

Abstract
Sulfoglucuronosyl paragloboside (SGPG), a minor glycosphingolipid (GSL) of endothelial cells, is a ligand for L-selectin and has been implicated in neuro-inflammatory diseases, such as Guillian-Barré syndrome. Inflammatory cytokines, such as TNFα and IL-1β, up-regulate SGPG expression by stimulating gene expression for glucuronosyltransferases, both P and S forms (GlcATp and GlcATs), and the HNK-1 sulfotransferase (HNK-1 ST). Transfection of a human cerebromicrovascular endothelial cell (SV-HCEC) line with HNK-1 ST siRNA down-regulated SGPG expression, inhibited cytokine-stimulated T cell adhesion, and offered protection against apoptosis. However, the precise mechanisms of SGPG elevation in endothelial cell death (apoptosis) and the maintenance of blood-brain or blood-nerve barrier (BBB or BNB) integrity in inflammation have not been elucidated. Blocking SGPG expression inhibited cytokine-mediated stimulation of NF-κB activity but stimulated MAP kinase (ERK) activity. Furthermore, elevation of SGPG by over-expression of GlcATp and GlcATs triggered endothelial cell apoptosis, with GlcATs being more potent than GlcATp. While SGPG-mediated endothelial cell apoptosis was preceded by inhibiting the intracellular NF-κB activity, interfering with Akt and ERK activation and stimulating caspase 3 in SV-HCECs, HNK-1ST siRNA transfection also interfered with IKB phosphorylation but stimulated ERK activation. Our data indicate that SGPG is a critical regulatory molecule for maintaining endothelial cell survival and BBB/BNB barrier function.
Keywords: Apoptosis, Cell signaling, Endothelial cells, Glycosphingolipid, Inflammation, Sulfoglucuronosyl paragloboside
Introduction
Glycosphingolipids (GSLs) are important constituents of the plasma membrane and are involved in regulating a variety of cellular functions (Dasgupta et al. 2007, Hakomori 2008, Kanda et al. 1995). A large number of glycoproteins, such as neural cell adhesion molecules (NCAMs) (Ong et al. 2002), L1, myelin-associated glycoprotein (MAG) (Kruse et al. 1985), tenascin-C, tenascin-R, and tissue plasminogen activator (Voshol et al. 1996), contain the human natural killer antigen (HNK-1) epitope, a carbohydrate antigen that modulates neurite outgrowth (Martini et al. 1992), cell adhesion, and synaptic plasticity (Dityatev & Schachner 2003). The minimal structural components of the HNK-1 epitope have been shown to consist of a sulfated disaccharide residue, 3-sulfoglucuronosyl (β1–3) galactosyl (β1-) (Tokuda et al. 1998). The HNK-1 epitope is also shared by two glucuronosyl glycosphingolipids (SGGLs), sulfated glucuronosyl paragloboside (SGPG) and sulfated glucuronosyl lactosaminyl paragloboside (SGLPG), whose structures were established independently by us and Jungalwala’s group (Chou et al. 1986, Ariga et al. 1987). The structures of these two SGGLs are represented as follows: SGPG, SO4-3GlcA(β1–3)Gal(β1-4)GlcNAc(β1–3)Gal(β1–4)Glc(β1–1) ceramide; and SGLPG, SO4-3GlcA(β1–3)Gal(β1–4)GlcNAc(β1–3) Gal(β1–4)GlcNAc(β1–3)Gal(β1–4)Glc(β1–1) ceramide. Both SGGLs are minor components of the total GSLs of central and peripheral nervous systems (CNS and PNS), with SGPG being the major component of the two (Ariga et al. 1987, Ariga & Yu 1987, Chou et al. 1986). In addition to their known biological functions in nervous system development, they are also involved as autoantigens in autoimmune peripheral neuropathies such as Guillian-Barré syndrome (GBS); however, their precise pathogenic roles in disease development have not yet been fully evaluated.
Previous studies from our laboratory and that of others have shown that GBS, is likely triggered by infection by Gram-negative bacteria, such as Campylobacter jejuni, through a molecular mimicry mechanism in which anti-glycolipid antibodies are generated against an oligosaccharide portion of the bacterial lipooligosaccharide coat (Yu et al. 2006). Clinical symptoms develop by two principal pathogenic mechanisms: a) the autoantibodies, in the present context, antibodies against SGPG, must enter from the circulation into the nerve parenchyma to cause neurodegeneration by an antibody-mediated and complement-dependent mechanism (Maeda et al. 1991a, Maeda et al. 1991b, Kohriyama et al. 1988, Kaida et al. 2009, Kohriyama et al. 1987), and b) by a cell-mediated process that entails the penetration of inflammatory T cells, elicited by bacterial infection, to enter into the nerve tissues (Ariga & Yu 1987, Dasgupta et al. 2007, Kanda et al. 1995, Ariga et al. 1987, Kohriyama et al. 1988). In either case, the blood-brain and blood-nerve barrier (BBB/BNB) function is compromised to allow immunoglobulins or immune cells to penetrate the nerve parenchyma to attack the nerve tissues ((Yu et al. 2006, Kohriyama et al. 1987). At present, although the precise etiology of disease onset is still not fully understood (Geleijns et al. 2005, Compston & Coles 2008), the detection of a large concentration of inflammatory cytokines, presence of lymphocytes in nervous tissues, and an elevated concentrations of autoantibodies in the patient serum and body fluid is a hallmark of GBS.
Our previous studies suggest that two inflammatory cytokines, TNFα and IL-1β, presumably elicited by bacterial infection, up-regulate SGPG expression in bovine brain endothelial cells (BMECs) and in human cerebromicrovascular endothelial cells (SV-HCECs). These cytokines promote CD4+ cell adhesion to endothelial cells with SGPG serving as a ligand for L-selectin (Dasgupta et al. 2009, Dasgupta et al. 2007) expressed on T cells. Subsequent studies from our laboratory revealed that both TNFα and IL-1β stimulated glucuronosyl-transferase genes, both the P and S forms, designated as GlcATp and GlcATs, respectively, and as such up-regulation was mediated via stimulation of NF-κB activity. Inhibition of HNK-1ST gene expression, using HNK-1 sulfotransferase siRNA or HNK-1STsiRNA, down-regulates NF-κB activity and, consequently, blocks cytokine-mediated SGPG elevation and T cell adhesion (Dasgupta et al. 2009) and penetration through the tight junction. The adhesion ultimately leads to the penetration of lymphocytes and other active agents into the brain and nerve compartments, which triggers phagocytosis, demyelination, and axonal degeneration. The regulation of cytokine-mediated changes in endothelial cells and the mechanism of penetration of a large number of T cells through the tight-gap junction is a subject of considerable scientific interest.
Although these studies indicate that SGPG is a direct participant in inflammatory processes as an adherent for T cells, the mechanism of action of SGPG up- and down-regulation in endothelial cell function, and as an active signaling mediator in endothelial cell death has not yet been examined. To further delineate the role of SGPG in regulation of endothelial cell function under inflammation and its precise mechanism, we have examined the cell signaling pathways of SGPG after inhibition SGPG expression by transfecting the cells with HNK-1ST gene siRNA (loss of function) or by promoting SGPG synthesis by overexpressing EGFP-GlcATp and EGFP-GlcATs (gain in function). Using dual luciferase assay, Western blot analysis, and immuno-fluorescence, we found that both up- and down-regulation of SGPG reduced NF-κB activity, which is mediated by IKB accumulation. Our data suggest that cellular SGPG and its expression is a critical factor for endothelial cell survival and involved in regulating the function of BBB/BNB (Armulik et al. 2010).
Materials and Methods
Human TNFα and IL-1β were purchased from PeproTech (Rocky Hill, NJ), siRNA HNK-1ST SMARTpool was purchased from Dharmacon Inc. (Thermo Fisher Scientific, Lafayette, CO), with the following primer sequences: Duplex 5: sense, GCU GAU UGU UCU AAA UGG AUU and anti-sense, 5′-P UCC AUU UAG AAC AAU CAG CUU; Duplex 6: sense, GUA AGA GAU CCC UUC GAA AUU and anti-sense: 5′-P UUU CGA AGG GAU CUC UUA CUU; Duplex 7: sense, UGA CAA CCA UGC CGG AGG UUU and anti-sense, 5′-P ACU UCC GGC AUG GUU GUC AUU; Duplex 8: sense, CUA GCA AGU UCA UCA CGU UUU and anti-sense, 5′-P AAC GUG AUG AAC UUG CUA GUU. A scrambled siRNA SMARTpool was used to compare the transfection performance and validity of the results. The siRNA sample was dissolved in 1 x siRNA buffer (0.5 ml ≡ 20 μM) and preserved into 10 tubes (50 μl each). The dual luciferase reporter assay system was purchased from Promega (Madison, WI). Cell culture media and growth factors were purchased from Invitrogen (Carlsbad, CA). Purified SGPG and its monoclonal antibody (NGR 50, mouse IgG) were received as generous gifts from Dr. Toshio Ariga, Institute of Molecular Medicine and Genetics, Georgia Health Sciences University, Augusta, GA. Antibodies against IKB, ERK, and caspase 3 were purchased from Cell Signaling Technology (Danvers, MA). In situ cell detection kit was purchased from Roche Applied Science (Indianapolis, IN). Heparin (Na-salt) was obtained from Sigma Chemical (St. Louis, MO). All reagents, buffers, and chemicals were of analytical grades.
Cell culture
Endothelial cells of human cerebro-microvascular origin (SV-HCECs) (Muruganandam et al. 1997) which were generously supplied by Dr. D. Stanimirovic, National Research Council of Canada, Ottawa, Canada, were grown in 0.5% gelatin-coated dish in media (Media 199, Invitrogen) containing insulin-transferrin- selenium, heparin, and penicillin-streptomycin (Cellgro) as described previously (Dasgupta et al. 2007, Muruganandam et al. 1997, Duvar et al. 2000).
Inhibition of HNK-1ST gene expression by HNK-1ST siRNA transfection
Cells were transfected in Amaxa Nucleofector equipment using T20 Program in an aseptic condition as described previously (Dasgupta et al. 2009, Dasgupta et al. 2007). Briefly, cells were grown in 3 × 100-mm dishes, collected by trypsinization, counted, and divided into 3 groups (1.0 × 106 cells per group). Cells were then suspended in 0.1 ml of tranfecting media in the presence of HNK-1ST siRNA (7.5 μl ≡ 150 pmol). Controls were prepared simultaneously using a scrambled siRNA mixture for comparison. The mixture containing the cell suspension was transferred into a sterile cuvette (2-mm gap) and zapped using the T20 Program. Five hundred μl of the pre-warmed medium (complete) was added immediately, and the cells were aspirated carefully. The transfected cells were dispersed in 3 × 60-mm dish, pre-coated with 0.5% gelatin. The media were changed after 4–6 h of transfection, and cells were incubated for 48 h before being exposed to the inflammatory cytokines. After incubation for another 18–24 h, cells were washed with cold PBS, and total proteins were dissolved in Lamelli buffer for examining cell signaling. Protein content was measured using RcDc reagents (Bio-Rad, Hercules, CA). A time-dependent ERK activation was studied using IL-1β (25 ng/ml) and TNFα (100 ng/ml) at 0 h, 2 h, 4 h, 8 h, 12 h and 24 h. Our data indicate that both IL-1β and TNFα stimulated ERK optimally at 24 h. Hence we have decided to expose the cells between 18–24 h.
Identification of signaling molecules by Western blot analysis
A defined amount of the protein (20–30 μg) was applied on a gradient polyacrylamide gel (Bio-Rad, Hercules, CA) and subjected to electrophoresis. The protein bands were transferred onto a PVDF membrane and visualized by Ponceau staining. The following specified proteins were identified using Western blot analysis, phospho- and total IKB (for NF-κB activity), phospho- and total ERK, phospho- and total Akt, and active caspase 3 (for apoptosis). Protein loading was normalized using α-tubulin or β-actin. Bands were identified using a chemilumniscence reagent, ECL (GE Health Care, Buckinghamshire, UK). In addition, we have also examined activation of two other MAP kinases, phospho-JUN and phospho-P38 for comparison.
Construction of EGFP-GlcATp and EGFP-GlcATs plasmids and transfection of SV-HCECs with EGFP cDNA-plasmid
For the construction of pcDNA3.1-GlcATp, and GlcATs, human GlcATp and GlcATs cDNA was amplified using following the primers: for GlcATp: sense, 5′-AA CTC GAG ATG CCG AAG AGA CGG GAC ATC CTA G -3′ (Xho-1 site) and antisense, 5′-AA AAG CTT GAT CTC CAC CGA GGG GTC AGT G-3′ (Hind III site), and for GlcATs: sense: 5′-AAA AGC TTT ACC TCA ATT TTC AGT GTG T-3′ (Xho-1 site) antisense: 5′-AAC TCG AGA TGA AGT CCG CGC TTT TCA C-3′ (Hind III site). The EGFP-GlcATp/-GlcATs vector was obtained by ligation of an Xho-1/Hind III fragment from pcDNA3.1-GlcATp/GlcATs into EGFP-N1. Transfection was performed using electroporation as described previously (≡1 μg of plasmid/1 × 106 cells). A group of cells was transfected with an equivalent amount of combined EGFP-GlcATp and EGFP- GlcATs plasmid (0.5 μg each). The rate of success of the procedure was observed by the expression of GFP in transfected cells after 24 h of incubation.
NF-κB activity assay
Along with the EGFP-GlcATp and EGFP-GlcATs plasmids, cells were co-transfected using 0.8 μg of pNF-κB luciferase, a multimerized κB-luciferase reporter gene plasmid, and 0.4 μg of pRL-CMV (Renilla luciferase) internal control plasmid to normalize the efficacy of the transfection procedure. After 24 h of incubation, cell lysate was prepared and the level of luciferase activity was determined using the dual luciferase reporter system in accordance with the instruction of the manufacturer (Promega, Madison, WI).
Immnuno-overlay analysis of the SGPG concentration in transfected cells
Since SGPG is a minor component of the total GSLs in SV-HCEC (Muruganandam et al. 1997, Duvar et al. 2000), we cultured the control (EGFP-transfection) and transfected cells separately in 150-mm dishes to obtain a sufficient number of cells for SGPG analysis by an immuno-overlay method. Control (EGFP-transfected) cells and EGFP-GlcATp/-GlcATs-transfected cells were grown for 48 h as described. Cells were then collected, washed with cold PBS, and preserved at −20°C before use. Lipids were extracted from cells using solvent mixtures (chloroform:methanol:water 2:4:1, v/v; followed by chloroform:methanol 2:1, v/v), and the SGPG fraction was purified from the lipid extract using DEAE A-25 (acetate form) as described previously (Dasgupta et al. 2007). Each purified fraction was dissolved in a defined volume of solvent (determined by the protein content) and an equal volume of the samples was applied to an HPTLC with standard SGPG. The plate was developed using the solvent system of chloroform:methanol:0.25% CaCl2 (55:45:10; v/v), coated, and exposed to MAb NGR 50 (specific for SGPG), followed by a mouse peroxidase conjugated-secondary antibody. After washing with PBS, a chemiluminescence reagent, ECL, was added to the plate, and the bands were revealed by exposing to an X-ray film (Dasgupta et al. 2007).
Fluorochrome inhibitor of caspases assay (FLICA)
Using FLICA, we then determined cell death induced by SGPG expression. Staining of active caspases using the FLICA assay was performed with EGFP-GlcAT-plasmids-transfected SV-HCECs using sulforhodamine-labeled fluoromethyl ketone peptide inhibitor (red) according to the manufacturer’s instructions (Immunochemistry Technologies). Cells were transfected with the respective plasmid, grown on a cover-slip for 24–48 h, and then stained for active caspases using FLICA. Briefly, the FLICA reagent was added to the medium and the cells incubated for 1 h at 37°C under 5% CO2. The cells were washed once with washing solution and then fixed with 4% p-formaldehyde (PFA) in phosphate-buffered saline (PBS) for further analysis.
Immunocytochemical localization of SGPG and caspase 3
To verify the effect of GlcATp/GlcATs transfection on SGPG expression, we probed immunocytochemically the localization of SGPG in control and GlcATp/GlcATs-transfected cells (Dasgupta et al. 2007). Cells were cultured on cover slips and grown 24–48 h, washed with 1 × Hank’s balanced salt solution, and fixed using 4% PFA. The fixed cells were permeabilized and then treated with MAb NGR 50 and caspase 3 antibody, followed by an appropriate secondary antibody (anti-mouse IgG or anti-rabbit IgG) conjugated with cy3 or cy5. Cells were further stained with Hoechst 33258 (nuclear stain) and visualized under a confocal microscope.
Statistical evaluation
Data are expressed as means ± standard deviations (SD) from 3–5 independent experiments. Statistical significance was determined using Student’s t-test for comparison between two means and by two way ANOVA in MS EXCEL v2007.
Results
Inhibition of the HNK-1ST gene reduced NF-κB activity by inhibiting IκB activation in SV-HCECs after cytokine stimulation but promoted ERK activation
We previously showed that transfection of cells with HNK-1ST siRNA reduced HNK-1ST gene expression and inhibited SGPG up-regulation by suppressing cytokine-stimulated NF-κB activity. To evaluate that mechanism, we examined the protein level of IKB and its phosphorylation level. The level of IKB, an inhibitor protein of NF-κB, is controlled by its phosphorylation. Phosphorylated IKB is released and degraded, thus and activating NF-κB (Hayden & Ghosh 2004). Consistent with our previous report (Dasgupta et al. 2009), the IKB protein level was increased by the down-regulation of SGPG through HNK-1ST siRNA and, at the same time the IKB phosphorylation was decreased, with or without cytokine treatment (Figure 1). It is noteworthy that the time course activation of ERK by IL-1β and TNFα showed an identical profile with optimum activation at 24 h. Hence our study measuring ERK activation using cytokine exposure after siHNK-1 transfection, is in compliance with our time-course study. A representative figure is shown in Figure 2.
Figure 1. Activation of ERK and inhibition of IKB and caspase 3 by HNK-1 siRNA transfection.
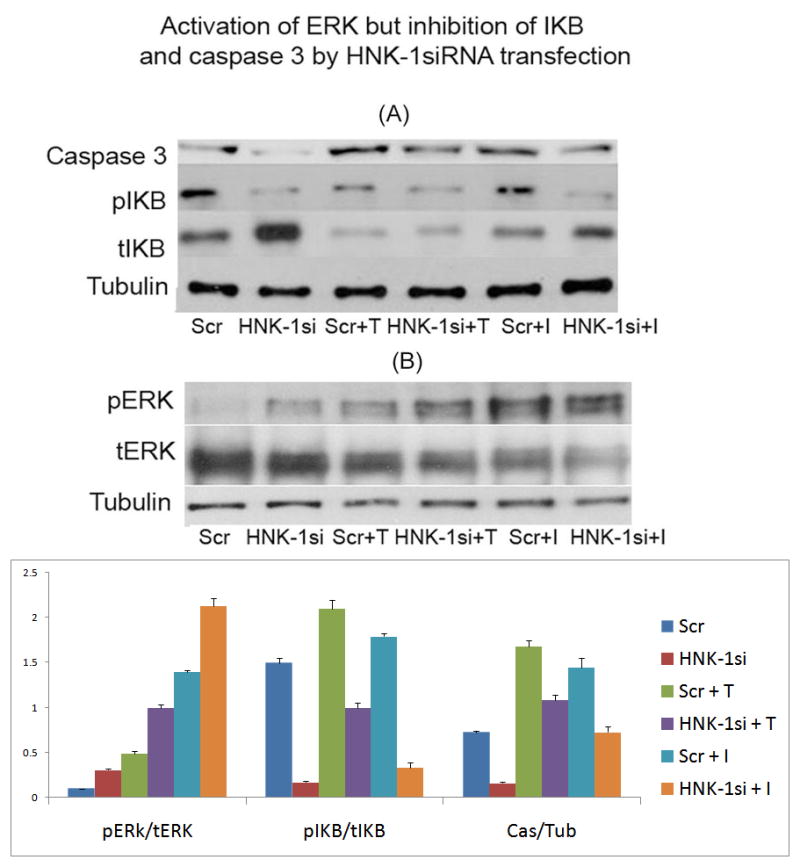
(A) Inhibition of caspase 3 and IKB, (B) Activation of ERK. Cells were exposed to cytokines overnight (between 18–24 h) and protein was dissolved using Lemmelli buffer. The protein bands were separated and identified using specified antibody.
Scr, Scrambled siRNA transfection; HNK-1si, HNK-1ST siRNA transfection; Scr + T, Scrambled siRNA + TNFα; HNK-1 si + T, HNK-1ST siRNA + TNFα; Scr + I, Scrambled siRNA + IL-1β; HNK-1 si + I, HNK-1ST siRNA + IL-1β.
Lower figure: Each band (area) was scanned and quantified using the ImageJ program. The ratio of two band area such as pERk/tERK, pIKB/tIKB, and caspase 3/tubulin was determined and plotted. Bar represents SD from at least 3 independent experiments, Comparison was made by T-test analysis between each set, Scr vs. HNK-1si, p < 0.005; Scr + T vs. HNK-1si + T, p < 0.0005; Scr + I vs. HNK-1si + I, p < 0.0005. Comparison between groups using two-way ANOVA indicates the significance with a p value of 1.59×10−10.
Figure 2. Time-course study of ERK activation in SV-HCECs by cytokine.
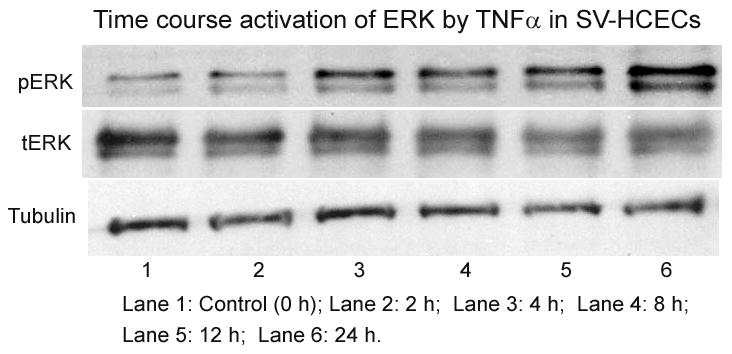
Cells were incubated with and TNFα (100 ng/ml) at different time period from 0 h (control) to 24 h. Cells were collected and protein was dissolved in Lemmili’s buffer. ERK activation was measured using Western blot as described in the text.
It is noteworthy that HNK-1ST down-regulation by siRNA rendered the cells more resistant to apoptosis, as measured by reduced caspase 3 activation (Figure 1) and less TUNEL staining (Figure 3). Because of the loose attachment of cells on the slide after HNK-1ST siRNA transfection, cytokine exposure time was reduced to 8 h (instead of 24 h). The exposed cells were fixed with PFA, and then subjected to TUNEL staining. Approximately 15% cell death was recorded with IL-1β treatment and more than 20–25% cell death after exposure to TNFα. To investigate the cell survival signaling pathway, we assayed for Akt and ERK (MAPK) activation. Our data indicated that ERK activation (Figure 1) could be the factor that protects the cells, leading to enhanced cell survival by SGPG inhibition. Akt and phosphorylated Akt were not affected, however (data not shown). Moreover, we have also measured the activation of two other kinases, JUN and P38 that were not activated by siHNK-1ST transfection. However, P38 kinase showed a mild activation (only 60–80% stimulation) with GATp/GATs transfection (result not shown).
Figure 3. Protection from IL-1β- and TNFα-mediated cell death (TUNEL) by HNK-1ST siRNA transfection.
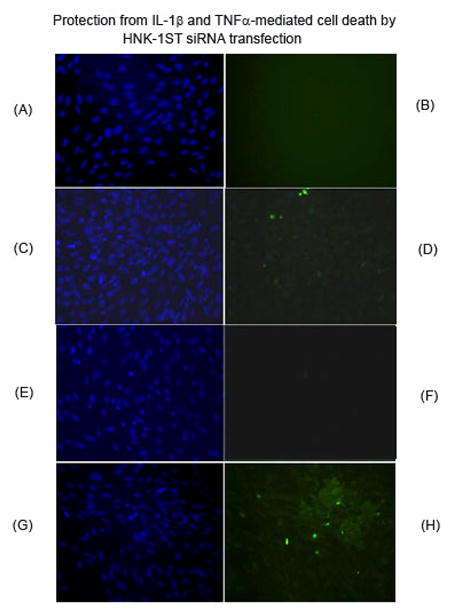
Cells were exposed to IL-1β (A–D) and TNFα (E–H) after scrambled and HNK-1ST siRNA transfection. A, C, E, and G present Hoechst staining; B, D, F, and H are TUNEL assays. A, B, E, and F are HNK-1ST siRNA transfection; C, D, G, and H represent scrambled siRNA transfection. Cell death was observed in scrambled transfection after cytokine exposure (D and H).
GlcATp/GlcATs-transfection stimulated SGPG expression
To gain further insight into the precise role of SGPG in endothelial cell functions, we performed a gain-of-function experiment by upregulating the SGPG expression level by inflammatory cytokines. cDNAs of GlcATp and GlcATs were cloned, and the cells were transfected with EGFP-GlcATp, EGFP-GlcATs, and the combined clones. To identify the efficacy of GlcATp and GlcATs transfection, we visualized the transfected cells under a fluorescent microscope for GFP expression and then identified GFP expression by Western blot analysis using GFP-antibody. One single protein band was detected in GFP transfection, two protein bands were detected in GlcATp/GlcATs-GFP transfection, and three protein bands were detected in the combined transfection experiment (results not shown). In addition, we measured the SGPG concentrations in all transfected cells to verify that SGPG expression was indeed stimulated. Briefly, the transfected cells were cultured for a defined period of incubation (24–48 h), and the cells were collected after mild trypsinization. SGPG concentration was measured using the purified lipid fraction in the lipid extract employing MAb NGR50 in a TLC-immuno-overlay method (Dasgupta et al. 2007). We found that the level of SGPG was enhanced by GlcATp and GlcATs transfection, and the efficacy of up-regulation was GlcATp + GlcATs > GlcATs > GlcATp, corresponding to a 20-, 12-, and 8-fold increase (Figure 4), respectively, in SGPG concentration compared to the control (EGFP).
Figure 4. TLC-immunooverlay of SGPG isolated from SV-HCECs after EGFP, EGFP-GlcATp, EGFP-GlcATs and EGFP-GlcATp + Ts transfection.
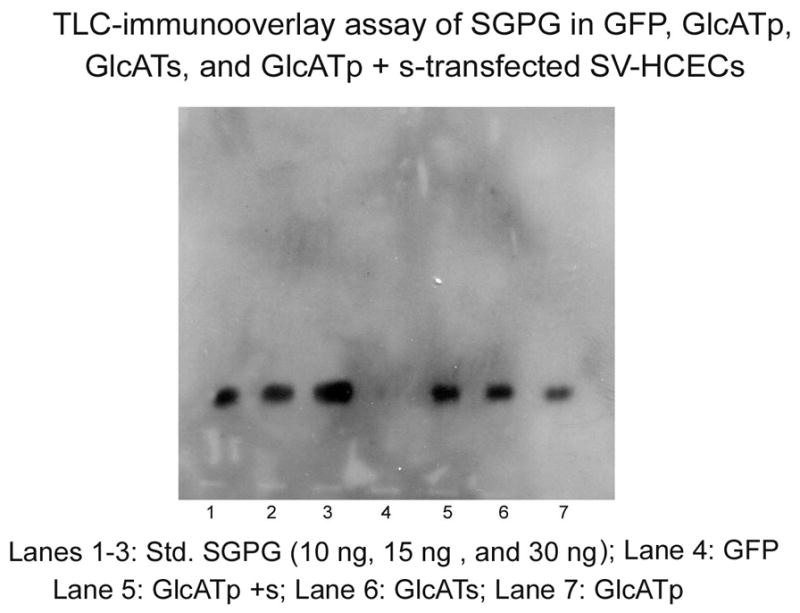
The transfected cells were incubated for 48 h. Lipids were extracted using solvent mixtures and SGPG was purified into a fraction using DEAE-Sephadex A25 column. The fraction was dissolved in a defined volume of solvent (chloroform:methanol:water 12:7:1, v/v) according to protein concentration, and a portion of the solution equivalent to an equal amount of protein was applied on an aluminum-backed HPLTC along with a reference standards. The plate was developed in chloroform;methanol:0.25% CaCl2 (50:45:10 v/v). After the coating with a isobutylmethacrate solution in hexane, the plate was incubated with the mAb NGR50 followed by a secondary HRP-conjugated anti-mouse IgG. The bands were identified using ECL. Band from each lane was scanned and quantitated using the ImageJ program.
GlcATp/GlcATs-transfection reduced NF-κB activity by inhibiting IKB activation
To determine the effect of SGPG expression on NF-κB activity in the transfected cells, we used the luciferase assay. The empty vector (EGFP-N1) was used as a control. To our surprise, the GlcATp and GlcATs transfected cells showed reduced NF-κB activity compared to that of the control cells (Figure 5), and that reduction is further supported by the inhibition of IKB activation (Figure 6A) as shown by Western blot analysis. This observation was unexpected, since SGPG inhibition also reduced NF-κB activity in HNK-1ST siRNA transfected cells, which down-regulated SGPG expression after cytokine stimulation (Dasgupta et al. 2009).
Figure 5. Assay of NF-κB activity in Vector (GFP), GlcATp, and GlcATs transfected endothelial cells.
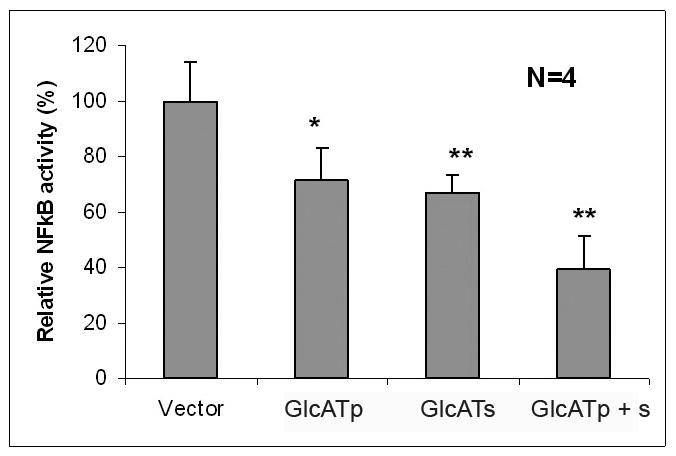
The procedure is described in detail in the text. Vector, green fluorescent protein; GlcATp, GlcATp transfection; GlcATs, GlcATs transfection, GlcATp + s, combined transfection of GlcATp and GlcATs. Bar represents SD from four independent determinations. * p < 0.05; ** p < 0.005
Comparison was made between vector vs. GlcATp, p < 0.05; vector vs. GlcATs, p < 0.005; vector vs. GlcATp + s, p < 0.005.
Figure 6.

(A) Caspase 3 activation and inhibition of IKB phosphorylated by SGPG in SV-HCECs; (B) Inhibition of ERK and Akt phosphorylation by SGPG in SV-HCECs. GFP, Green fluorescent protein; GlcATp, GlcATp transfection; GlcATs, GlcATs transfection, GlcATp + s, Combined transfection of GlcATp and GlcATs.
Lower figure: Each band (area) was scanned and quantified using ImageJ program. The ratio of two band areas such as caspase 3/tubulin, pIKB/tIKB, pERk/tERK, and pAKt/tAkt was determined and plotted as the graph. Bar represents SD from four independent assays. *** p < 0.0005. Comparison was made between GFP vs. GlcATp; GFP vs. GlcATs; and, GFP vs. GlcATp + Ts; in all cases p < 0.0005. Note: The expression of caspase 3/tubulin in GFP transfected cells was negligible.
GlcATp/GlcATs-transfection promoted cell apoptosis
Since NF-κB is involved in cell survival pathways, we next examined the apoptosis level in the GlcATp- and GlcATs-transfected cells by measuring caspase 3 activity. Figure 6A shows that caspase 3 was activated in GlcATp- and GlcATs-tranfected cells, for which GlcATs exhibited a stronger effect than GlcATp. The double transfection (GlcATp + GlcATs), however, shows a strong synergistic effect compared to GlcATp or GlcATs transfection alone (Figure 5A). Taken together, our data indicate that increased SGPG expression induced cell apoptosis in SV-HCECs. To understand the apoptotic mechanism relevant to cell survival signals, we determined the PI3 kinase (Akt) and MAP kinase (ERK) activation using Western blot analysis and found that the transfected cells showed a down-regulation of phospho-Akt and phospho-ERK activity (Figure 6B).
Modulating SGPG expression inhibits NF-κB activity through TNFα-receptor signaling
To further investigate SGPG regulation in inhibiting NF-κB activity, we examined TNFα-receptor (TNFR1 and TNFR2) expression after HNK-1ST gene silencing and in SGPG elevation by GlcATp/GlcATs transfected cells as these receptors are involved in regulation of NF-κB activity and are associated with both cell death and cell survival pathways, respectively (McCoy & Tansey 2008). Our data indicated that silencing SGPG expression activated TNFR2 expression, and that elevation of SGPG expression stimulated TNFR1 and reduced TNFR2 expression (Figures 7A and 7B).
Figure 7.
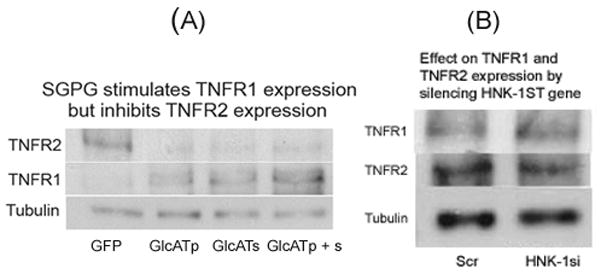
(A) SGPG stimulates TNFR1 expression but inhibits TNFR2 expression; (B) Effect on TNFR1 and TNFR2 expression by silencing HNK-1ST gene.
GFP, Green fluorescent protein; GlcATp, GlcATp transfection; GlcATs, GlcATs transfection, GlcAT p + s, Combined transfection of GlcATp and GlcATs; Scr, Scrambled siRNA transfection; HNK-1si, HNK-1ST siRNA transfection.
Further determination of cell apoptosis induced by SGPG
To further evaluate the effect of SGPG expression on cell death, we performed immunofluorescence and FLICA. Immunofluorescence of SGPG and activated caspase 3 showed that cells which expressed EGFP-GlcATp (green), EGFP-GlcATs (green), or both, had higher SGPG expression (red) and stained positive for active caspase 3 (Figure 8), which confirms our lipid measurement and Western blot analysis data. Using FLICA, we found that GlcATp and GlcATs transfection promoted cell death by 15–20% and 35–40%, respectively, while reaching approximately 50% or more cell death in cells transfected with a combination of GlcATp and GlcATs (Figure 9).
Figure 8. Immunocytochemical localization of GFP, SGPG, and caspase 3 expression in GFP, GlcATp, GlcATs and GlcATp + s-transfected SV-HCECs.
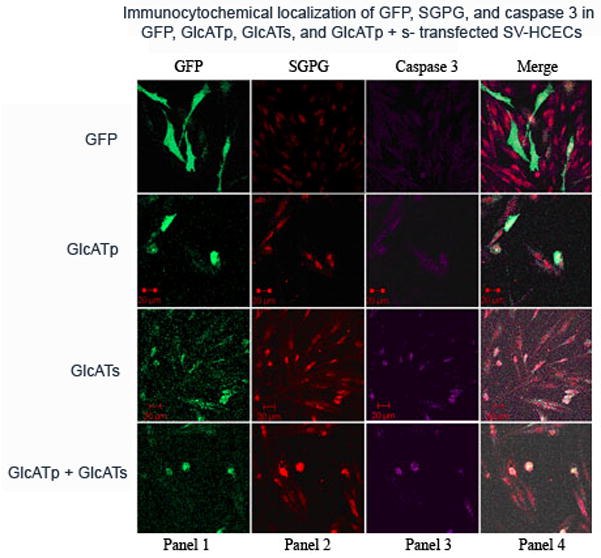
Cells were stained with MAb NGR50 (SGPG, cy3) and caspase 3 antibody (cy 5) and visualized under a confocal microscope. Panel 1: GFP expression, Panel 2: SGPG expression, Panel 3: Caspase 3 expression, Panel 4 represents the merging of panel 2 and panel 3.
GFP, Green fluorescent protein; GlcATp, GlcATp transfection; GlcATs, GlcATs transfection, GlcATp + s, combined transfection of GlcATp and GlcATs.
Figure 9. Determination of cell viability by FLICA in SV-HCECs.
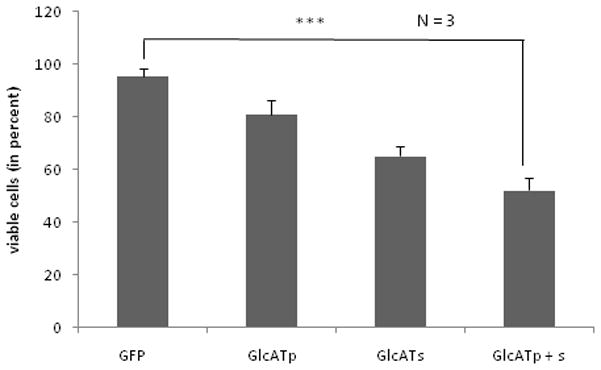
Cells were grown on coated cover-slips in a 12-well plate, stained with FLICA and Hoechst, respectively, fixed and visualized under a fluorescent microscope. FLICA-positive cells (red) were counted compared to total cells (Hoechst).
GFP, Green fluorescent protein; GlcATp, GlcATp transfection; GlcATs, GlcATs transfection, GlcATp + s, Combined transfection with GlcATp and GlcATs. Bar means the SD from a set of three independent assays. *** p < 0.0005
Comparison was made between GFP vs. GlcATp, p < 0.05; GFP vs. GlcATs, p < 0.005; and, GFP vs. GlcATp + s, p < 0.0005.
Discussion
Our previous studies indicate that SGPG expression in endothelial cells is stimulated by inflammatory cytokines promoting T cell attachment as an early inflammatory response. To further delineate the precise role of SGPG under inflammatory conditions, we have adopted a two-prong approach: (a) inhibiting SGPG expression using HNK-1ST siRNA, and (b) over-expressing SGPG in endothelial cells using GlcAT plasmids. We have demonstrated, for the first time that SGPG is a messenger for endothelial cell death and may be implicated as a player in progression of certain neuro-degenerative diseases.
Cytokine-mediated SGPG up-regulation preceded via NF-κB activation, as evidenced by phosphorylation of IKB, leading to apoptosis by stimulating caspase 3 activity and inhibiting ERK activation. Inhibition of HNK-1ST gene expression down-regulated the NF-κB activity by inhibiting IKB phosphorylation, and reducing caspase 3 activity. At the same time, the ERK survival pathway, but not Akt, was also activated (Figure 1). Therefore, we hypothesize that this combined effect offers the cell protection from undergoing apoptosis and resistance in cytokine-stimulated cell permeability changes.
By regulating the expression of SGPG (gain-of-function and loss-of function studies), we have established a novel role of SGPG in cell apoptosis. Since down-regulation for SGPG expression by HNK-1ST siRNA reduced the NF-κB activity, we predicted that over-expression of SGPG would stimulate such activity. To our surprise, we found that NF-κB activity was also inhibited in both GlcATp and GlcATs-transfected cells, and the efficacy of inhibition is GlcATp < GlcATs < GlcATp+GlcATs. Our results of NF-κB inhibition were further confirmed by inhibition of IKB phosphorylation. These observations further underscore the complexity of NF-κB activation, as has been documented in the literature (Moscat & Diaz-Meco 2011). Thus, our finding raises an important issue regarding the pricise mechanism for NF-κB activity regulation in relevant to SGPG expression. To delineate the mechanism of NF-κB inhibition by silencing SGPG (by HNK-1ST siRNA) expression, we examined TNFα-receptors 1 and 2 (TNFR1 and TNFR2) expression employing Western blot analysis. Transfection of siRNA led to the reduction of TNFR1, but showed no effect on TNFR2 (Figure 7B). SGPG over-expression by GlcATp/GlcATs transfection, however, resulted in a reduction of the level of TNFR2 and an elevation of the level of TNFR1 (Figure 7A). TNFR1 is expressed in most cell types and can be activated by binding of either soluble or trans-membrane TNF, with a preference for soluble TNF. By contrast, TNFR2 is expressed primarily by microglia and endothelial cells and is preferentially activated by trans-membrane TNF (McCoy & Tansey 2008). Elevation of soluble TNF is a hallmark of conditions of certain chronic neuro-inflammation, including multiple sclerosis, amyotrophic lateral sclerosis, and Parkinson’s disease (McCoy & Tansey 2008).
TNFR1 signaling has been reported to stimulate cell apoptosis via complex II in NF-κB-mediated signaling (Micheau & Tschopp 2003), and its inhibition by silencing the HNK-1ST gene is consistent with down-regulation of caspase 3 activity by HNK-1ST siRNA transfection with reduction of NF-κB activity (pro-apoptotic). Signaling through TNFR2 activates inflammatory and pro-survival signaling pathways through recruitment of TRAF1 and TRAF2 adaptor proteins and subsequent activation of the NF-κB pathway (Rothe et al. 1995, McCoy & Tansey 2008, Rothe et al. 1994, Rao et al. 1995). TNFR2 does not contain a death domain and, thus, unlike signaling through TNFR1, TNFR2 activation does not lead to caspase activation (McCoy & Tansey 2008). Overall, TNFR2 activation is believed to initiate primarily pro-inflammatory and pro-survival signaling (McCoy & Tansey 2008). Our data indicate that SGPG expression mediates cell apoptosis by inhibiting TNFR2 and by stimulating TNFR1 expression and caspase 3-activation (cell death signaling). This activity is reflected by a reduction of NF-κB activity (cell survival); by contrast, SGPG inhibition by HNK-1ST reduces NF-κB activity (apoptotic signal) by inhibiting the TNFR1 expression that leads to cell survival. However, further study is necessary to delineate the precise mechanism.
The GlcATp/GlcATs-transfected cells apparently showed enhanced cell death; we found that the cultures contained fewer viable cells with the increasing time of incubation. More viable cells were observed after 24 h of incubation as compared to 48 h of incubation, and this observation was confirmed by FLICA assay. To correlate apoptosis and SGPG expression, we used TLC-immunooverlay assay to quantitate SGPG concentration. Additionally, we examined SGPG regulation by immuno-cytochemistry along with GFP expression and caspase 3 activity assay. SGPG concentration was up-regulated in the transfected cells. The order of expression level was GlcATp < GlcATs < GlcATp+GlcATs, a similar profile to that observed in caspase 3 activation as well as in NF-κB inhibition. It is noteworthy that active caspase 3 expression was elevated specifically in cells that showed a higher level of SGPG expression (also identified by GFP expression), while cells with only GFP (control) transfection showed neither SGPG over-expression nor cell death (Figure 8). After 48 h of incubation, approximately 45%–50% or more of cell death was observed in cells transfected with combined EGFP-GlcATs and EGFP-GlcATp, while transfection with either one, independently prompted 30%–35% and 15%–20% cell death, respectively. Western blot analysis of the cell lysate indicated that caspase 3 was activated to a large extent, although the other survival pathways, Akt and ERK, were inhibited by SGPG over-expression. Again, the number of cell death was proportional to the degree of NF-κB inhibition. In addition, we have previously demonstrated that GlcATs is the predominant gene in SV-HCECs; its expression was more highly stimulated by cytokines than by GlcATp (Dasgupta et al. 2007). The present study further extended that observation and showed that over-expression of GlcATs had a detrimental effect on cell viability. Hence, we have unequivocally established that in addition to SGPG’s other cellular functions, its expression under inflammatory conditions is a death signal for endothelial cells, and an inhibition of their expression prevents T cell adhesion and protects against cell death (Figure 10).
Figure 10. Diagrammatic presentation of the SGPG-mediated novel signaling pathway for endothelial cell death and survival.
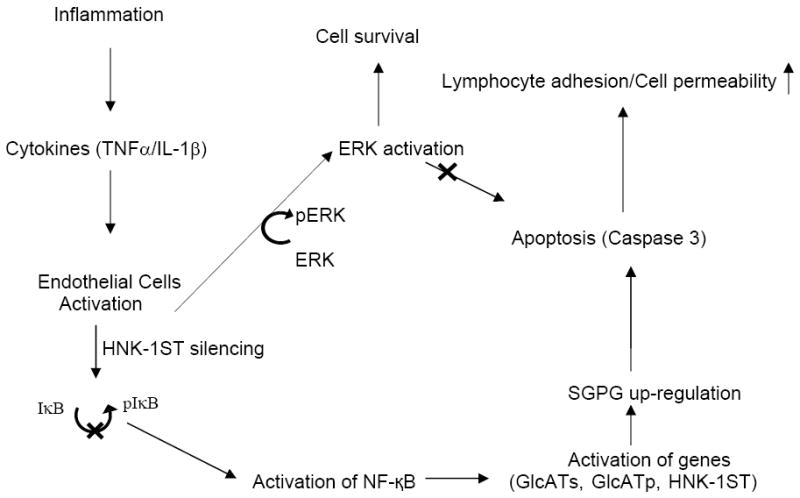
Inflammatory cytokines up-regulate SGPG expression by stimulating GlcATp, GlcATs, and HNK-1ST genes via NF-κB activation. SGPG over-expression stimulates lymphocyte (T cell) adhesion (Dasgupta et al. 2009) and promotes endothelial cell death, and thereby increases the cell permeability. Silencing of SGPG expression (by HNK-1ST siRNA) inhibits NF-κB activity, stimulates ERK activation, and prevents endothelial cell death (details in Discussion).
Evidence suggests that T cells routinely survey the BBB by infiltrating the barrier under a normal conditions to maintain homeostasis of the nervous system (Hickey 2001) along with their apparent ability to repair the nervous system (Schwartz & Cohen 2000, Hohlfeld et al. 2000). However, the endothelial cell death may affect the integrity of BBB/BNB and increase cellular permeability by loosening tight junctions, as involvement of endothelial cell death/dysfunction has been implicated in pathogenesis of many neurological disorders such as stroke, focal cerebral ischemia, and Alzheimer disease (Nagasawa & Kogure 1989, Zipfel et al. 2009, Deininger et al. 2002, Cheng et al. 2004, Jimenez et al. 2000, Hossmann 1994, Mahad et al. 2003). Endothelial cell apoptosis may cause the breakdown of the barrier, leading to vasogenic edema (Rizzo & Leaver 2010). In addition, the elimination or prevention of endothelial cell dysfunction and death is critical for tissue homeostasis (Wyllie et al. 1980) and, due to inappropriate regulation, apoptosis can also promote, contribute to, and even exacerbate the disease process (Hetts 1998). Hence, elucidating the mechanism of endothelial cell death and/or dysfunction to disease processes could help in advancing our knowledge and in developing new therapies for certain neurological disorders (Rizzo & Leaver 2010). Although we have used brain microvascular endothelial cells for our study, we believe that a similar mechanism also applies to BNB, as endothelial cells are also integral component of the BNB in maintaining the barrier function, which is critical in peripheral neuropathological conditions, such as GBS.
In an in vivo model, an inflammatory signal proceeds via infiltration of T cells, phagocytic cells, cytokines, and chemokines through the BBB/BNB. These cells and macromolecules then gain access to the nerve tissues, initiating the cell-mediated degenerative process. At any stage, an auto-immune response can also be triggered by an auto-antibody and malfunction of the immune system. This auto-antibody can easily penetrate a damaged BBB/BNB barrier, leading to destruction of the CNS/PNS and propagating the disease progression. Hence, the passage of the invading molecules through the BBB/BNB is one of the most important criteria for the onset and development of the degenerative process. A classical example of such failure of auto-antibody penetration has already been documented from our laboratory indicating that a very high titer auto-antibody raised in rabbits did not initiate any CNS/PNS demyelination (Dasgupta et al. 2004). Hence, we conclude that SGPG concentration in endothelial cells may regulate the attachment and penetration of activated T cells and phagocytes under inflammation, and in maintaining normal barrier function. We are highly encouraged by the success of our siRNA (loss-of-function) study, which indicates that inhibition of SGPG expression may be a viable strategy for designing a suitable in vivo cell-permeability inhibitor. Such an inhibitor can be used as a potential therapeutic agent in neuro-inflammatory diseases by preventing endothelial cell death and protecting the nervous system from invasion by circulating immune cells, pathogenic immunoglobulins, or other bio-degrading macromolecules.
Acknowledgments
This work was supported by grants from NIH-NINDS NS11853 and NS26994 to RKY. We thank Dr. Erhard Bieberich, Institute of Molecular Medicine and Genetics, Georgia Health Sciences University, for helpful comments and criticisms.
Abbreviations
- BBB
Blood-brain barrier
- BNB
Blood-nerve barrier
- FLICA
Fluorochrome inhibitor of caspases assay
- GBS
Guillian-Barré syndrome
- GlcATp
Glucuronosyltransferase P
- GlcATs
Glucuronosyltransferase S
- GSL
Glycosphingolipid
- HNK-1
human natural killer antigen
- SGPG
Sulfated glucuronosylparagloboside
- SV-HCEC
Human cerebromicrovascular endothelial cell
Footnotes
There is no conflict of interest with present work.
References
- Ariga T, Kohriyama T, Freddo L, et al. Characterization of sulfated glucuronic acid containing glycolipids reacting with IgM M-proteins in patients with neuropathy. J Biol Chem. 1987;262:848–853. [PubMed] [Google Scholar]
- Ariga T, Yu RK. Isolation and characterization of ganglioside GM1b from normal human brain. J Lipid Res. 1987;28:285–291. [PubMed] [Google Scholar]
- Armulik A, Genove G, Mae M, et al. Pericytes regulate the blood-brain barrier. Nature. 2010;468:557–561. doi: 10.1038/nature09522. [DOI] [PubMed] [Google Scholar]
- Cheng YD, Al-Khoury L, Zivin JA. Neuroprotection for ischemic stroke: two decades of success and failure. NeuroRx. 2004;1:36–45. doi: 10.1602/neurorx.1.1.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou DK, Ilyas AA, Evans JE, Costello C, Quarles RH, Jungalwala FB. Structure of sulfated glucuronyl glycolipids in the nervous system reacting with HNK-1 antibody and some IgM paraproteins in neuropathy. J Biol Chem. 1986;261:11717–11725. [PubMed] [Google Scholar]
- Compston A, Coles A. Multiple sclerosis. Lancet. 2008;372:1502–1517. doi: 10.1016/S0140-6736(08)61620-7. [DOI] [PubMed] [Google Scholar]
- Dasgupta S, Li D, Yu RK. Lack of apparent neurological abnormalities in rabbits sensitized by gangliosides. Neurochem Res. 2004;29:2147–2152. doi: 10.1007/s11064-004-6888-7. [DOI] [PubMed] [Google Scholar]
- Dasgupta S, Silva J, Wang G, Yu RK. Sulfoglucuronosyl paragloboside is a ligand for T cell adhesion: regulation of sulfoglucuronosyl paragloboside expression via nuclear factor kappaB signaling. J Neurosci Res. 2009;87:3591–3599. doi: 10.1002/jnr.22153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasgupta S, Yanagisawa M, Krishnamurthy K, Liour SS, Yu RK. Tumor necrosis factor-alpha up-regulates glucuronosyltransferase gene expression in human brain endothelial cells and promotes T-cell adhesion. J Neurosci Res. 2007;85:1086–1094. doi: 10.1002/jnr.21214. [DOI] [PubMed] [Google Scholar]
- Deininger MH, Fimmen BA, Thal DR, Schluesener HJ, Meyermann R. Aberrant neuronal and paracellular deposition of endostatin in brains of patients with Alzheimer’s disease. J Neurosci. 2002;22:10621–10626. doi: 10.1523/JNEUROSCI.22-24-10621.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dityatev A, Schachner M. Extracellular matrix molecules and synaptic plasticity. Nat Rev Neurosci. 2003;4:456–468. doi: 10.1038/nrn1115. [DOI] [PubMed] [Google Scholar]
- Duvar S, Suzuki M, Muruganandam A, Yu RK. Glycosphingolipid composition of a new immortalized human cerebromicrovascular endothelial cell line. J Neurochem. 2000;75:1970–1976. doi: 10.1046/j.1471-4159.2000.0751970.x. [DOI] [PubMed] [Google Scholar]
- Geleijns K, Schreuder GM, Jacobs BC, Sintnicolaas K, van Koningsveld R, Meulstee J, Laman JD, van Doorn PA. HLA class II alleles are not a general susceptibility factor in Guillain-Barre syndrome. Neurology. 2005;64:44–49. doi: 10.1212/01.WNL.0000148727.02732.01. [DOI] [PubMed] [Google Scholar]
- Hakomori SI. Structure and function of glycosphingolipids and sphingolipids: recollections and future trends. Biochim Biophys Acta. 2008;1780:325–346. doi: 10.1016/j.bbagen.2007.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayden MS, Ghosh S. Signaling to NF-kappaB. Genes Dev. 2004;18:2195–2224. doi: 10.1101/gad.1228704. [DOI] [PubMed] [Google Scholar]
- Hetts SW. To die or not to die: an overview of apoptosis and its role in disease. JAMA. 1998;279:300–307. doi: 10.1001/jama.279.4.300. [DOI] [PubMed] [Google Scholar]
- Hickey WF. Basic principles of immunological surveillance of the normal central nervous system. Glia. 2001;36:118–124. doi: 10.1002/glia.1101. [DOI] [PubMed] [Google Scholar]
- Hohlfeld R, Kerschensteiner M, Stadelmann C, Lassmann H, Wekerle H. The neuroprotective effect of inflammation: implications for the therapy of multiple sclerosis. J Neuroimmunol. 2000;107:161–166. doi: 10.1016/s0165-5728(00)00233-2. [DOI] [PubMed] [Google Scholar]
- Hossmann KA. Viability thresholds and the penumbra of focal ischemia. Ann Neurol. 1994;36:557–565. doi: 10.1002/ana.410360404. [DOI] [PubMed] [Google Scholar]
- Jimenez B, Volpert OV, Crawford SE, Febbraio M, Silverstein RL, Bouck N. Signals leading to apoptosis-dependent inhibition of neovascularization by thrombospondin-1. Nat Med. 2000;6:41–48. doi: 10.1038/71517. [DOI] [PubMed] [Google Scholar]
- Kaida K, Ariga T, Yu RK. Antiganglioside antibodies and their pathophysiological effects on Guillain-Barre syndrome and related disorders--a review. Glycobiology. 2009;19:676–692. doi: 10.1093/glycob/cwp027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanda T, Yamawaki M, Ariga T, Yu RK. Interleukin 1 beta up-regulates the expression of sulfoglucuronosyl paragloboside, a ligand for L-selectin, in brain microvascular endothelial cells. Proc Natl Acad Sci U S A. 1995;92:7897–7901. doi: 10.1073/pnas.92.17.7897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohriyama T, Ariga T, Yu RK. Preparation and characterization of antibodies against a sulfated glucuronic acid-containing glycosphingolipid. J Neurochem. 1988;51:869–877. doi: 10.1111/j.1471-4159.1988.tb01823.x. [DOI] [PubMed] [Google Scholar]
- Kohriyama T, Kusunoki S, Ariga T, Yoshino JE, DeVries GH, Latov N, Yu RK. Subcellular localization of sulfated glucuronic acid-containing glycolipids reacting with anti-myelin-associated glycoprotein antibody. J Neurochem. 1987;48:1516–1522. doi: 10.1111/j.1471-4159.1987.tb05694.x. [DOI] [PubMed] [Google Scholar]
- Kruse J, Keilhauer G, Faissner A, Timpl R, Schachner M. The J1 glycoprotein--a novel nervous system cell adhesion molecule of the L2/HNK-1 family. Nature. 1985;316:146–148. doi: 10.1038/316146a0. [DOI] [PubMed] [Google Scholar]
- Maeda Y, Bigbee JW, Maeda R, Miyatani N, Kalb RG, Yu RK. Induction of demyelination by intraneural injection of antibodies against sulfoglucuronyl paragloboside. Exp Neurol. 1991a;113:221–225. doi: 10.1016/0014-4886(91)90178-f. [DOI] [PubMed] [Google Scholar]
- Maeda Y, Brosnan CF, Miyatani N, Yu RK. Preliminary studies on sensitization of Lewis rats with sulfated glucuronyl paragloboside. Brain Res. 1991b;541:257–264. doi: 10.1016/0006-8993(91)91026-w. [DOI] [PubMed] [Google Scholar]
- Mahad DJ, Lawry J, Howell SJ, Woodroofe MN. Longitudinal study of chemokine receptor expression on peripheral lymphocytes in multiple sclerosis: CXCR3 upregulation is associated with relapse. Mult Scler. 2003;9:189–198. doi: 10.1191/1352458503ms899oa. [DOI] [PubMed] [Google Scholar]
- Martini R, Xin Y, Schmitz B, Schachner M. The L2/HNK-1 Carbohydrate Epitope is Involved in the Preferential Outgrowth of Motor Neurons on Ventral Roots and Motor Nerves. Eur J Neurosci. 1992;4:628–639. doi: 10.1111/j.1460-9568.1992.tb00171.x. [DOI] [PubMed] [Google Scholar]
- McCoy MK, Tansey MG. TNF signaling inhibition in the CNS: implications for normal brain function and neurodegenerative disease. J Neuroinflammation. 2008;5:45. doi: 10.1186/1742-2094-5-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micheau O, Tschopp J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell. 2003;114:181–190. doi: 10.1016/s0092-8674(03)00521-x. [DOI] [PubMed] [Google Scholar]
- Moscat J, Diaz-Meco MT. Fine tuning NF-kappaB: new openings for PKC-zeta. Nat Immunol. 2011;12:12–14. doi: 10.1038/ni0111-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muruganandam A, Herx LM, Monette R, Durkin JP, Stanimirovic DB. Development of immortalized human cerebromicrovascular endothelial cell line as an in vitro model of the human blood-brain barrier. FASEB J. 1997;11:1187–1197. doi: 10.1096/fasebj.11.13.9367354. [DOI] [PubMed] [Google Scholar]
- Nagasawa H, Kogure K. Correlation between cerebral blood flow and histologic changes in a new rat model of middle cerebral artery occlusion. Stroke. 1989;20:1037–1043. doi: 10.1161/01.str.20.8.1037. [DOI] [PubMed] [Google Scholar]
- Ong E, Suzuki M, Belot F, Yeh JC, Franceschini I, Angata K, Hindsgaul O, Fukuda M. Biosynthesis of HNK-1 glycans on O-linked oligosaccharides attached to the neural cell adhesion molecule (NCAM): the requirement for core 2 beta 1,6-N-acetylglucosaminyltransferase and the muscle-specific domain in NCAM. J Biol Chem. 2002;277:18182–18190. doi: 10.1074/jbc.M201312200. [DOI] [PubMed] [Google Scholar]
- Rao P, Hsu KC, Chao MV. Upregulation of NF-kappa B-dependent gene expression mediated by the p75 tumor necrosis factor receptor. J Interferon Cytokine Res. 1995;15:171–177. doi: 10.1089/jir.1995.15.171. [DOI] [PubMed] [Google Scholar]
- Rizzo MT, Leaver HA. Brain endothelial cell death: modes, signaling pathways, and relevance to neural development, homeostasis, and disease. Mol Neurobiol. 2010;42:52–63. doi: 10.1007/s12035-010-8132-6. [DOI] [PubMed] [Google Scholar]
- Rothe M, Pan MG, Henzel WJ, Ayres TM, Goeddel DV. The TNFR2-TRAF signaling complex contains two novel proteins related to baculoviral inhibitor of apoptosis proteins. Cell. 1995;83:1243–1252. doi: 10.1016/0092-8674(95)90149-3. [DOI] [PubMed] [Google Scholar]
- Rothe M, Wong SC, Henzel WJ, Goeddel DV. A novel family of putative signal transducers associated with the cytoplasmic domain of the 75 kDa tumor necrosis factor receptor. Cell. 1994;78:681–692. doi: 10.1016/0092-8674(94)90532-0. [DOI] [PubMed] [Google Scholar]
- Schwartz M, Cohen IR. Autoimmunity can benefit self-maintenance. Immunol Today. 2000;21:265–268. doi: 10.1016/s0167-5699(00)01633-9. [DOI] [PubMed] [Google Scholar]
- Tokuda A, Ariga T, Isogai Y, Komba S, Kiso M, Hasegawa A, Tai T, Yu RK. On the specificity of anti-sulfoglucuronosyl glycolipid antibodies’. J Carbohyd Chem. 1998;17:535–546. [Google Scholar]
- Voshol H, van Zuylen CW, Orberger G, Vliegenthart JF, Schachner M. Structure of the HNK-1 carbohydrate epitope on bovine peripheral myelin glycoprotein P0. J Biol Chem. 1996;271:22957–22960. doi: 10.1074/jbc.271.38.22957. [DOI] [PubMed] [Google Scholar]
- Wyllie AH, Kerr JF, Currie AR. Cell death: the significance of apoptosis. Int Rev Cytol. 1980;68:251–306. doi: 10.1016/s0074-7696(08)62312-8. [DOI] [PubMed] [Google Scholar]
- Yu RK, Usuki S, Ariga T. Ganglioside molecular mimicry and its pathological roles in Guillain-Barre syndrome and related diseases. Infect Immun. 2006;74:6517–6527. doi: 10.1128/IAI.00967-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zipfel GJ, Han H, Ford AL, Lee JM. Cerebral amyloid angiopathy: progressive disruption of the neurovascular unit. Stroke. 2009;40:S16–19. doi: 10.1161/STROKEAHA.108.533174. [DOI] [PMC free article] [PubMed] [Google Scholar]