

Abstract
During the appearance of the signaling lipid PI(3,4,5)P3, an important subset of pleckstrin homology (PH) domains target signaling proteins to the plasma membrane. To ensure proper pathway regulation, such PI(3,4,5)P3-specific PH domains must exclude the more prevalant, constitutive plasma membrane lipid PI(4,5)P2 and bind the rare PI(3,4,5)P3 target lipid with sufficiently high affinity. Our previous study of the E17K mutant of protein kinase B (AKT1) PH domain, together with evidence from Carpten et al (1), revealed that the native AKT1 E17 residue serves as a sentry glutamate that excludes PI(4,5)P2, thereby playing an essential role in specific PI(3,4,5)P3 targeting (2). The sentry glutamate hypothesis proposes that an analogous sentry glutamate residue is a widespread feature of PI(3,4,5)P3-specific PH domains, and that charge reversal mutation at the sentry glutamate position will yield both increased PI(4,5)P2 affinity and constitutive plasma membrane targeting.
To test this hypothesis the present study investigates the E345 residue, a putative sentry glutamate, of General Receptor for Phosphoinositides 1 (GRP1) PH domain. The results show that incorporation of the E345K charge reversal mutation into GRP1 PH domain enhances PI(4,5)P2 affinity 8-fold and yields constitutive plasma membrane targeting in cells, reminiscent of the effects of the E17K mutation in AKT1 PH domain. Hydrolysis of plasma membrane PI(4,5)P2 releases E345K GRP1 PH domain into the cytoplasm and the efficiency of this release increases when target Arf6 binding is disrupted. Overall, the findings provide strong support for the sentry glutamate hypothesis and suggest that the GRP1 E345K mutation will be linked to changes in cell physiology and human pathologies, as demonstrated for AKT1 E17K (1, 3). Analysis of available PH domain structures suggests that a lone glutamate residue (or, in some cases an aspartate) is a common, perhaps ubiquitous, feature of PI(3,4,5)P3-specific binding pockets that functions to lower PI(4,5)P2 affinity.
Keywords: peripheral membrane protein; protein kinase B; pleckstrin homology domain; phosphatidylinositol-3,4,5-trisphosphate signaling; phosphoinositide
Signaling events at the cytoplasmic surface of the plasma membrane play a central role in a wide array of signaling cascades, and the signaling lipid PI(3,4,5)P3 serves as an important second messenger at that membrane surface. Upon activation by upstream signals, the regulatory enzyme phosphoinositide-3-kinase (PI3K) phosphorylates the constitutive plasma membrane lipid PI(4,5)P2 to generate PI(3,4,5)P3 (4–6). The resulting appearance of a PI(3,4,5)P3 signal in the plasma membrane recruits an array of signaling proteins to the membrane surface. Typically these proteins possess a pleckstrin homology (PH) domain that specifically targets PI(3,4,5)P3. Upon recruitment to the plasma membrane, such proteins are activated and regulate various essential cell processes including growth, chemotaxis, DNA synthesis, cytoskeletal rearrangements, vesicle trafficking, and apoptosis (4, 6–16).
Over 561 human proteins contain PH domains and a significant fraction of these PH domains are believed to bind PIP lipids with high affinity (17). PH domains that are recruited to the plasma membrane specifically during a PI(3,4,5)P3 signaling event face a significant target recognition challenge: they must selectively bind PI(3,4,5)P3, which is a rare membrane component, while excluding the constitutive plasma membrane PI(4,5)P2 which is at least 100-fold more prevalent even during a peak PI(3,4,5)P3 signal (4, 13).
PH domains share a common core structure consisting of two antiparallel β-sheets forming a β-sandwich (7, 18). At one edge of the β-sandwich up to three connecting loops provide basic side chains and other contacts that form the binding pocket for the negatively charged target lipid headgroup. The length and sequence of these loops provide each PH domain with its appropriate headgroup specificity (19) and, in principle, loop mutations could alter membrane recognition yielding dramatic cellular consequences. For example, Carpten et al. (1) found that the E17K mutation in the PH domain of the regulatory kinase AKT1 causes constitutive plasma membrane targeting, hyperactivation of AKT1 kinase, transformation of tissue culture cells. This mutation is linked to multiple human pathologies including cancers and Proteus syndrome (1, 3).
Our previous study of the E17K AKT1 PH domain revealed the mechanistic basis of its constitutive plasma membrane targeting, thereby uncovering a new molecular mechanism of carcinogenesis and leading to the sentry glutamate hypothesis of PI(3,4,5)P3 specificity (2). For proper silencing of AKT1 kinase (also termed protein kinase B or PKB), it is crucial that the PH domain remain docked to its inhibitory site on the kinase domain until a PI(3,4,5)P3 signal recruits the PH domain and relieves autoinhibition, thereby activating the kinase (20–23). Consequently, it is crucial that the PH domain exclude constitutive plasma membrane PI(4,5)P2 to prevent kinase activation in the absence of a PI(3,4,5)P3 signal. We found that the E17K mutation greatly enhances PH domain affinity for PI(4,5)P2, thereby explaining its known biological effects (including constitutive plasma membrane targeting, kinase hyperactivation, cell transformation, carcinogenicity and tissue overgrowth (2)). In addition, we proposed that other PI(3,4,5)P3-specific PH domains may also possess sentry glutamate residues.
The sentry glutamate hypothesis predicts that a glutamate-to-lysine charge reversal mutation at the sentry glutamate position of an arbitrary PI(3,4,5)P3-specific PH domain will, like the AKT1 E17K mutation, both enhance PI(4,5)P2 affinity and drive constitutive plasma membrane targeting (2). The present study tests this hypothesis by introducing the E345K charge reversal mutation at the putative sentry glutamate residue of General Receptor for Phosphoinositides 1 (GRP1). GRP1 is an Arf6 guanidine-nucleotide exchange factor (GEF) that catalyzes the activation of Arf6-GDP to Arf6-GTP at the plasma membrane surface. The GRP1 PH domain possesses a PI(3,4,5)P3-specific binding pocket as well as an Arf6 binding site that includes residues I307 and K340 (Figure 1) (24, 25). Previous findings have concluded that Arf6 and PI(3,4,5)P3 binding both contribute to plasma membrane affinity (25). The putative sentry glutamate residue of GRP1 PH domain, E345 (Figure 1), is located adjacent to the PI(3,4,5)P3 headgroup binding pocket. Notably, the GRP1 E345 residue is found on a different loop and on the opposite side of the PI(3,4,5)P3 inositol ring relative to the AKT1 E17 residue (18, 26, 27), but both glutamates are close enough to directly contact the bound headgroup. The varying locations of putative sentry glutamates (see Discussion) is not surprising given the structural diversity of PH domain PI(3,4,5)P3 binding pockets.
FIGURE 1.
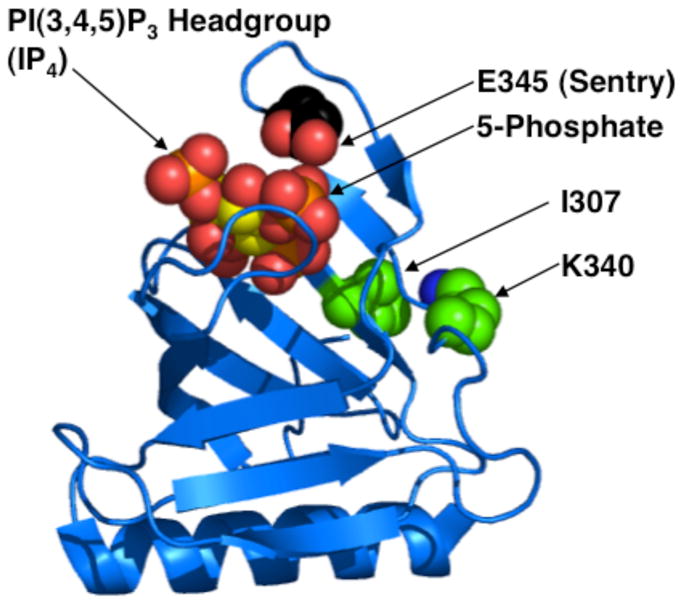
Locations of relevant residues in the crystal structure of GRP1 PH domain (18, 26). The proposed sentry glutamate E345 is located adjacent to binding pocket for PI(3,4,5)P3 headgroup (IP4), near the 5-phosphate. The Arf6 docking surface includes residues I307 and K340 (24, 25). The present study employs the E345K charge reversal mutation to test the sentry glutamate hypothesis, and the I307A mutation to weaken the interaction between GRP1 PH domain and Arf6.
As predicted by the sentry glutamate hypothesis, the present findings indicate that the GRP1 E345K mutation increases PH domain affinity for PI(4,5)P2 and triggers constitutive plasma membrane targeting. The effects of this mutation on PI(3,4,5)P3 and PI(4,5)P2 binding, both in vitro and in live cells, provide strong evidence that the native E345 side chain is a sentry glutamate ensuring minimal plasma membrane association until appearance of a PI(3,4,5)P3 signal. More broadly, analysis of the available crystal structures of other PI(3,4,5)P3-specific PH domains reveals analogous acidic side chains adjacent to the headgroup binding pocket that likely serve as sentry glutamate (or asparate) residues to decrease PI(4,5)P2 affinity, thereby ensuring plasma membrane targeting and pathway activation occurs only during a PI(3,4,5)P3 signal.
Materials and Methods
Reagents
All lipids were synthetic unless otherwise indicated. 1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (phosphatidylcholine, POPC, PC); 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (phosphatidylethanolamine, POPE, PE); 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoserine (phosphatidylserine, POPS, PS); L-α-phosphatidylinositol natural from bovine liver; L-α-phosphatidylinositol-4,5-bisphosphate (PI(4,5)P2, PIP2) natural from porcine brain; and sphingomyelin natural from porcine brain were all purchased from Avanti Polar Lipids. 1,2-dipalmitoyl-phosphatidylinositol-3,4,5-trisphosphate (phosphatidylinositol-3,4,5-trisphosphate, PI(3,4,5)P3) was from Cayman Chemical. N-[5-(Dimethylamino)naphthalene-1-sulfonyl]-1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine (dansyl-PE, dPE) was from Molecular Probes. Cholesterol (CH), inositol-1,2,3,4,5,6-hexakisphosphate (IP6) were from Sigma. Lipofectamine 2000 transfection reagent was from Invitrogen. Dithiothreitol (DTT) was purchased from Research Products International. Human recombinant PDGF-BB was purchased from PeproTech.
Preparation of Purified PH domains
For in vitro binding studies the PH domains of human GRP1 (residues 255–392) and mouse PLCδ1 (residues 12–142) were cloned by PCR as previously described into the EcoRI/XbaI site of a glutathione S-transferase (GST)-fusion vector (13). Mutagenesis of GRP1 PH was carried out using a QuickChangeXLII site directed mutagenesis kit (Stratagene). The GST-fusion constructs were overexpressed in BL21 Escherichia coli cells and isolated on a glutathione affinity column as previously described (13).
Preparation of Lipid Mixtures and Phospholipid Vesicles
Phospholipid bilayer vesicles were created using three lipid compositions (see Table 1) to mimic the plasma membrane inner leaflet either lacking or containing a PIP lipid: PM (−) PIP; PM (+) PI(4,5)P2; and PM (+) PI(3,4,5)P3. All lipids were dissolved in chloroform except PI(3,4,5)P3, which was dissolved in chloroform/methanol/water (1/2/0.8). Subsequently, each given lipid mixture was created in the indicated mole ratios and dried under vacuum until all solvent was removed, then hydrated by vortexing with binding buffer (25 mM N-(2-hydroxyethyl)piperazine-N′-2-ethanesulfonic acid (HEPES) (pH 7.4 with KOH), 140 mM KCl, 15 mM NaCl, 0.5 mM MgCl2). Finally, sonicated unilamellar phospholipids vesicles (SUVs) were prepared using a Misonix XL2020 probe sonicator as previously described (2, 13), yielding a total lipid concentration of 3 mM.
Table 1.
Lipid composition of plasma membrane mimics
| Name | Lipid Mixture | Lipid mol % |
|---|---|---|
| PM(+)PIPn | PE/PC/PS/PI/SM/CH/dPE/PIPn | 27.5/10.5/21/4.5/4.5/25/5/2 |
| PM | PE/PC/PS/PI/SM/CH/dPE | 29.5/10.5/21/4.5/4.5/25/5 |
Fluorescence Spectroscopy
Equilibrium binding experiments were carried out on a Photon Technology International QM-2000-6SE steady state fluorescence spectrometer at 25°C in binding buffer (above) plus 10 mM dithiothreitol (DTT) as previously described (2, 13). Excitation and emission slitwidths were 1 nm and 8 nm, respectively. Wavelengths used for specific experiments are indicated below.
Quantitative Measurement of PIP Lipid Affinity and Specificity
Our previously described competitive displacement assay was used to quantitate the high affinity binding of each PH domain to the selected PIP lipids (2, 13). In this approach, soluble inositol-1,2,3,4,5,6-hexakisphosphate (IP6) was employed as a competitive inhibitor to drive equilibrium displacement of PH domain from PIP-lipid containing membranes, while monitoring displacement by protein-to-membrane FRET. To determine the equilibrium dissociation constant for PIP lipid binding KD (PIPn) it was necessary to measure both the affinity of the free PH domain for the competitive inhibitor IP6 as well as the competitive inhibition constant for IP6 displacement of the PH domain from the target membrane.
The equilibrium dissociation constant of the free PH domain for the competitive inhibitor, KD (IP6), was measured using an increase in intrinsic Trp fluorescence to monitor binding of IP6 to the protein. Increasing concentrations of IP6 were titrated into a sample containing free PH domain protein (0.5 μM), 10mM DTT, and binding buffer. Intrinsic tryptophan fluorescence was measured using excitation and emission wavelengths λex = 284 nm and λem = 322 nm (for GRP1 PH) or λem = 340 nm (for PLCδ1 PH). Sodium phosphate, at six times the corresponding IP6 concentration, was also added to a control sample that was run parallel to the sample experiments to determine the effect of a highly anionic molecule on the Trp emission. The raw data was then corrected for dilution and the control cuvette data was subsequently subtracted, yielding the final titration data. Finally, a nonlinear least square curve fit employing Eq. 1 determined the equilibrium dissociation constant for IP6 [(KD(IP6)]:
| (1) |
where F is the observed fluorescence at a given IP6 concentration [IP6] and ΔFmax is the total magnitude of the fluorescence change at saturation with IP6.
The equilibrium apparent inhibition constant, KI(IP6)app, was measured by titrating IP6 into a sample containing the PH domain bound to a membrane-associated PIP lipid, using a protein-to-membrane FRET assay to monitor the membrane-bound PH domain. The solution contained 0.5 μM PH domain, 10 mM DTT, 3 μM accessible PIP lipid, 300 μM total lipid, and binding buffer. The IP6 competitively displaces the PH domain from the membrane, yielding a decrease in FRET. IP6 was also added to a control sample that was run parallel to the sample experiments to determine the effect of free IP6 on the FRET emission of free membranes. The control sample contained 10mM DTT, 3 μM accessible PIP lipid, 300 μM total lipid, and binding buffer. The raw data was corrected for dilution and then the control was subtracted from each sample. Finally, a nonlinear least square curve fit utilizing Eq. 2 determined the equilibrium apparent inhibition constant for IP6 [(KI(IP6)app]:
| (2) |
where F is the observed fluorescence and ΔFmax is the total magnitude of the fluorescence change at saturation with IP6. Using the (KD (IP6)) and (KI (IP6) app) and employing Eq. 3 the desired equilibrium dissociation constant for the binding of the PH domain to the membrane-associated PIP lipid (KD (PIPn)) was deduced:
| (3) |
For additional details see our previous publications (2, 13).
Quantitative Measurement of Association and Dissociation Kinetics
All kinetic experiments were carried out using Applied Photophysics SX.17 stopped-flow fluorescence instrument to monitor changes in protein-to-membrane FRET induced by membrane association or dissociation, always in binding buffer plus 10 mM DTT, at 25°C as previously described (2, 13). Association kinetics were measured by rapidly mixing PH domain (0.5 μM) with SUVs (300 μM total lipid) containing excess PIPn target lipid (3 μM accessible). Under these conditions the forward binding reaction is virtually irreversible due to the high affinity of the PH domain for PIP lipid, and best fit analysis of the binding timecourse with a double exponential yielded a predominant, fast component used to calculate the on-rate constant (kon). Dissociation kinetics were measured by rapidly mixing preformed, PH domain-membrane complex (0.5 μM protein, 300 μM total lipid) with excess IP6 (10 mM for GRP1 WT PH domain with PIP3; 20 mM for GRP1 E345K or E345K/I307A PH domain with PIP3; 8 mM for GRP1 E345K or E345K/I307A or PLCδ1 PH domain with PIP2). Best fitting of a double exponential function to the dissociation timecourse typically yielded fast and predominant slow components, and the latter was used to calculate the off-rate constant (koff) as previously described (13).
Fluorescent Fusion Proteins
The pEGFP-GRP1 PH construct was a kind gift to our lab from Ben Tycko (Columbia University Medical Center, New York) (28), and pRFP-PLCδ1 PH was a kind gift from M. Katan (Cancer Research UK Centre for Cell and Molecular Biology, London). In each plasmid, the fluorescent protein was replaced with Citrine fluorescent protein (Cit) by subcloning. Mutagenesis of Cit-GRP1 PH to generate the indicated mutations was carried out using a QuickChangeXLII site directed mutagenesis kit (Stratagene).
Transfections into Live Cells
NIH 3T3 cells (American Type Culture Collection) were cultured in DMEM containing 10% fetal bovine serum, 100 U/ml penicillin, 100 μg/ml streptomycin, 0.292 mg/ml glutamine in 5% CO2 at 37°C. Cells were plated in 6 cm culture dishes at 4.2 × 105 cells/dish and let sit overnight. Transfections were carried out with 4 μg total DNA using lipofectamine 2000 (Invitrogen) with OptiMEM (Invitrogen) according to manufacturer’s protocol. Prior to imaging, transfected cells (24–48 hours after transfection) were plated in 35 mm glass bottom dishes (MatTek) at 1 × 105 cells/dish and adhered for one to two hours. Where indicated, serum starvation of cells was carried out in DMEM containing 0.2% BSA and 0.292 mg/ml glutamine and incubated for 6 hours in 5% CO2 at 37°C.
Cell Imaging Studies
Transfected NIH 3T3 cells were rinsed and incubated in HBSS buffered with 25 mM HEPES at pH 7.4 (HHBSS) containing 0.01% endotoxin-free BSA. In experiments involving Ca2+-stimulated PI(4,5)P2 hydrolysis by phospholipase C, the HBSS was purchased Ca2+- and Mg2+-free and subsequently supplemented with 0.493 mM magnesium chloride and 0.407 mM magnesium sulfate to provide an initial Ca2+-free extra cellular environment. Immediately prior to imaging, ionomyosin was added to 10 μM. Subsequently, at time zero Ca2+ was added at the indicated extracellular concentration to trigger Ca2+ influx as previously described (2, 29, 30). Citrine images were acquired on a Nikon inverted microscope equipped with a 60 × 1.4 N.A. oil immersion objective, a CFP/YFP-Cit/RFP dichroic mirror, single band excitation and emission filters (Chroma Technology), a CoolSNAP ES camera (Photometrics), and a mercury lamp. For the kinetic time courses, time-lapse imaging involved 300 msec acquisitions with a total of 30 sec between image sets analyzed in ImageJ (NIH, http://rsb.info.nih.gov/ij/).
Results
Strategy and PH Domains Employed
The present study of the putative sentry glutamate E345 of GRP1 began with the construction of a set of PH domains possessing side chains of varying charge at the 345 position. Our previous study (2) of the AKT1 sentry glutamate E17K employed three sentry position variants: E17(WT), E17Q and E17K. To facilitate comparisons with this previous study we constructed and isolated the corresponding three GRP1 PH domains: E345(WT), E345Q and E345K. The relative affinities of these three GRP1 PH domains for membrane-bound PI(3,4,5)P3 and PI(4,5)P2, each embedded in bilayers mimicking the lipid composition of plasma membrane, were initially measured in a qualitative assay (2). The resulting relative affinities were E345K > E345Q > E345(WT) for PI(3,4,5)P3, and the same order E345K > E345Q > E345(WT) for PI(4,5)P2. These trends matched those previously observed for AKT1 PH domain, namely E17K > E17Q > E17(WT) for both PI(3,4,5)P3 and PI(4,5)P2 (2). Thus, as predicted by the sentry glutamate hypothesis and previously noted for AKT1 PH domain (2), side chains of different charge at the putative GRP1 sentry position alter the PIP lipid affinity in the simple fashion expected for direct electrostatic interaction with a negatively charged headgroup. Relative to the neutral sentry glutamine, the negatively charged glutamate lowers affinity for the anionic PIP lipid, while the positively charged lysine increases affinity for PIP lipid.
To more rigorously test the predictions and generality of the sentry glutamate hypothesis, the remainder of the present study focused on the native E345(WT) and E345K GRP1 PH domains. These two domains exhibit opposite extremes of PIP lipid affinity; moreover, E345K is analogous to the well characterized, disease-linked AKT1 E17K mutation. The sentry glutamate hypothesis proposes the native E345 residue plays a key role in maintaining specific PI(3,4,5)P3 targeting in cells by reducing affinity for constitutive plasma membrane PI(4,5)P2 (2, 31). The hypothesis therefore predicts that the E345K mutation, by significantly increasing the affinity for PI(4,5)P2, will enable the mutant protein to bind constitutively to plasma membrane in resting cells. Further experiments were carried out to quantitate the PI(3,4,5)P3 and PI(4,5)P2 affinities of E345(WT) and E345K GRP1 PH domains in vitro, and to compare their plasma membrane targeting in cells. As these experiments proceeded it became clear that Arf6 binding contributed significantly to GRP1 PH domain targeting in cells as previously reported (24, 25), thus the E345K/I307A mutant was constructed to clarify the role of Arf6 interactions in the E345K background. Finally, the PI(4,5)P2-specific PLCδ1 PH domain was included as a control displaying a target lipid preference opposite to that of the PI(3,4,5)P3-specific E345(WT) GRP1 PH domain (7, 30, 32, 33). Figure 1 shows the locations in the GRP1 PH domain structure of the E345 and I307 positions targeted for mutation, and of the bound PI(3,4,5)P3 headgroup (inositol-1,3,4,5-P4, or IP4).
E345K Mutation Enhances GRP1 PH Domain Affinity for Both PI(4,5)P2 and PI(3,4,5)P3, with Important Implications for Target Lipid Specificity
A competitive displacement assay previously developed in our laboratory (13) was used to quantify the affinities of PH domains for PI(3,4,5)P3 and PI(4,5)P2 in sonicated unilamellar membrane vesicles. The competitive displacement assay enables determination of equilibrium dissociation constants for membrane-bound target lipids in the range 0.1 to 10,000 nM, well-suited for measuring the nanomolar affinity of native GRP1 PH domain for target lipid PI(3,4,5)P3 in bilayers composed of seven lipids mimicking the lipid composition of the plasma membrane inner leaflet (Table 1) (2). The first step of the assay measured the dissociation constant KD(IP6) for PH domain binding to the competitive inhibitor inositol hexakisphosphate (IP6) in solution by titrating inhibitor into a sample of free PH domain while monitoring the increase in intrinsic tryptophan fluorescence triggered by inhibitor binding (Eq. 1 in Methods). The second step measured the apparent inhibition constant KI(IP6) for competitive displacement of the PH domain from membrane-associated PIP lipid by titrating inhibitor into a suspension of PH domains bound to vesicles while monitoring the decrease in protein-to-membrane FRET (Eq. 2). Finally, the standard equation for competitive inhibition (Eq. 3) was used to calculate the dissociation constant KD(PIPn) for PH domain binding to the membrane-associated target lipid.
Figure 2 summarizes the equilibrium titrations for (i) the native, wild type GRP1 PH domain, WT; (ii) the single mutant PH domain, E345K; and (iii) the double mutant PH domain, E345K/I307A. Shown for each mutant are both the titration quantifying competitive inhibitor IP6 binding to free PH domain (Figs. 2A–C) and the titration quantifying competitive displacement of PH domain from membrane-associated PI(3,4,5)P3 or PI(4,5)P2 (Fig. 2D–F) by IP6. Table 2 summarizes the resulting equilibrium constants KD(IP6), KI(IP6), and KD(PIPn) for the three GRP1 PH domains, as well as for the PLCδ1 PH domain (titrations not shown).
FIGURE 2.

Quantitative equilibrium binding and specificity measurements. (A–C) Direct measurement of IP6 binding to free PH domain in which 0.5 μM of each protein was titrated with IP6 while quantitating the increase in tryptophan emission resulting from complex formation (see Methods). (D–F) Competitive displacement assay for the indicated PH domain bound to either PI(3,4,5)P3 (squares) or PI(4,5,)P2 (circles) on plasma membrane-like sonicated unilamellar vesicles. To form the protein-membrane complex, protein (0.5 μM) was mixed with target vesicles (300 μM total lipid, 3 μM accessible PIP lipid). Subsequently, protein was displaced from target vesicles by titration with the competitive inhibitor IP6 while monitoring the decrease in protein-to-membrane FRET (see Methods). In each panel, assays were carried out in triplicate at 25º C in a physiological buffer, data points are averages (±1 standard deviation), and best fit curves were defined by Eq. 1 (A–C) or 2 (D–F). Insets (D–F) show full titration for proteins bound to PI(3,4,5)P3. Table 2 presents parameters defined by averaging best-fit values defined by three or more independent measurements.
Table 2.
Equilibrium and Kinetic Parameters for PH domains Binding to Plasma Membrane Mimics Containing PIP Lipids
| PH Domain | KD(IP6) (μM) | Plasma Membrane Mimica | Equilibrium
|
Kinetics
|
|||
|---|---|---|---|---|---|---|---|
| KI(IP6) (mM) | KD(PIPn) (nM) | kon (×106 M−1 s−1) | koff (s−1) | KD (koff/kon) (nM) | |||
| WT | 20 ± 3 | PM(+)PI(3,4,5)P3 | 5 ± 2 | 14 ± 8 | 14 ± 1 | 0.79 ± 0.01 | 55 ± 1 |
| PM(+)PI(4,5)P2 | 0.04 ± 0.01 | 3000 ± 2000 | NDb | NDb | — | ||
| E345K | 4.1 ± 0.1 | PM(+)PI(3,4,5)P3 | 7 ± 2 | 1.7 ± 0.1 | 20 ± 1 | 0.042 ± 0.007 | 2.1 ± 0.3 |
| PM(+)PI(4,5)P2 | 0.026 ± 0.005 | 400 ± 50 | 53 ± 1 | 23 ± 1 | 430 ± 20 | ||
| E345K/I307A | 8 ± 2 | PM(+)PI(3,4,5)P3 | 14 ± 4 | 1.7 ± 0.7 | 35 ± 1 | 0.16 ± 0.01 | 4.6 ± 0.1 |
| PM(+)PI(4,5)P2 | 0.035 ± 0.005 | 800 ± 100 | 32 ± 1 | 18 ± 1 | 550 ± 20 | ||
| PLCδ1 | 4 ± 1 | PM(+)PI(3,4,5)P3 | NDb | > 104c | NDb | NDb | — |
| PM(+)PI(4,5)P2 | 0.07 ± 0.01 | 150 ± 20 | 180 ± 10 | 15 ± 1 | 81 ± 5 | ||
Comparison of the equilibrium dissociation constants for competitive inhibitor IP6 reveals that the E345K mutation significantly increases the affinity of the free GRP1 PH domain for IP6. Thus, the free E345K domain exhibits a nearly 5-fold higher IP6 affinity than the WT domain (KD(IP6) of 4.1 ± 0.1 μM and 20 ± 3 μM, respectively). By contrast, the I307A mutation at the Arf6 interaction site has little or no effect on inhibitor binding, such that the IP6 affinity of the E345K/I307A mutant (KD(IP6) of 8 ± 2 μM) is within 2-fold that of E345K.
Comparison of the equilibrium dissociation constants for PH domain binding to PI(3,4,5)P3 or PI(4,5)P2 embedded in the plasma membrane-like bilayer reveals that the E345K mutation significantly increases the affinity of GRP1 PH domain for both of these PIP lipids (Table 2). The native WT domain binds its membrane-associated target PI(3,4,5)P3 with high affinity (KD of 14 ± 8 nM) and binds membrane-bound PI(4,5)P2 over 200-fold more weakly (KD of 3000 ± 2000 nM). At the opposite extreme, the PLCδ1 PH domain binds its target PI(4,5)P2 with a relatively high affinity (KD of 150 ± 20 nM) while exhibiting much weaker affinity for PI(3,4,5)P3 (KD > 104 nM). In GRP1 PH domain, the E345K charge reversal mutation yields 8-fold greater affinity for the native target PI(3,4,5)P3 (KD of 1.7 ± 0.1 nM) relative to WT (Table 2). Strikingly, the E345K mutation also generates a nearly 8-fold higher affinity for PI(4,5)P2 (KD of 400 ± 50 nM) compared to WT (Table 2), yielding a PI(4,5)P2 affinity within 2.6-fold that of the PLCδ1 PH domain (Table 2). Since the latter domain is widely used as a PI(4,5)P2 sensor and binds to constitutive plasma membrane PI(4,5)P2 in cells, the E345K mutant is expected to target constitutively to plasma membrane. Finally, the Arf6 interaction mutant E345K/I307A exhibits PI(3,4,5)P3 and PI(4,5)P2 affinities (KD of 1.7 ± 0.7 and 800 ± 100 nM, respectively) that are indistinguishable, within a factor of 2, of the corresponding PI(3,4,5)P3 and PI(4,5)P2 affinities observed for the E345K mutant (Table 2). It follows that, in the absence of Arf6, the I307A mutation has little effect on binding of the E345K mutant to target lipids.
Kinetic Analysis Reveals that E345K Triggers PIP Lipid Affinity Increases Primarily by Slowing Off-Rates
In principle, the E345K mutation could increase the equilibrium affinities of GRP1 PH domain for PI(3,4,5)P3 and PI(4,5)P2 by speeding association kinetics or slowing dissociation kinetics. To resolve these possibilities, where feasible the on- and off-rates were measured for the wild type and E345K mutant PH domains binding to membrane-embedded PI(3,4,5)P3 and PI(4,5)P2 by stopped-flow fluorescence, using protein-to-membrane FRET to monitor membrane association or dissociation. Such kinetic measurements were straightforward for the interaction of wild type and E345K domains with PI(3,4,5)P3, since both proteins bind this lipid tightly. (Kinetic parameters could not be determined for wild type protein binding to PI(4,5)P2 due to the low affinity).
Association reactions were carried out by rapidly mixing the free PH domain with target membranes then monitoring the approach-to-equilibrium. Under the saturating conditions employed, the off-rate is negligible and the approach-to-equilibrium yields the true on-rate. Figure 3A and Table 2 show that all three of the GRP1 PH domains, regardless of whether they lack or possess the E345K mutation or the I307A Arf6 interaction mutation, exhibit similar on-rates for PI(3,4,5)P3 within a narrow 2.5-fold range (kon from (14 ± 1) × 106 M−1 s−1 to (35 ± 1) × 106 M−1 s−1). Moreover, the on-rates of the E345K mutant and the E345K/I307A double mutant for both PI(3,4,5)P3 and PI(4,5)P2 fall within a similar 3-fold range (kon from (20 ± 1) × 106 M−1 s−1 to (53 ± 1) × 106 M−1 s−1, Table 2). The observed similarities in the measurable on-rates of GRP1 PH domains for PI(3,4,5)P3 and PI(4,5)P2 likely arise from the rate-determining electrostatic search mechanism that the PH domain is known to employ to find its rare target lipid, since this search mechanism relies on interactions between the protein and background PS lipids (13, 34). Such PS-based searching is expected to yield similar on-rates for different PIP target lipids as long as they are present at similar mole percents, as in the present study where PI(3,4,5)P3 and PI(4,5)P2 were always 2 mole percent, and PS was always 21 mole percent.
FIGURE 3.

Association and dissociation rate measurements. Shown are association and dissociation reactions of indicated PH domains binding to PI(3,4,5)P3 (A) or PI(4,5,)P2 (B) embedded in plasma membrane-like bilayers. In all cases the final protein and lipid concentrations were identical to the equilibrium competitive displacement assays (Fig. 2). Reactions were initiated by rapid mixing of either the free PH domain with membranes (association) or the prebound PH domain-membrane complex with IP6 (dissociation) as described in Methods. Note that the time axis is optimized for each panel. For each reaction, at least five shots were carried out at 25º C in a physiological buffer and then averaged to give the representative trace. Bold curves indicate best-fit double exponential functions, which yielded dominant fast (association) and slow (dissociation) components used to determine on- and off-rate constants as previously described (13). For each rate constant, at least 3 independent measurements were averaged to produce the parameters summarized in Table 2. The inset shows the full timecourses of the two mutant dissociation reactions.
Dissociation reactions were carried out by stopped-flow mixing pre-formed PH domain–membrane complexes with excess competitive inhibitor, IP6, thereby ensuring that membrane dissociation is essentially irreversible. As shown in Figure 3A (IV) the E345K mutation slows the dissociation of GRP1 PH domain from PI(3,4,5)P3-containing membranes by a factor of 18-fold relative to wild type (koff of 0.042 ± 0.007 s−1 and 0.79 ± 0.01 s−1, respectively) (Table 2). The rate of dissociation of the E345K mutant from PI(4,5)P2 is well within the measurable range, but is 540-fold faster than its dissociation from PI(3,4,5)P3 (koff of 23 ± 1 s−1 and 0.042 ± 0.007 s−1, respectively) (Table 2). Interestingly, the E345K mutant exhibits a PI(4,5)P2 off-rate nearly the same, within 1.5-fold, as that of PLCδ1 PH domain (koff of 23 ± 1 s−1 and 15 ± 1 s−1, respectively), helping to explain the similar PI(4,5)P2 affinities of the two PH domains (Table 2). The effect of I307A on the dissociation kinetics of the E345K mutant is minimal, such that the E345K/I307A off-rates for PI(3,4,5)P3 and PI(4,5)P2 are within 3.8-fold of the corresponding E345K off-rates (Table 2).
Wherever possible, the measured on- and off- rate constants were used to calculate a kinetic dissociation constant for each protein-target lipid combination (KD(PIPn) = koff/kon). As shown in Table 2, the KD values calculated from kinetic constants are within 4-fold of the KD values measured in equilibrium studies. Such good agreement validates both the equilibrium and kinetic measurements.
Examination of the on- and off-rate constants of the E345K mutant reveals that its higher affinity for PI(3,4,5)P3 over PI(4,5)P2 arises primarily from a 540-fold slower PI(3,4,5)P3 off-rate. Notably, however, the major functional difference between wild type GRP1 PH domain and the E345K mutant (see cell studies below) arises from the increased affinity of the mutant for PI(4,5)P2. While it is not possible to directly determine the kinetic mechanism of this 8-fold affinity increase due to inability to measure the WT PI(4,5)P2 on-rate, the PI(4,5)P2 on-rates of the wild type and E345K PH domains are likely to be similar. Recall that the PI(3,4,5)P3 on-rates of wild type and E345K, as well as the PI(3,4,5)P3 and PI(4,5)P2 on-rates of the E345K and E345K/I307A mutants, are all similar owing to the rate limiting electrostatic search mechanism (see above). Assuming the WT PI(4,5)P2 on-rate falls in the same range due to its standard rate-limiting search process, it would follow that the 8-fold higher affinity of the E345K mutant for PI(4,5)P2 arises from an approximately 8-fold slower dissociation from PI(4,5)P2 relative to WT.
Together the in vitro equilibrium and kinetic studies indicate the E345K putative sentry glutamate mutation in GRP1 PH domain behaves similarly to the oncogenic, E17K sentry glutamate mutation in AKT1 PH domain that we characterized previously (2). In both the E345K GRP1 and the E17K AKT1 PH domain mutants, the glutamate to lysine charge reversal mutation significantly increases both the PI(3,4,5)P3 and PI(4,5)P2 affinities. Notably, the resulting PI(4,5)P2 affinity of E345K GRP1 PH domain, like that of E17K AKT1 PH domain, is remarkably similar to the PI(4,5)P2 affinity of PLCδ1 PH domain – the standard PI(4,5)P2 sensor employed in many in vitro and live cell studies (2, 30). It was previously observed that the E17K AKT1 PH domain is bound to plasma membrane in the absence of a PI(3,4,5)P3 signal (1) due to its interaction with PI(4,5)P2 (2). The present in vitro evidence led us to predict that the E345K GRP1 PH domain would display a similar constitutive targeting to plasma membrane PI(4,5)P2 in resting cells.
Live Cell Studies Confirm the GRP1 E345K PH Domain is Targeted to the Plasma Membrane by PI(4,5)P2
Live cell imaging was used to visualize the intracellular targeting patterns of the PH domains studied in vitro while varying the plasma membrane levels of PI(3,4,5)P3 and PI(4,5)P2. Each PH domain was fused at its N-terminus to citrine fluorescent protein (Cit), expressed in serum-starved NIH 3T3 fibroblasts, and imaged in live cells by wide field fluorescence microscopy. Resting cells were serum-starved to minimize stimulation by attractants and growth factors in the media, ensuring low basal plasma membrane levels of the signaling lipid PI(3,4,5)P3.
Figure 4A summarizes the cellular distributions of PH domains in resting cells, and in activated cells with high levels of plasma membrane PI(3,4,5)P3. In resting cells, the wild type GRP1 PH domain fusion Cit–WT is cytoplasmic with no detectable plasma membrane binding (Fig 4A, B). In contrast, the E345K mutant fusion protein Cit–E345K is significantly targeted to plasma membrane, consistent with its increased in vitro affinity for constitutive plasma membrane PI(4,5)P2 (Fig. 4A, B; Table 2). As expected, the PLCδ1 PH domain is also targeted to plasma membrane in resting cells due to its high affinity for constitutive PI(4,5)P2 (Fig. 4A, B; Table 2). Global extracellular addition of PDGF activates phosphatidylinositol-3-kinase (PI3K) and generates plasma membrane PI(3,4,5)P3 (35). The resulting PI(3,4,5)P3 signal recruits the wild type PH domain to the plasma membrane, and drives additional plasma membrane recruitment of the E345K mutant (Fig. 4A). The observed PI(3,4,5)P3-stimulated recruitment of the wild type and E345K PH domains to plasma membrane is consistent with the high PI(3,4,5)P3 affinities observed for both domains in vitro (Table 2). The E345K/I307A mutant behaves similarly to the E345K mutant, exhibiting both detectable plasma membrane binding in resting cells (Fig. 4A, B) and enhanced membrane targeting during a PI(3,4,5)P3 signal (Fig. 4A).
FIGURE 4.

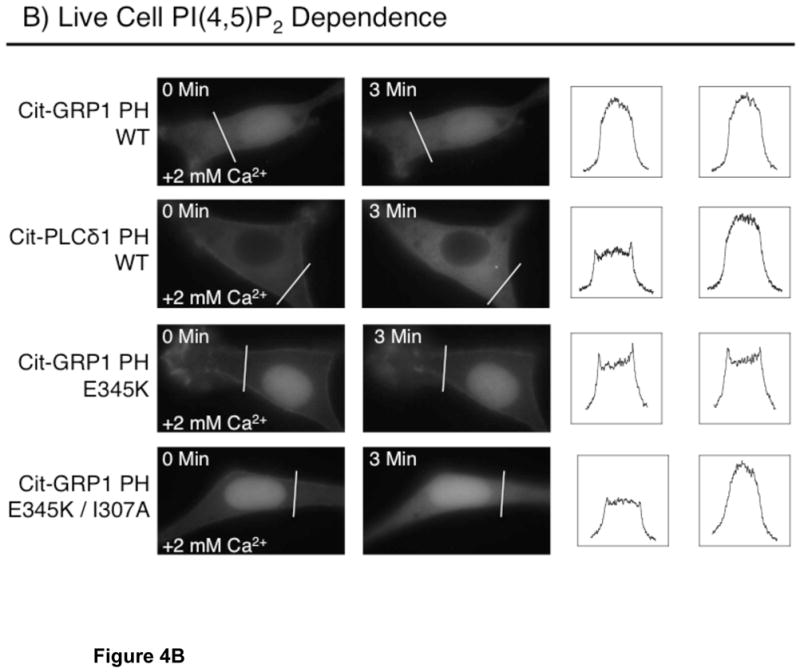
PIP lipid dependence of plasma membrane targeting in NIH 3T3 cells. (A) Dependence of membrane targeting on PI(3,4,5)P3. Shown are the zero time and one minute images for the indicated citrine-coupled PH domains. Serum-starved NIH 3T3 cells were stimulated with 5 nM PDGF at time zero and imaged at multiple timepoints (see Methods). (B) Dependence of membrane targeting on PI(4,5)P2 for the indicated citrine-coupled PH domains. Shown are the zero time and three minute images for the indicated citrine-coupled PH domains. Serum-starved NIH 3T3 cells were incubated in 20 μM ionomycin in Ca2+-free buffer, then stimulated at time zero with 2 mM extracellular Ca2+ to trigger cytoplasmic Ca2+ influx and Ca2+-activated PI(4,5)P2 hydrolysis (45) with imaging at multiple timepoints (see Methods). Each set of images are representative examples of responses observed for 12 or more cells. To assist visualization of the protein movements between the plasma membrane and cytoplasm, fluorescence intensities for pixels defined by the indicated line across each cell are plotted for both before (left box) and after (right box) treatment.
To directly test the hypothesis that the E345K mutant targets to plasma membrane PI(4,5)P2 in resting cells, Ca2+ and ionomycin were added globally to increase cytoplasmic Ca2+ and activate PLCδ1 hydrolysis, thereby destroying the plasma membrane PI(4,5)P2 (29, 30). Figure 4B shows the effect of the resulting PI(4,5)P2 loss on the cellular distribution of each PH domain. The Ca2+-ionomycin treatment fully relocates the PLCδ1 PH domain from the plasma membrane to the cytoplasm, confirming that the treatment destroys the PI(4,5)P2 lipid this domain targets (Fig. 4B). By contrast, the treatment only partially relocates the E345K domain to the cytoplasm (Fig. 4B) unless the I307A mutation is known to weaken the PH domain-Arf6 interaction (24, 25) is present. Thus, the E345K/I307A double mutant is fully relocated to cytoplasm by the Ca2+-ionomycin treatment (Fig. 4B). These observations demonstrate that PI(4,5)P2 binding and Arf6 interactions both make important contributions to the plasma membrane affinity and bound state lifetime of the E345K mutant, consistent with the previous conclusion that native GRP1 PH domain is a dual PI(3,4,5)P3 and Arf6 sensor (24, 25).
Overall, the live cell findings strongly support the conclusion that the E345K sentry glutamate mutation in GRP1 PH domain, like the corresponding E17K mutation in AKT1 PH domain, drives constitutive plasma membrane targeting via enhanced affinity for PI(4,5)P2. As a result, the E345K GRP1 PH domain, like the E17K AKT1 PH domain, localizes to plasma membrane even in resting cells lacking a PI(3,4,5)P3 signal.
Discussion
The present study extends our previous analysis of the molecular mechanism of the E17K mutation in AKT1 PH domain to the analogous E345K mutation in GRP1 PH domain (2). Together, these two studies strongly support the conclusion that a sentry glutamate adjacent to the PI(3,4,5)P3 binding pocket plays a crucial role in reducing the affinities of both PH domains for constitutive plasma membrane PI(4,5)P2, thereby ensuring that PH domain recruitment to plasma membrane occurs only during a PI(3,4,5)P3 signal. Like the AKT1 E17K mutation, the GRP1 E345K mutation at the sentry glutamate position (i) increases the affinity for soluble IP6 and for membrane-bound PI(3,4,5)P3 and PI(4,5)P2 in vitro, (ii) yields a PI(4,5)P2 affinity in vitro similar to that of PLCδ1 PH domain, the prototypical PI(4,5)P2 sensor, and (iii) yields binding to constitutive plasma membrane PI(4,5)P2 in resting cells, as observed for control PLCδ1 PH domain but not for WT GRP1 PH domain.
The constitutive plasma membrane targeting observed for the sentry glutamate mutants AKT1 E17K and GRP1 E345K arises from their increased PI(4,5)P2 affinities, and not from their increased PI(3,4,5)P3 affinities. Thus, in resting cells, starvation to minimize growth factors in the media, thereby suppressing plasma membrane PI(3,4,5)P3, does not block the constitutive plasma membrane targeting of either PH domain mutant ((2) and Fig. 4, this article). Instead, in vitro binding measurements reveal that the PI(4,5)P2 affinities of AKT1 E17K and GRP1 E345K PH domains are raised to a level sufficient to bind the constitutive plasma membrane PI(4,5)P2 in resting cells, like the PLCδ1 PH domain ((2) and Table 2). The AKT1 E17K mutation increases the PH domain PI(4,5)P2 affinity at least 80-fold, from a native KD > 104 nM to a KD of 130 ± 30 nM for the mutant, indistinguishable from the PI(4,5)P2 affinity of PLCδ1 PH domain (2). The GRP1 E345K mutation increases PI(4,5)P2 affinity approximately 8-fold, yielding a KD of 400 ± 50 nM in vitro (Table 2), again similar to the PI(4,5)P2 affinity of PLCδ1 PH domain. Moreover, in cells the PI(4,5)P2 affinity of GRP E345K PH domain is expected to be further enhanced by interactions with plasma membrane-associated Arf6 ((24, 25) and below). Confirming this picture, the constitutively plasma-membrane targeted AKT1 E17K, GRP1 E345K and PLCδ1 PH domains are displaced from the plasma membrane upon introduction of high cytoplasmic [Ca2+] to activate phospholipase C activity and thereby hydrolyze the PI(4,5)P2 pool ((2) and Fig. 4B), although the GRP1 E345K PH domain is fully displaced by PI(4,5)P2 hydrolysis only when its interaction with Arf6 is weakened by incorporation of the I307A mutation (Fig. 4B). In short, the evidence indicates that the primary physiological role of the native sentry glutamate of AKT1 and GRP1 is to minimize binding to constitutive plasma membrane PI(4,5)P2 in resting cells. Secondarily, the sentry glutamate also reduces the effective PI(3,4,5)P3 affinity, and this affinity tuning could further optimize cellular responses to PI(3,4,5)P3 signals.
More broadly, the present findings have widespread implications for PI(3,4,5)P3-specific PH domains. Currently, there are seven PI(3,4,5)P3-specific PH domains for which structures are available bound to the PI(3,4,5)P3 headgroup. Figure 5 illustrates the confirmed sentry glutamates of AKT1 (E17, PDB 1H10 (27)) and GRP1 (E345, PDB 1FGY and 1FHX (18, 26)) PH domains, and shows that the other five known structures also possess a putative sentry glutamate (or, in two cases, a putative sentry aspartate): (i) Bruton’s tyrosine kinase (BTK, E41, PDB 1B55 (36)); (ii) Arf nucleotide-binding-site opener (ARNO, E341, PDB 1U27 (37)); (iii, iv) centaurin α1 PH1 and PH2 (CENTA1, D142 and E268, respectively, PDB 3LJU(38)); and (v) 3-phosphanositide-dependent kinase 1 (PDK1, D488, PDB 1W1D (39)). It follows that an acidic sentry side chain is a common, perhaps ubiquitous, feature of PI(3,4,5)P3-specific PH domains. However, these acidic sentry residues may have arisen from convergent evolution, since their locations in the PH domain architecture are not conserved. Examination of Figure 5 reveals that the proposed sentry residues are distributed among different loops and are disposed differently relative to the PI(3,4,5)P3 headgroup. In five cases (AKT1, GRP1, ARNO, CENTA1 PH1 and PH2) the sentry side chain carboxylate is predicted to have the strongest repulsive electrostatic interaction with the 5-phosphate of the PI(3,4,5)P3 headgroup due to proximity or, in certain cases, due to screening of other proximal phosphates by intervening basic side chains. In the remaining cases (BTK, PDK1) the side chain carboxylate is predicted to interact most strongly with the 1-phosphate.
FIGURE 5.

Comparison of all six PI(3,4,5)P3-specific PH domains for which co-crystal structures with bound PI(3,4,5)P3 headgroup are currently available (26,17,25,35–38). Highlighted are the putative sentry residue and the headgroup phosphate predicted to have the strongest electrostatic interactions with the sentry side chain carboxylate (see Discussion).
The different locations of sentry glutamates are likely to yield subtle mechanistic differences as illustrated by a comparison of AKT1 and GRP1. In both cases, the mobile side chain carboxylate of the sentry glutamate can closely approach the 5-phosphate, but AKT E17 is on loop β1/β2 (27) and GRP1 E345 is on loop β6/β7 (18) on opposite sides of the inositol ring. The E17K AKT1 PH domain mutation increases the PI(3,4,5)P3 affinity by a factor of 8-fold, and increases the affinity for PI(4,5)P2 by a much greater 80-fold factor. By contrast, the E345K GRP1 PH domain mutation increases the affinity for both PI(3,4,5)P3 and PI(4,5)P2 by the same factors of 8-fold. The simplest explanation for the more dramatic effect of the E17K mutation on PI(4,5)P2 affinity is that the sentry glutamate of AKT1 PH domain prevents the binding of the PI(4,5)P2 headgroup in an alternative orientation that would yield significantly tighter binding, while this alternative PI(4,5)P2 orientation is not accessible in GRP1 PH domain.
The strikingly similar, 8-fold affinity increases observed for the interactions of E17K AKT1 PH domain with PI(3,4,5)P3, and for GRP1 PH domain with both PI(3,4,5)P3 and PI(4,5)P2, may share similar mechanisms. In principle, at least three electrostatic interactions could be involved, but only one is favored by the data. (i) In crystal structures of the apo state, the AKT1 or GRP1 sentry glutamate forms a salt bridge to a nearby basic side chain required for PIP headgroup coordination (E17 to K14 and E345 to R349, respectively). This salt bridge must be broken for the PIP headgroup to bind, yielding an energy barrier for binding that is eliminated by the E17K or E345K mutation. However, the kinetic analysis indicates this barrier is insignificant since the measured target lipid on-rates (Table 2) are largely unaffected by the sentry mutations. The findings are consistent with the previously documented rate-determining, electrostatic search process that involves membrane-embedded PS and defines the target lipid association kinetics (13, 34). It follows that an apo-state salt bridge does not play a central role in sentry glutamate function. (ii) When bound to its target lipid on the membrane surface, the AKT1 or GRP1 PH domain interacts electrostatically with negatively charged lipids, primarily phosphatidylserine (PS). This interaction would be repulsive for the native sentry glutamate, but the E17K or E345K mutation would convert this repulsive interaction into an attractive interaction, thereby stabilizing the bound state and increasing target lipid affinity. Such a picture is disfavored by the observation that a membrane is not required for the affinity increase, since both mutations trigger affinity increases for soluble IP6 similar in magnitude to the affinity increases observed for membrane-embedded PIP lipids ((2) and Table 2). (iii) Instead, the molecular basis of sentry function appears to be the electrostatic interaction between the sentry side chain carboxylate and the PIP lipid headgroup. The sentry glutamate side chain is presumably dynamic, and the AKT1 and GRP1 co-crystal structures with a PI(3,4,5)P3 headgroup analogue both indicate the side chain carboxylate can approach the inositol 5-phosphate of the target headgroup well within the ~ 8 Å Debye limit for significant electrostatic interactions at physiological ionic strength. The resulting carboxylate-phosphate charge repulsion would generate an energy barrier for PIP headgroup binding that would be converted to a favorable electrostatic interaction by the E17K or E345K mutation. This picture explains the higher PI(3,4,5)P3, PI(4,5)P2 and IP6 affinities observed for the AKT1 and GRP1 sentry lysine mutants, as well as the slower PI(3,4,5)P3 dissociation kinetics observed for these mutants ((2) and Table 2).
Thus, the native sentry glutamate is a negative regulator of PI(4,5)P2 binding, and charge reversal to lysine enhances both the PI(4,5)P2 and PI(3,4,5)P3 affinities. These observations may provide a clue to the evolution of PI(3,4,5)P3-specific PH domains. If ancestor cells possessed sufficiently low constitutive levels of PI(4,5)P2 and generated smaller PI(3,4,5)P3 signals, the original PH domains may have required lysine or arginine residues at the position now occupied by the sentry glutamate. Modern PI(3,4,5)P3-specific PH domains that encounter high levels of PI(4,5)P2, however require the sentry glutamate to ensure that plasma membrane targeting occurs only during a PI(3,4,5)P3 signal. The glutamate also moderates the high PI(3,4,5)P3 affinity generated by the multiple basic residues that coordinate the PI(3,4,5)P3 in the headgroup binding pocket. In the case of GRP1 PH domain, one of these residues is a His sidechain (His 355) that may further modulate PI(3,4,5)P3 targeting by introducing a strong pH sensitivity (40).
The physiological impact of the glutamate to lysine charge reversal mutation at the sentry glutamate position has been previously demonstrated for full length AKT1 E17K, since this mutant is an oncogenic kinase ((1, 41) and recently confirmed in (42)). The constitutive plasma membrane targeting of AKT1 E17K leads to hyperactivation of its kinase activity, transformation of tissue culture cells, multiple human cancers and, in the case of Proteus syndrome, tissue overgrowth (1, 3). Similarly, while the BTK E41 residue has not been directly confirmed as a sentry glutamate due to lack of quantitative equilibrium and kinetic analysis, the BTK E41K mutation does lead to PI(4,5)P2 binding in vitro as well as constitutive targeting to plasma membrane and transformation of tissue culture cells (36, 43, 44). Thus, the available evidence suggests that BTK E41 is likely a sentry glutamate and its primary role is to minimize PI(4,5)P2 binding in resting cells, as demonstrated for AKT1 E17 and GRP1 E345. While to date only the AKT1 E17K mutation has been directly linked to disease, the ability of charge reversal mutations at sentry glutamate positions to trigger constitutive plasma membrane targeting suggests that future studies may well uncover diseases linked to the GRP1 E345K or BTK E41K mutation, or to sentry charge reversals in other PH domains.
Acknowledgments
†Support provided by NIH R01 GM-063235 (to JJF) and NIH T32 GM-065103 (to KEL)
The authors thank Drs. Amy Palmer for technical advice on cell imaging, Brian Ziemba for critical reading of the manuscript, and Biochemistry reviewers for helpful suggestions.
Abbreviations
- AKT1
protein kinase B, isoform 1
- PDK1
phosphoinositide dependent protein kinase 1
- PLCδ-1
phospholipase C delta, isoform 1
- PH
pleckstrin homology
- PC
phosphatidylcholine
- PS
phosphatidylserine
- PE
phosphatidylethanolamine
- dPE
dansyl-PE
- PI(3,4,5)P3
phosphatidylinositol-3,4,5-trisphosphate
- PI(4,5)P2
phosphatidylinositol-4,5-bisphosphate
- DTT
dithiothreitol
- IP6
inositol-hexakisphosphate
References
- 1.Carpten JD, Faber AL, Horn C, Donoho GP, Briggs SL, Robbins CM, Hostetter G, Boguslawski S, Moses TY, Savage S, Uhlik M, Lin A, Du J, Qian YW, Zeckner DJ, Tucker-Kellogg G, Touchman J, Patel K, Mousses S, Bittner M, Schevitz R, Lai MH, Blanchard KL, Thomas JE. A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature. 2007;448:439–444. doi: 10.1038/nature05933. [DOI] [PubMed] [Google Scholar]
- 2.Landgraf KE, Pilling C, Falke JJ. Molecular mechanism of an oncogenic mutation that alters membrane targeting: Glu17Lys modifies the PIP lipid specificity of the AKT1 PH domain. Biochemistry. 2008;47:12260–12269. doi: 10.1021/bi801683k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lindhurst MJ, Sapp JC, Teer JK, Johnston JJ, Finn EM, Peters K, Turner J, Cannons JL, Bick D, Blakemore L, Blumhorst C, Brockmann K, Calder P, Cherman N, Deardorff MA, Everman DB, Golas G, Greenstein RM, Kato BM, Keppler-Noreuil KM, Kuznetsov SA, Miyamoto RT, Newman K, Ng D, O’Brien K, Rothenberg S, Schwartzentruber DJ, Singhal V, Tirabosco R, Upton J, Wientroub S, Zackai EH, Hoag K, Whitewood-Neal T, Robey PG, Schwartzberg PL, Darling TN, Tosi LL, Mullikin JC, Biesecker LG. A mosaic activating mutation in AKT1 associated with the Proteus syndrome. N Engl J Med. 2011;365:611–619. doi: 10.1056/NEJMoa1104017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vanhaesebroeck B, Leevers SJ, Ahmadi K, Timms J, Katso R, Driscoll PC, Woscholski R, Parker PJ, Waterfield MD. Synthesis and function of 3-phosphorylated inositol lipids. Annu Rev Biochem. 2001;70:535–602. doi: 10.1146/annurev.biochem.70.1.535. [DOI] [PubMed] [Google Scholar]
- 5.Hawkins PT, Anderson KE, Davidson K, Stephens LR. Signalling through Class I PI3Ks in mammalian cells. Biochemical Society Transactions. 2006;34:647–662. doi: 10.1042/BST0340647. [DOI] [PubMed] [Google Scholar]
- 6.Newton AC. Lipid activation of protein kinases. J Lipid Res. 2009;50(Suppl):S266–271. doi: 10.1194/jlr.R800064-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lemmon MA, Ferguson KM. Signal-dependent membrane targeting by pleckstrin homology (PH) domains. Biochem J. 2000;350(Pt 1):1–18. [PMC free article] [PubMed] [Google Scholar]
- 8.Czech MP. PIP2 and PIP3: complex roles at the cell surface. Cell. 2000;100:603–606. doi: 10.1016/s0092-8674(00)80696-0. [DOI] [PubMed] [Google Scholar]
- 9.Insall RH, Weiner OD. PIP3, PIP2, and cell movement--similar messages, different meanings? Dev Cell. 2001;1:743–747. doi: 10.1016/s1534-5807(01)00086-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang F, Herzmark P, Weiner OD, Srinivasan S, Servant G, Bourne HR. Lipid products of PI(3)Ks maintain persistent cell polarity and directed motility in neutrophils. Nat Cell Biol. 2002;4:513–518. doi: 10.1038/ncb810. [DOI] [PubMed] [Google Scholar]
- 11.Czech MP. Dynamics of phosphoinositides in membrane retrieval and insertion. Annu Rev Physiol. 2003;65:791–815. doi: 10.1146/annurev.physiol.65.092101.142522. [DOI] [PubMed] [Google Scholar]
- 12.DiNitto JP, Cronin TC, Lambright DG. Membrane recognition and targeting by lipid-binding domains. Sci STKE. 2003:re16. doi: 10.1126/stke.2132003re16. [DOI] [PubMed] [Google Scholar]
- 13.Corbin JA, Dirkx RA, Falke JJ. GRP1 pleckstrin homology domain: activation parameters and novel search mechanism for rare target lipid. Biochemistry. 2004;43:16161–16173. doi: 10.1021/bi049017a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hennessy BT, Smith DL, Ram PT, Lu Y, Mills GB. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat Rev Drug Discov. 2005;4:988–1004. doi: 10.1038/nrd1902. [DOI] [PubMed] [Google Scholar]
- 15.Hurley JH. Membrane binding domains. Biochim Biophys Acta. 2006;1761:805–811. doi: 10.1016/j.bbalip.2006.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fayard E, Xue G, Parcellier A, Bozulic L, Hemmings BA. Protein kinase B (PKB/Akt), a key mediator of the PI3K signaling pathway. Curr Top Microbiol Immunol. 2010;346:31–56. doi: 10.1007/82_2010_58. [DOI] [PubMed] [Google Scholar]
- 17.Finn RD, Mistry J, Tate J, Coggill P, Heger A, Pollington JE, Gavin OL, Gunasekaran P, Ceric G, Forslund K, Holm L, Sonnhammer EL, Eddy SR, Bateman A. The Pfam protein families database. Nucleic Acids Res. 2010;38:D211–222. doi: 10.1093/nar/gkp985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lietzke SE, Bose S, Cronin T, Klarlund J, Chawla A, Czech MP, Lambright DG. Structural basis of 3-phosphoinositide recognition by pleckstrin homology domains. Mol Cell. 2000;6:385–394. doi: 10.1016/s1097-2765(00)00038-1. [DOI] [PubMed] [Google Scholar]
- 19.Park WS, Heo WD, Whalen JH, O’Rourke NA, Bryan HM, Meyer T, Teruel MN. Comprehensive identification of PIP3-regulated PH domains from C. elegans to H. sapiens by model prediction and live imaging. Mol Cell. 2008;30:381–392. doi: 10.1016/j.molcel.2008.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Calleja V, Laguerre M, Larijani B. 3-D structure and dynamics of protein kinase B-new mechanism for the allosteric regulation of an AGC kinase. J Chem Biol. 2009;2:11–25. doi: 10.1007/s12154-009-0016-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Calleja V, Alcor D, Laguerre M, Park J, Vojnovic B, Hemmings BA, Downward J, Parker PJ, Larijani B. Intramolecular and intermolecular interactions of protein kinase B define its activation in vivo. PLoS Biol. 2007;5:e95. doi: 10.1371/journal.pbio.0050095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Okuzumi T, Fiedler D, Zhang C, Gray DC, Aizenstein B, Hoffman R, Shokat KM. Inhibitor hijacking of Akt activation. Nat Chem Biol. 2009;5:484–493. doi: 10.1038/nchembio.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu WI, Voegtli WC, Sturgis HL, Dizon FP, Vigers GP, Brandhuber BJ. Crystal structure of human AKT1 with an allosteric inhibitor reveals a new mode of kinase inhibition. PLoS One. 2010;5:e12913. doi: 10.1371/journal.pone.0012913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jackson TR, Kearns BG, Theibert AB. Cytohesins and Centaurins: Mediators of PI 3-Kinase Regulated Arf Signaling. Trends Biochem Sci. 2000;25:489–495. doi: 10.1016/s0968-0004(00)01644-3. [DOI] [PubMed] [Google Scholar]
- 25.Cohen LA, Honda A, Varnai P, Brown FD, Balla T, Donaldson JG. Active Arf6 Recruits ARNO/Cytohesin GEFs to the PM by Binding Their PH Domains. Mol Biol Cell. 2007;18:2244–2253. doi: 10.1091/mbc.E06-11-0998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ferguson KM, Kavran JM, Sankaran VG, Fournier E, Isakoff SJ, Skolnik EY, Lemmon MA. Structural basis for discrimination of 3-phosphoinositides by pleckstrin homology domains. Mol Cell. 2000;6:373–384. doi: 10.1016/s1097-2765(00)00037-x. [DOI] [PubMed] [Google Scholar]
- 27.Thomas CC, Deak M, Alessi DR, van Aalten DM. High-resolution structure of the pleckstrin homology domain of protein kinase b/akt bound to phosphatidylinositol (3,4,5)-trisphosphate. Curr Biol. 2002;12:1256–1262. doi: 10.1016/s0960-9822(02)00972-7. [DOI] [PubMed] [Google Scholar]
- 28.Saxena A, Morozov P, Frank D, Musalo R, Lemmon MA, Skolnik EY, Tycko B. Phosphoinositide binding by the pleckstrin homology domains of Ipl and Tih1. J Biol Chem. 2002;277:49935–49944. doi: 10.1074/jbc.M206497200. [DOI] [PubMed] [Google Scholar]
- 29.Allen V, Swigart P, Cheung R, Cockcroft S, Katan M. Regulation of inositol lipid-specific phospholipase cdelta by changes in Ca2+ ion concentrations. Biochem J. 1997;327 (Pt 2):545–552. doi: 10.1042/bj3270545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van der Wal J, Habets R, Varnai P, Balla T, Jalink K. Monitoring agonist-induced phospholipase C activation in live cells by fluorescence resonance energy transfer. J Biol Chem. 2001;276:15337–15344. doi: 10.1074/jbc.M007194200. [DOI] [PubMed] [Google Scholar]
- 31.Falke JJ. Membrane Recruitment as a Cancer Mechanism: A Case Study of Akt PH Domain. Cell science. 2007;4:25–30. [PMC free article] [PubMed] [Google Scholar]
- 32.Garcia P, Gupta R, Shah S, Morris AJ, Rudge SA, Scarlata S, Petrova V, McLaughlin S, Rebecchi MJ. The pleckstrin homology domain of phospholipase C-delta 1 binds with high affinity to phosphatidylinositol 4,5-bisphosphate in bilayer membranes. Biochemistry. 1995;34:16228–16234. doi: 10.1021/bi00049a039. [DOI] [PubMed] [Google Scholar]
- 33.Singh SM, Murray D. Molecular modeling of the membrane targeting of phospholipase C pleckstrin homology domains. Protein Sci. 2003;12:1934–1953. doi: 10.1110/ps.0358803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Knight JD, Falke JJ. Single-molecule fluorescence studies of a PH domain: new insights into the membrane docking reaction. Biophys J. 2009;96:566–582. doi: 10.1016/j.bpj.2008.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Toker A, Cantley LC. Signalling through the lipid products of phosphoinositide-3-OH kinase. Nature. 1997;387:673–676. doi: 10.1038/42648. [DOI] [PubMed] [Google Scholar]
- 36.Baraldi E, Djinovic Carugo K, Hyvonen M, Surdo PL, Riley AM, Potter BV, O’Brien R, Ladbury JE, Saraste M. Structure of the PH domain from Bruton’s tyrosine kinase in complex with inositol 1,3,4,5-tetrakisphosphate. Structure. 1999;7:449–460. doi: 10.1016/s0969-2126(99)80057-4. [DOI] [PubMed] [Google Scholar]
- 37.Cronin TC, DiNitto JP, Czech MP, Lambright DG. Structural determinants of phosphoinositide selectivity in splice variants of Grp1 family PH domains. EMBO J. 2004;23:3711–3720. doi: 10.1038/sj.emboj.7600388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tong Y, Tempel W, Wang H, Yamada K, Shen L, Senisterra GA, MacKenzie F, Chishti AH, Park HW. Phosphorylation-independent dual-site binding of the FHA domain of KIF13 mediates phosphoinositide transport via centaurin alpha1. Proc Nat Acad Sci USA. 2010;107:20346–20351. doi: 10.1073/pnas.1009008107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Komander D, Fairservice A, Deak M, Kular GS, Prescott AR, Peter Downes C, Safrany ST, Alessi DR, van Aalten DM. Structural insights into the regulation of PDK1 by phosphoinositides and inositol phosphates. EMBO J. 2004;23:3918–3928. doi: 10.1038/sj.emboj.7600379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.He J, Haney RM, Vora M, Verkhusha VV, Stahelin RV, Kutateladze TG. Molecular mechanism of membrane targeting by the GRP1 PH domain. J Lipid Res. 2008;49:1807–1815. doi: 10.1194/jlr.M800150-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Malanga D, Scrima M, De Marco C, Fabiani F, De Rosa N, De Gisi S, Malara N, Savino R, Rocco G, Chiappetta G, Franco R, Tirino V, Pirozzi G, Viglietto G. Activating E17K mutation in the gene encoding the protein kinase AKT1 in a subset of squamous cell carcinoma of the lung. Cell Cycle. 2008;7:665–669. doi: 10.4161/cc.7.5.5485. [DOI] [PubMed] [Google Scholar]
- 42.Dannemann N, Hart JR, Ueno L, Vogt PK. Phosphatidylinositol 4,5-bisphosphate-specific AKT1 is oncogenic. Int J Cancer. 2010;127:239–244. doi: 10.1002/ijc.25012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li T, Tsukada S, Satterthwaite A, Havlik MH, Park H, Takatsu K, Witte ON. Activation of Bruton’s tyrosine kinase (BTK) by a point mutation in its pleckstrin homology (PH) domain. Immunity. 1995;2:451–460. doi: 10.1016/1074-7613(95)90026-8. [DOI] [PubMed] [Google Scholar]
- 44.Varnai P, Rother KI, Balla T. Phosphatidylinositol 3-kinase-dependent membrane association of the Bruton’s tyrosine kinase pleckstrin homology domain visualized in single living cells. J Biol Chem. 1999;274:10983–10989. doi: 10.1074/jbc.274.16.10983. [DOI] [PubMed] [Google Scholar]
- 45.Varnai P, Balla T. Visualization of phosphoinositides that bind pleckstrin homology domains: calcium- and agonist-induced dynamic changes and relationship to myo-[3H]inositol-labeled phosphoinositide pools. J Cell Biol. 1998;143:501–510. doi: 10.1083/jcb.143.2.501. [DOI] [PMC free article] [PubMed] [Google Scholar]