activation through ERα.
One role for Ral-dependent recruitment of ERα to the AP-1 binding site may be to stimulate JNK1 enzymatic activity
Our studies have revealed a new role for JNK1 in determining gene regulatory outcomes by ERα.
Acknowledgments
The authors would like to thank the following: (1) Dr. Gary Isaacs and Dr. Nina Heldring for technical advice and helpful discussions about the directions of this work, (2) Shrikanth Gadad, Miao Sun, and Jane Bowman for critical reading of this manuscript, (3) Dr. Steve Nordeen for the MMP1 reporter plasmid, (4) Dr. Miltos Kininis for the PRUNE-Luc reporter plasmid, (5) Dr. David Shapiro for the HeLa-ERα cells, (6) Dr. Charles Vinson for the dominant negative A-Fos construct, and (7) Dr. Benita Katzenellenbogen for the RSV-hERα expression plasmid. This work was supported by a grant from the NIH/NIDDK (DK058110) to W.L.K. and the Cecil H. and Ida Green Reproductive Biology Sciences endowments.
Footnotes
Conflict of Interest
The authors have no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Acevedo ML, Kraus WL. Transcriptional activation by nuclear receptors. Essays Biochem. 2004;40:73–88. doi: 10.1042/bse0400073. [DOI] [PubMed] [Google Scholar]
- Angel P, Imagawa M, Chiu R, Stein B, Imbra RJ, Rahmsdorf HJ, Jonat C, Herrlich P, Karin M. Phorbol ester-inducible genes contain a common cis element recognized by a TPA-modulated trans-acting factor. Cell. 1987;49:729–739. doi: 10.1016/0092-8674(87)90611-8. [DOI] [PubMed] [Google Scholar]
- Barr RK, Bogoyevitch MA. The c-Jun N-terminal protein kinase family of mitogen-activated protein kinases (JNK MAPKs) Int J Biochem Cell Biol. 2001;33:1047–1063. doi: 10.1016/s1357-2725(01)00093-0. [DOI] [PubMed] [Google Scholar]
- Bennett BL, Sasaki DT, Murray BW, O’Leary EC, Sakata ST, Xu W, Leisten JC, Motiwala A, Pierce S, Satoh Y, Bhagwat SS, Manning AM, Anderson DW. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc Natl Acad Sci U S A. 2001;98:13681–13686. doi: 10.1073/pnas.251194298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruna A, Nicolas M, Munoz A, Kyriakis JM, Caelles C. Glucocorticoid receptor-JNK interaction mediates inhibition of the JNK pathway by glucocorticoids. Embo J. 2003;22:6035–6044. doi: 10.1093/emboj/cdg590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bungard D, Fuerth BJ, Zeng PY, Faubert B, Maas NL, Viollet B, Carling D, Thompson CB, Jones RG, Berger SL. Signaling kinase AMPK activates stress-promoted transcription via histone H2B phosphorylation. Science. 2010;329:1201–1205. doi: 10.1126/science.1191241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung E, Acevedo ML, Cole PA, Kraus WL. Altered pharmacology and distinct coactivator usage for estrogen receptor-dependent transcription through activating protein-1. Proc Natl Acad Sci U S A. 2005;102:559–564. doi: 10.1073/pnas.0407113102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung E, Kraus WL. Genomic analyses of hormone signaling and gene regulation. Annu Rev Physiol. 2010;72:191–218. doi: 10.1146/annurev-physiol-021909-135840. [DOI] [PubMed] [Google Scholar]
- Dawson MA, Bannister AJ, Gottgens B, Foster SD, Bartke T, Green AR, Kouzarides T. JAK2 phosphorylates histone H3Y41 and excludes HP1alpha from chromatin. Nature. 2009;461:819–822. doi: 10.1038/nature08448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn C, Wiltshire C, MacLaren A, Gillespie DA. Molecular mechanism and biological functions of c-Jun N-terminal kinase signalling via the c-Jun transcription factor. Cell Signal. 2002;14:585–593. doi: 10.1016/s0898-6568(01)00275-3. [DOI] [PubMed] [Google Scholar]
- Edmunds JW, Mahadevan LC. MAP kinases as structural adaptors and enzymatic activators in transcription complexes. J Cell Sci. 2004;117:3715–3723. doi: 10.1242/jcs.01346. [DOI] [PubMed] [Google Scholar]
- Glass CK, Rosenfeld MG. The coregulator exchange in transcriptional functions of nuclear receptors. Genes Dev. 2000;14:121–141. [PubMed] [Google Scholar]
- Heldring N, Pike A, Andersson S, Matthews J, Cheng G, Hartman J, Tujague M, Strom A, Treuter E, Warner M, Gustafsson JA. Estrogen receptors: how do they signal and what are their targets. Physiol Rev. 2007;87:905–931. doi: 10.1152/physrev.00026.2006. [DOI] [PubMed] [Google Scholar]
- Heldring N, Isaacs GD, Diehl AG, Sun M, Cheung E, Ranish JA, Kraus WL. Multiple sequence-specific DNA-binding proteins mediate estrogen receptor signaling through a tethering pathway. Mol Endocrinol. 2011;25:564–574. doi: 10.1210/me.2010-0425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess J, Angel P, Schorpp-Kistner M. AP-1 subunits: quarrel and harmony among siblings. J Cell Sci. 2004;117:5965–5973. doi: 10.1242/jcs.01589. [DOI] [PubMed] [Google Scholar]
- Hu S, Xie Z, Onishi A, Yu X, Jiang L, Lin J, Rho HS, Woodard C, Wang H, Jeong JS, Long S, He X, Wade H, Blackshaw S, Qian J, Zhu H. Profiling the human protein-DNA interactome reveals ERK2 as a transcriptional repressor of interferon signaling. Cell. 2009;139:610–622. doi: 10.1016/j.cell.2009.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karin M. The regulation of AP-1 activity by mitogen-activated protein kinases. J Biol Chem. 1995;270:16483–16486. doi: 10.1074/jbc.270.28.16483. [DOI] [PubMed] [Google Scholar]
- Karin M, Liu Z, Zandi E. AP-1 function and regulation. Curr Opin Cell Biol. 1997;9:240–246. doi: 10.1016/s0955-0674(97)80068-3. [DOI] [PubMed] [Google Scholar]
- Kim MY, Woo EM, Chong YT, Homenko DR, Kraus WL. Acetylation of estrogen receptor alpha by p300 at lysines 266 and 268 enhances the deoxyribonucleic acid binding and transactivation activities of the receptor. Mol Endocrinol. 2006;20:1479–1493. doi: 10.1210/me.2005-0531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kininis M, Chen BS, Diehl AG, Isaacs GD, Zhang T, Siepel AC, Clark AG, Kraus WL. Genomic analyses of transcription factor binding, histone acetylation, and gene expression reveal mechanistically distinct classes of estrogen-regulated promoters. Mol Cell Biol. 2007;27:5090–5104. doi: 10.1128/MCB.00083-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kininis M, Kraus WL. A global view of transcriptional regulation by nuclear receptors: gene expression, factor localization, and DNA sequence analysis. Nucl Recept Signal. 2008;6:e005. doi: 10.1621/nrs.06005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuiper GG, van den Bemd GJ, van Leeuwen JP. Estrogen receptor and the SERM concept. J Endocrinol Invest. 1999;22:594–603. doi: 10.1007/BF03343616. [DOI] [PubMed] [Google Scholar]
- Kushner PJ, Agard DA, Greene GL, Scanlan TS, Shiau AK, Uht RM, Webb P. Estrogen receptor pathways to AP-1. J Steroid Biochem Mol Biol. 2000;74:311–317. doi: 10.1016/s0960-0760(00)00108-4. [DOI] [PubMed] [Google Scholar]
- Lalmansingh AS, Uht RM. Estradiol regulates corticotropin-releasing hormone gene (crh) expression in a rapid and phasic manner that parallels estrogen receptor-alpha and -beta recruitment to a 3′,5′-cyclic adenosine 5′-monophosphate regulatory region of the proximal crh promoter. Endocrinology. 2008;149:346–357. doi: 10.1210/en.2007-0372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonard DM, O’Malley BW. The expanding cosmos of nuclear receptor coactivators. Cell. 2006;125:411–414. doi: 10.1016/j.cell.2006.04.021. [DOI] [PubMed] [Google Scholar]
- Madak-Erdogan Z, Lupien M, Stossi F, Brown M, Katzenellenbogen BS. Genomic collaboration of estrogen receptor alpha and extracellular signal-regulated kinase 2 in regulating gene and proliferation programs. Mol Cell Biol. 2011;31:226–236. doi: 10.1128/MCB.00821-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonnell DP, Chang CY, Norris JD. Capitalizing on the complexities of estrogen receptor pharmacology in the quest for the perfect SERM. Ann N Y Acad Sci. 2001;949:16–35. doi: 10.1111/j.1749-6632.2001.tb03999.x. [DOI] [PubMed] [Google Scholar]
- Nordeen SK. Luciferase reporter gene vectors for analysis of promoters and enhancers. Biotechniques. 1988;6:454–458. [PubMed] [Google Scholar]
- Olive M, Krylov D, Echlin DR, Gardner K, Taparowsky E, Vinson C. A dominant negative to activation protein-1 (AP1) that abolishes DNA binding and inhibits oncogenesis. J Biol Chem. 1997;272:18586–18594. doi: 10.1074/jbc.272.30.18586. [DOI] [PubMed] [Google Scholar]
- Paech K, Webb P, Kuiper GG, Nilsson S, Gustafsson J, Kushner PJ, Scanlan TS. Differential ligand activation of estrogen receptors ERalpha and ERbeta at AP1 sites. Science. 1997;277:1508–1510. doi: 10.1126/science.277.5331.1508. [DOI] [PubMed] [Google Scholar]
- Pascual-Ahuir A, Struhl K, Proft M. Genome-wide location analysis of the stress-activated MAP kinase Hog1 in yeast. Methods. 2006;40:272–278. doi: 10.1016/j.ymeth.2006.06.007. [DOI] [PubMed] [Google Scholar]
- Pokholok DK, Zeitlinger J, Hannett NM, Reynolds DB, Young RA. Activated signal transduction kinases frequently occupy target genes. Science. 2006;313:533–536. doi: 10.1126/science.1127677. [DOI] [PubMed] [Google Scholar]
- Reese JC, Katzenellenbogen BS. Differential DNA-binding abilities of estrogen receptor occupied with two classes of antiestrogens: studies using human estrogen receptor overexpressed in mammalian cells. Nucleic Acids Res. 1991;19:6595–6602. doi: 10.1093/nar/19.23.6595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang Y, Brown M. Molecular determinants for the tissue specificity of SERMs. Science. 2002;295:2465–2468. doi: 10.1126/science.1068537. [DOI] [PubMed] [Google Scholar]
- Stender JD, Kim K, Charn TH, Komm B, Chang KC, Kraus WL, Benner C, Glass CK, Katzenellenbogen BS. Genome-wide analysis of estrogen receptor alpha DNA binding and tethering mechanisms identifies Runx1 as a novel tethering factor in receptor-mediated transcriptional activation. Mol Cell Biol. 2010;30:3943–3955. doi: 10.1128/MCB.00118-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suganuma T, Mushegian A, Swanson SK, Abmayr SM, Florens L, Washburn MP, Workman JL. The ATAC acetyltransferase complex coordinates MAP kinases to regulate JNK target genes. Cell. 2010;142:726–736. doi: 10.1016/j.cell.2010.07.045. [DOI] [PubMed] [Google Scholar]
- Turjanski AG, Vaque JP, Gutkind JS. MAP kinases and the control of nuclear events. Oncogene. 2007;26:3240–3253. doi: 10.1038/sj.onc.1210415. [DOI] [PubMed] [Google Scholar]
- Vicent GP, Ballare C, Nacht AS, Clausell J, Subtil-Rodriguez A, Quiles I, Jordan A, Beato M. Induction of progesterone target genes requires activation of Erk and Msk kinases and phosphorylation of histone H3. Mol Cell. 2006;24:367–381. doi: 10.1016/j.molcel.2006.10.011. [DOI] [PubMed] [Google Scholar]
- Warner M, Nilsson S, Gustafsson JA. The estrogen receptor family. Curr Opin Obstet Gynecol. 1999;11:249–254. doi: 10.1097/00001703-199906000-00003. [DOI] [PubMed] [Google Scholar]
- Webb P, Lopez GN, Uht RM, Kushner PJ. Tamoxifen activation of the estrogen receptor/AP-1 pathway: potential origin for the cell-specific estrogen-like effects of antiestrogens. Mol Endocrinol. 1995;9:443–456. doi: 10.1210/mend.9.4.7659088. [DOI] [PubMed] [Google Scholar]
- Webb P, Nguyen P, Valentine C, Lopez GN, Kwok GR, McInerney E, Katzenellenbogen BS, Enmark E, Gustafsson JA, Nilsson S, Kushner PJ. The estrogen receptor enhances AP-1 activity by two distinct mechanisms with different requirements for receptor transactivation functions. Mol Endocrinol. 1999;13:1672–1685. doi: 10.1210/mend.13.10.0357. [DOI] [PubMed] [Google Scholar]
- Webb P, Nguyen P, Kushner PJ. Differential SERM effects on corepressor binding dictate ERalpha activity in vivo. J Biol Chem. 2003;278:6912–6920. doi: 10.1074/jbc.M208501200. [DOI] [PubMed] [Google Scholar]
- Zhang CC, Krieg S, Shapiro DJ. HMG-1 stimulates estrogen response element binding by estrogen receptor from stably transfected HeLa cells. Mol Endocrinol. 1999;13:632–643. doi: 10.1210/mend.13.4.0264. [DOI] [PubMed] [Google Scholar]