

Abstract
Background
This study describes the functional interaction between the putative Ca2+ channel TRP4 and the cystic fibrosis transmembrane conductance regulator, CFTR, in mouse aorta endothelium (MAEC).
Results
MAEC cells express CFTR transcripts as shown by RT-PCR analysis. Application of a phosphorylating cocktail activated a Cl- current with characteristics similar to those of CFTR mediated currents in other cells types (slow activation by cAMP, absence of rectification, block by glibenclamide). The current is present in trp4 +/+ MAEC, but not in trp4 -/- cells, although the expression of CFTR seems unchanged in the trp4 deficient cells as judged from RT-PCR analysis.
Conclusions
It is concluded that TRP4 is necessary for CFTR activation in endothelium, possibly by providing a scaffold for the formation of functional CFTR channels.
Background
The cystic fibrosis transmembrane conductance regulator (CFTR) is well described as a low-conductance, cyclic nucleotide-regulated Cl- channel in epithelial cells [1]. Only recently, CFTR has also been detected in vascular endothelium [2]. Endothelial cells (EC) form an anticoagulative barrier but also control many other functions, such as regulation of the vascular tone by secretion of vasoactive compounds such as bradykinin, and autocoids, such as nitric oxide and prostacyclin [3]. These functions are modulated by a diversity of ion channels among which Cl- channels [4, 5]. Endothelial Cl- channels, the volume-regulated anion channel, VRAC, and Ca2+ activated Cl- channels, CaCC, have been shown to modulate EC electrogenesis, are possible mechano-sensors, serve as permeation pathways for amino acids and organic osmolytes and may be involved in regulation of the driving force for Ca2+ entry [for a review, see 6]. This list of Cl- channels has been extended with CFTR, which is functional in human umbilical vein endothelium and in human lung microvascular endothelial cells [1], but not in bovine pulmonary artery endothelial cells [6]. As we show in this work, it is also functional in mouse aorta endothelial cells.
MAEC express different types of putative ion channel transcripts which are encoded by genes of the trp family, trp1, 2, 3, 4, and 6 [7, 8]. TRP4 forms part of a store operated Ca2+ entry channel which is involved in the control of NO-dependent relaxation of the mouse aorta [8]. In addition, TRP4 has been shown to interact via a VTTRL motif in its C-terminal region with the first PDZ domain of the regulatory factor of the Na+- H+exchanger NHERF, which also interacts with PLCβ [9]. The two PDZ domain protein NHERF associates also with the actin cytoskeleton via members of the ezrin/radixin/moesin family [10, 11]. It is also well established that the C terminus of CFTR constitutes a PDZ-interacting domain (QDTRL for the last five C-terminal amino acids) that is required for CFTR polarization to the apical plasma membrane and interaction with the PDZ domain-containing protein NHERF [12]. Thus, both TRP4 and CFTR may bind to similar PDZ-domain proteins.
We have studied the functional expression of CFTR in both trp4 wild type and in trp4 deficient MAEC cells. We show here that CFTR is present in both cell types, but is not functional in trp4 deficient endothelial cells. These data may hint to a more general function of trp4 as regulator of other ion channels and to a novel regulatory mechanism for CFTR.
Results
Expression of CFTR in mouse aorta endothelium
We have been unable to detect CFTR in bovine pulmonary endothelial cells [6], but its expression has recently been described in endothelium [1]. We have therefore assessed the expression of CFTR in mouse aorta EC (MAEC) by means of two sets of primers, the one detecting exon 5 through exon 9 of CFTR transcripts, and the other one detecting exons 23 and 24 of CFTR transcripts (figure 1A, B). The data show that CFTR is expressed in both wild-type and trp4 deficient MAEC cells, and are consistent with the recent detection of CFTR expression in human umbilical vein endothelium and human lung microvascular endothelial cells.
Figure 1.
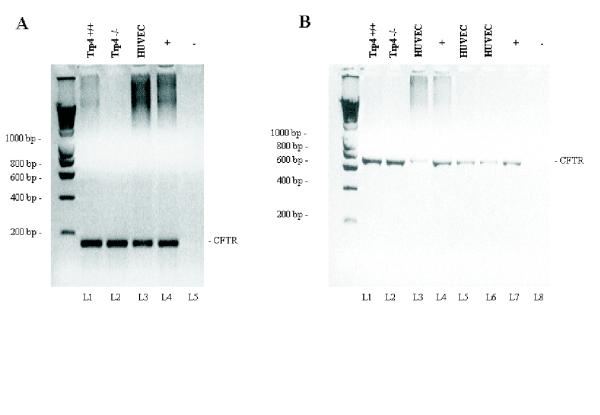
RT-PCR showing the expression of CFTR in mouse aorta endothelial cells A) cDNA from murine TRP4 +/+ and TRP4 -/- MAEC (lanes 1 and 2), human umbilical vein cells (HUVEC, lane 3) and human nasal epithelium cells (+, for a positive control) were amplified using primers P.CFTR-3249.5 and P.CFTR-3428.3, generating a 180 base pair fragment encoding partial sequences of exons 17a and 17b. Lane 5 is a negative control. B) cDNA from murine TRP4 +/+ and TRP4 -/- MAEC (lanes 1 and 2), from three different preparations of cDNA from HUVEC cells (lanes 3, 5 and 6) and human nasal epithelial cDNA as a positive control (lanes 4 and 7) were amplified using primers PF.CFTR-661.5 and P.CFTR-1360.3, generating a 700 bp fragment harboring exons 5 through 9. Lane 8 is the negative control of amplification.
Functional characterization of CFTR in MAEC
Subsequently, we have investigated the functional expression of CFTR in wild type MAEC. Application of the phosphorylating cocktail, containing10 μM forskolin and 100 μM IBMX, activated in these cells a current without any apparent rectification and voltage-independent kinetics, which reversed at -26 ± 6 mV (n = 6), i.e. close to the Cl-equilibrium potential, ECl (figure 2A). Its phenotype was completely different from that of Cl- currents activated by loading the cells with Ca2+ (figure 2B), which are outwardly rectifying, slowly activate at positive potentials and inactivate at potentials negative to ECl [13, 14]. Challenging the MAEC with a 25 % hypotonic extracellular solution activated another type of Cl- current that inactivates at positive potentials and shows a less pronounced outward rectification (figure 2C). The latter currents have been described in detail in other EC as VRAC, volume-regulated anion currents [6, 15, 16]. The cAMP-activated current reached a stationary value approximately 2 minutes after application of the phosphorylating cocktail (figure 3A), and disappeared after wash-out of the phosphorylation cocktail. The cAMP-activated current was observed in 16 out of 22 cells, but its density was rather small (4.9 ± 1.1 pA/pF at +50 mV, obtained from voltage ramps, n = 16) compared to that of the other Cl- currents. It was activated without any change in cell volume or elevation of intracellular Ca2+. Glibenclamide (50 μM) blocked the cAMP-activated current by 62 ± 4 % (n = 6) (figure 3D). Obviously, the profile of this current is similar to that of CFTR currents in other tissues, i.e. slow activation, linear I-V curve, time-independent kinetics and inhibition by glibenclamide, indicating that CFTR channels are also functionally expressed in MAEC, and coexist with at least two other types of Cl-channels.
Figure 2.
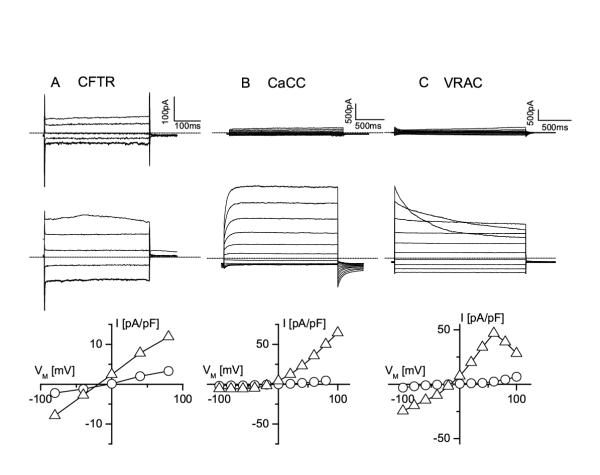
At least three different Cl- channels exist in MAEC A. Current traces in a non-stimulated MAEC cell (upper traces) and in the same cell stimulated with the phosphorylation cocktail (lower traces) in response to voltage steps from +80 to -80 mV (decrement = 40 mV, VH = 0 mV). The bottom panel shows the corresponding I-V curves from the current amplitudes recorded at the end of each voltage step (open circles for the resting cell and open triangles for the stimulated cell). Note the voltage-independent kinetics of the current, the lack of rectification and the reversal potential close to ECl B. Current traces in an MAEC cell immediately after breaking into the cell and before it is loaded with Ca2+ (upper traces) and after equilibration with pipette Ca2+ (1 μM). Voltage steps form -100 to +100 mV (increment +20 mV), VH is -20 mV. Note the slow activation at positive potentials, and the inactivation at negative potentials, which are typical for CaCC currents. The corresponding I-V curves in the bottom panel illustrate the strong outward rectification of the CaCC current. C. Current traces from an MAEC cell before and during cell swelling induced by challenging the cell with a 25 % hypotonic solution. Same step protocol as in B. Note the inactivation of the current at positive potentials, which is a feature of volume-regulated anion channels (VRAC). The corresponding I-V curves at the bottom illustrate the weak outward rectification of the VRAC currents.
Figure 3.
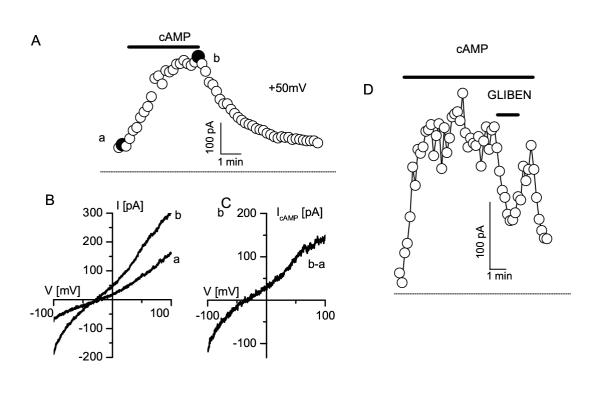
cAMP activated Cl- currents in MAEC A. Slow activation of the CFTR currents. Currents values were obtained form linear voltage ramps and were measured at +50 mV (holding potential, VH = 0 mV). B. Instantaneous current-voltage relationships measured from voltage ramps at the times indicated by solid circles (a and b) in panel A. C. Difference current, b-a, from the current measured after and before stimulation with the cAMP elevating cocktail. From such protocols current density was measured for the wild type and the trp4-deficient MAEC (see figure 4). D. Block of the cAMP activated current by 50 μM glibenclamide (measured from voltage ramps at +50 mV, VH = 0 mV)
Down-regulation of CFTR currents in trp4-deficient MAEC
The phosphorylating cocktail failed to activate a similar current in trp4-deficient MAEC. Figure 4 shows an example for stimulating wild-type and trp4- deficient MAEC. On the other hand, the CaCC current activated by loading MAEC with 1 μM [Ca2+]i was not significantly different from that in wild type MAEC, i.e. 16 ± 4 pA/pF in WT(n = 11) compared to 24 ± 6 pA/pF (n = 8) in trp4 -/- cells at +100 mV. Also VRAC was not significantly different in both cell types (peak currents at +100 mV: 58 ± 8 pA/pF, n = 7, in WT cells; 55 ± 11 pA/pF, n = 6 in trp4 -/- MAEC).
Figure 4.
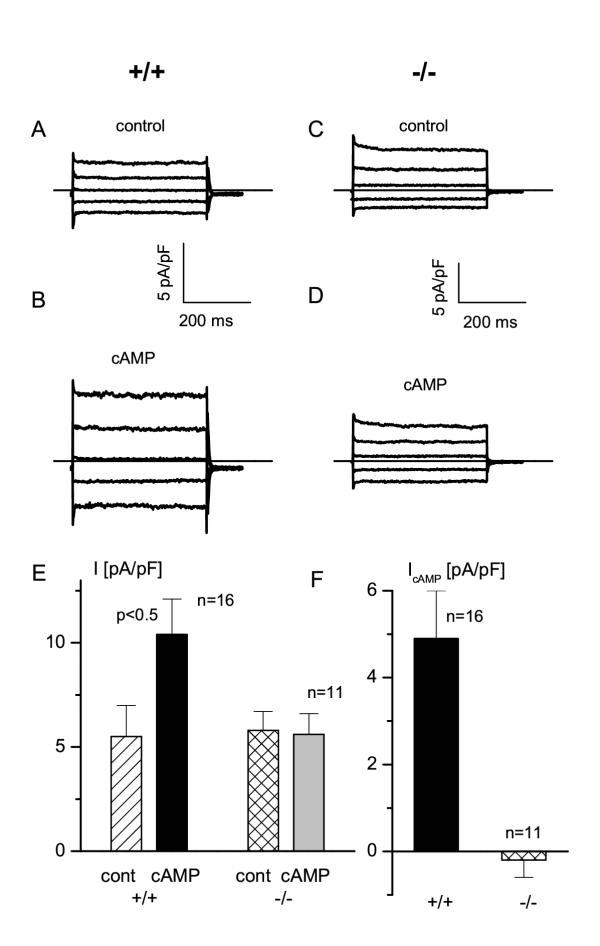
Failure of CFTR activation in trp4 deficient MAEC A. Current traces from a wild type MAEC obtained from steps protocols (voltage steps from +80 to -80 mV, decrement 40 mV, holding potential was VH = 0 mV). B. Same protocol as in panel A, but after application of the phosphorylating cocktail C. Same protocol as in A, but for a trp4 deficient cell. D. Same protocol as in C for the trp4 -/- cell after stimulation with the phosphorylating cocktail. E. Pooled current densities at +50 mV (obtained from voltage ramps) in wild type and trp4 deficient MAEC under resting conditions (cont) and after stimulation with the phosphorylating cocktail (cAMP). Note the lack of current activation by the phosphorylating cocktail in the trp4 deficient MAEC. Amplitude of the cAMP activated current in wild type (+/+) and trp4 deficient (-/-) endothelial cells, i.e. the difference between the current amplitudes in stimulated and unstimulated cells in panel E.
Discussion
We show that mouse aorta endothelial cells express functional CFTR channels, and that their activation is defective in mouse aorta endothelium of trp4-deficient mice, although these cells still expressed the CFTR transcripts.
CFTR expression has already been demonstrated in human umbilical vein (HUVEC) and lung microvascular endothelial cells (HLMVEC) [1], but was not detectable in bovine pulmonary artery endothelial cells [6]. We show here that CFTR is also expressed as a functional channel in mouse aorta endothelial cells. It has been suggested that the endothelial CFTR has 100% identity with the corresponding epithelial cDNA from exon 3 to exon 6, including exon 5 which is absent in cardiac CFTR [1]. CFTR is also present in corneal, non-vascular endothelium [17]. In MAEC cells, the first arterial cell type described to express CFTR, cAMP activated Cl- currents are smaller than Ca2+ activated and volume-regulated Cl- currents in the same cell type. Activation of CFTR in endothelial cells could be of functional interest in transendothelial transport, in pH regulation because of the CFTR permeability for bicarbonate, and intriguingly also as part of the NO signaling cascade because of their sensitivity to cGMP, which might be elevated during endothelial NO production.
Besides its role as a Cl- channel, CFTR also regulates the activity of various ion channels and transporters, mainly via direct protein-protein interactions [18, 19]. Some of these interactions might be mediated by the association of CFTR via its C-terminal PDZ-binding motif (D-T-R-L) with the PDZ-binding domains of other proteins, such as NHERF [11, 12, 20, 21]. CFTR also functions as regulator of ATP release [22, 23], which might be important for EC function because the released ATP could in turn bind to endothelial P2Y2 and P2X4 receptors [for a review, see 6].
The finding that CFTR, although detectable in RT-PCR, cannot be activated in trp4-defiecint mouse aorta endothelium might be of a special interest, since it shows that the functional activity of CFTR may be connected to the expression of trp4, a member of the trp-family that is involved in capacitative Ca2+ (CCE). CCE, mediated via store-operated channels (SOC), may affect CFTR activity indirectly via modulation of adenylyl or guanyl cyclases. It has indeed been shown that block of CCE by micromolar concentrations of La3+ ions inhibits cyclic AMP synthesis [24, 25]. In endothelial cells, CCE is the principle stimulus for sustained activation of eNOS [26], and the concomitant elevation of the cGMP concentration may in turn activate CFTR [27, 28].
Alternatively, TRP4 may interact more directly with CFTR by forming a signaling complex via binding of their PDZ-binding motifs to PDZ-domain proteins. The Na+/H+ exchanger regulatory factor, NHERF, has been identified as a protein that binds both TRP4 and CFTR [10]. Also light activated TRP channels in the Drosophila photoreceptor cluster with several other signaling proteins by associating with the PDZ-domain protein INAD. It is therefore not unlikely that TRP4 and CFTR associate in MAEC via similar PDZ-domain proteins, such as NHERF or the human INAD-Like (hINAD-L) protein [29].
Conclusions
This is the first report describing a functional interaction between a member of the TRP family and CFTR. It is therefore tempting to speculate that TRPs might be either regulators of CFTR or targets of CFTR regulating proteins.
Materials and Methods
Cell isolation
Gene targeting for the construction of the trp4 deficient mouse has been described in detail elsewhere[8]. In short, part of the exon encoding transmembrane domain 4 and 5 and part of the linker between S5 and S6 was replaced by the neomycin-resistance gene and the deletion of the trp4 gene and lack of expression was confirmed [8].
The "primary explant technique" was used to study freshly isolated endothelial cells from mouse aorta of trp4 +/+ (wild type) and trp4 -/- mice [30]. In short, the aorta was removed form anesthetized and heparinized mice. Small pieces of the aorta were placed with their intimal side on matrigel-coated six well plates containing a very small volume of growth medium (mix for 100 ml: 80 ml DMEM (GIBCO BRL 41965) with in addition 10 ml FCS, 7.5 mg ECGF, 200 μl heparin (10 U/ml final), 2 ml penicillin/streptomycin (100 U/ml final, GIBCO BRL 15070), 1 ml L-glutamine (100x, GIBCO BRL 25030-024), and 1 ml Minimal Essential Amino acids so that the aortic pieces adhere to the substratum. Matrigel-coated plates contained TGFβ, FGF, and tPA. Additional endothelial cell growth factors were added (75 μg/ml, ECGS, endothelial cell growth factor supplement, E2759, Sigma). Addition of heparin induced an antiproliferative effect on smooth muscle cells and fibroblasts. After approximately 6 days, we removed the aortic pieces and let the endothelial cells that have migrated from the aortic segments grow to confluence. In this state, explanted endothelial cells can be studied on the matrigel support. For further investigations, cells were passaged by using dispase (Becton Dickinson, Two Oak Park, Bedford, MA, USA). Thereafter, growth medium was added to stop the dispase activity by dilution. Cells were then harvested, centrifuged and resuspended with DMEM.
Reverse Transcriptase Polymerase Chain Reaction (RT-PCR)
Total RNA was isolated using TRIzol Reagent (Gibco BRL Life Technologies), according to the protocol provided by the manufacturer. After priming with oligo-dT oligonucleotides, cDNA was synthesized with MLV reverse-transcriptase (Life Technologies) according to the recommendations of the supplier. The cDNA was used as template in a polymerase chain reaction (PCR), using 5 μl of a cDNA synthesis reaction in a 50 μl amplification reaction. The amplification was performed in a DNA Thermal Cycler (Perkin Elmer). The temperature cycling conditions were: denaturation for 1 minute at 95°C, annealing for 1 minute at a temperature that is specific for each pair of primers (55°C for the primer couple P.CFTR-661.5 (5'-AAG TAT TGG ACA ACT TGT TAG TC-3' corresponding to nucleotides 661 to 683 of mouse CFTR cDNA, Accession number M69298) and P.CFTR-1360.3 (5'-TAA TTC CCC AAA TCC CTC CTC-3' 3' corresponding to nucleotides 1360 to 1340 of mouse CFTR cDNA, Accession number M69298), which amplify a 700 base pair fragment encompassing exon 5 (partial) through exon 9 (partial); and 60°C for the primer couple P.CFTR-3249.5 (5'-TGG AAT CTG AAG GCA GGA GTC-3' corresponding to nucleotides 3249 to 3269 of mouse CFTR cDNA, Accession number M69298) and P.CFTR-3428.3 (5'-TTC TCA TTT GGA ACC AGC GCA-3' corresponding to nucleotides 3428 to 3408 of mouse CFTR cDNA, Accession number M69298), which amplify a 180 base pair fragment excompassing exons 17a and 17b partially), extension at 72°C for 1 minute for fragments smaller than 1 kb for a total of 35-45 cycles, with a final extension step of 10 minutes to fully extend any remaining single stranded DNA. The first denaturation step was done for 6 minutes at 95°C.
Solutions and electrophysiology
For measurement of CFTR currents, we started the experiment by using a bath solution that contained (in mM): 150 NaCl, 6 KCl, 1 MgCl2, 1.5 CaCl2, 10 glucose, 10 HEPES, titrated with NaOH to pH 7.4. The Cl- equilibrium potential, ECl, is -36 mV. We then switched to a solution in which KCl had been substituted by CsCl. CFTR-channels were activated by a cocktail containing 100 μM IBMX (3-isobutyl-1-methylxanthine) and 10 μM forskolin (both from Sigma-Aldrich Chemie) dissolved in the bath solution. The pipette solution contained (in mM): 40 CsCl, 100 Cs-aspartate, 1 MgCl2, 0.1 EGTA, 4 Na2ATP, 10 HEPES, pH 7.2 with CsOH. Experiments were done at room temperature, ∼ 22°C.
Ca2+ activated Cl- currents were measured as described earlier [13, 14]. The bath solution contained (mM): 150 NMDG-chloride, 1 MgCl2, 1.5 CaCl2, 10 glucose, 50 mannitol, 50 nM charybdotoxin, 10 HEPES, titrated with NaOH to pH 7.4. Mannitol was used to suppress co-activation of volume-regulated anion channels (VRAC). Charybdotoxin (Sigma) was added to inhibit the big-conductance Ca2+ activated K+ channels, BKCa which is also present in MAEC cells [30]. The pipette solution contained (mM): 100 Cs-aspartate, 40 CsCl, 1 MgCl2, 4 Na2ATP, 1 μM Ca2+ buffered with 10 mM EGTA (CaBuf program, G. Droogmans, Leuven), 10 Hepes, pH 7.4 with CsOH.
Activation of VRAC has also been described in detail [16, 30]. In short, at the beginning of the patch-clamp recording, the Krebs solution was replaced by an isotonic Cs+ solution to suppress K+ currents, containing (in mM): 105 NaCl, 6 CsCl, 1 MgCl2, 1.5 CaCl2, 10 glucose, 90 mannitol, 10 HEPES, pH 7.4 with NaOH (320 ± 5 mOsm). Hypotonic solutions were obtained by omitting 90 mM mannitol from this solution (240 ± 5 mOsm). A pipette solution was used containing (in mM): 40 CsCl, 100 Cs-aspartate, 1 MgCl2, 1.93 CaCl2, 5 EGTA, 4 Na2ATP, 10 HEPES, pH 7.2 with CsOH (290 mOsm). This solution is slightly hypotonic compared to the Krebs' solution to avoid spontaneous activation of volume-sensitive Cl- currents. The concentration of free Ca2+ in this solution was buffered at 100 nM, which is below the threshold for activation of Ca2+ activated K+ and Cl- currents and also prevents activation of non-selective cation channels.
MAEC cells were patch clamped as described previously [8, 30]. Currents were monitored with an EPC-7 patch clamp amplifier (List Electronic, Germany). Patch electrodes had a resistance between 3 and 5 MΩ. An Ag-AgCl wire was used as reference electrode. VRAC and CaCC whole-cell membrane currents were measured in ruptured patches, sampled at 2 ms intervals (1024 points per record, filtered at 200 Hz), unless otherwise mentioned. All experiments were performed at room temperature (20-23°C).
The I-V curve of CFTR currents was derived from 400 ms linear voltage ramps from -100 mV to +100 mV or from 400 ms voltage steps to potentials ranging from +80 to -80 mV (in decrements of 40 mV) applied from a holding potential of 0 mV [31, 32]. Currents were sampled at 1 ms intervals (512 points) and filtered at 5 kHz.
Data analysis
Electrophysiological data were analyzed using the WinASCD software (G. Droogmans, Leuven). Pooled data are given as mean ± S.E.M. from n cells. Significance was tested using Student's paired t test (P < 0.05 are marked with an asterisk).
Acknowledgments
Acknowledgements
This work was supported by the Belgian Federal Government, the Flemish Government and the Onderzoeksraad KU Leuven (GOA 99/07, F.W.O. G.0237.95, F.W.O. G.0214.99, F.W.O. G. 0136.00; Interuniversity Poles of Attraction Program, Prime Ministers Office IUAP Nr.3P4/23), by "Levenslijn" (7.0021.99), a grant from the "Alphonse and Jean Forton Fonds - Koning Boudewijn Stichting" R7115 B0.
Contributor Information
Lin Wei, Email: Wei.Lin@med.kuleuven.ac.be.
Marc Freichel, Email: Marc.Freichel@med-rz.uni-saarland.de.
Martine Jaspers, Email: Martine.Jaspers@med.kuleuven.ac.be.
Harry Cuppens, Email: Harry.Cuppens@med.kuleuven.ac.be.
Jean-Jacques Cassiman, Email: jean-jacques.cassiman@med.kuleuven.ac.be.
Guy Droogmans, Email: guy.droogmans@med.kuleuven.ac.be.
Veit Flockerzi, Email: veit.flockerzi@med-rz.uni-sb.de.
Bernd Nilius, Email: bernd.nilius@med.kuleuven.ac.be.
References
- Tousson A, Van Tine BA, Naren AP, Shaw GM, Schwiebert LM. Characterization of CFTR expression and chloride channel activity in human endothelia. Am J Physiol. 1998;275:C1555–1564. doi: 10.1152/ajpcell.1998.275.6.C1555. [DOI] [PubMed] [Google Scholar]
- Nilius B, Casteels R. Biology of the vascular wall and its interaction with migratory and blood cells. In Comprehensive human physiology (Edited by R Greger and U Windhorst) Berlin Heidelberg, Springer Verlag. 1996. pp. 1981–1994.
- Nilius B, Eggermont J, Voets T, Buyse G, Manolopoulos VG, Droogmans G. Properties of volume-regulated anion channels in mammalian cells. Prog Bipohys Molec Biol. 1997;68:69–119. doi: 10.1016/s0079-6107(97)00021-7. [DOI] [PubMed] [Google Scholar]
- Nilius B, Eggermont J, Droogmans G. Chloride channels in endothelium: The role of mechano-stimulation and changes in cell volume. In Mechanical forces and the endothelium (Edited by PF Lelkes) London, Harvard Academic Publisher. 1999. pp. 33–54.
- Nilius B, Droogmans G. Functional role of ion channels in vascular endothelium. Physiol Rev. 2001. [DOI] [PubMed]
- Nilius B, Szücs G, Heinke S, Voets T, Droogmans G. Multiple types of chloride channels in bovine pulmonary artery endothelial cells. J Vasc Res. 1997;34:220–228. doi: 10.1159/000159226. [DOI] [PubMed] [Google Scholar]
- Freichel M, Schweig U, Stauffenberger S, Freise D, Schorb W, Flockerzi V. Store-operated cation channels in the heart and cells of the cardiovascular system. Cell Physiol Biochem. 1999;9:270–283. doi: 10.1159/000016321. [DOI] [PubMed] [Google Scholar]
- Freichel M, Suh SH, Pfeifer A, Schweig U, Trost C, Weissgerber P, Phillip S, Freise D, Droogmans G, Hofmann , et al. Lack of an endothelial store-operated Ca2+-current impairs agonist-dependent Ca2+ entry and vasorelaxation in TRP4 (CCE1) -/- mice. Nature - Cell Biol. 2001;3:121–127. doi: 10.1038/35055019. [DOI] [PubMed] [Google Scholar]
- Tang Y, Tang J, Chen ZG, Trost C, Flockerzi V, Lin M, Ramesh V, Zhu MX. Association of mammalian Trp4 and phospholipase C isozymes with a PDZ domain-containing protein, NHERF. J Biol Chem. 2000;275:37559–37564. doi: 10.1074/jbc.M006635200. [DOI] [PubMed] [Google Scholar]
- Reczek D, Berryman M, Bretscher A. Identification of EBP50: A PDZ-containing phosphoprotein that associates with members of the ezrin-radixin-moesin family. J Cell Biol. 1997;139:169–179. doi: 10.1083/jcb.139.1.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murthy A, Gonzalez Agosti C, Cordero E, Pinney D, Candia C, Solomon F, Gusella J, Ramesh V. NHE-RF, a regulatory cofactor for Na+-H+ exchange, is a common interactor for merlin and ERM (MERM) proteins. J Biol Chem. 1998;273:1273–1276. doi: 10.1074/jbc.273.3.1273. [DOI] [PubMed] [Google Scholar]
- Moyer BD, Duhaime M, Shaw C, Denton J, Reynolds D, Karlson KH, Pfeiffer J, Wang SS, Mickle JE, Milewski M, et al. The PDZ-interacting domain of cystic fibrosis transmembrane conductance regulator is required for functional expression in the apical plasma membrane. J Biol Chem. 2000;275:27069–27074. doi: 10.1074/jbc.M004951200. [DOI] [PubMed] [Google Scholar]
- Nilius B, Prenen J, Voets T, Vandenbremt K, Eggermont J, Droogmans G. Kinetic and Pharmacological Properties of the Calcium Activated Chloride Current in Macrovascular Endothelial Cells. Cell Calcium. 1997;22:53–63. doi: 10.1016/s0143-4160(97)90089-0. [DOI] [PubMed] [Google Scholar]
- Nilius B, Prenen J, Szucs G, Wei L, Tanzi F, Voets T, Droogmans G. Calcium-activated chloride channels in bovine pulmonary artery endothelial cells. J Physiol. 1997;498:381–396. doi: 10.1113/jphysiol.1997.sp021865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilius B, Oike M, Zahradnik I, Droogmans G. Activation of a Cl- current by hypotonic volume increase in human endothelial cells. J Gen Physiol. 1994;103:787–805. doi: 10.1085/jgp.103.5.787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilius B, Eggermont J, Voets T, Droogmans G. Volume-activated Cl--channels. Gen Pharmacol. 1996;27:67–77. doi: 10.1016/s0306-3623(96)00061-4. [DOI] [PubMed] [Google Scholar]
- Bonanno JA, Srinivas SP. Cyclic AMP activates anion channels in cultured bovine corneal endothelial cells. Exp Eye Res. 1997;64:953–962. doi: 10.1006/exer.1997.0290. [DOI] [PubMed] [Google Scholar]
- Kunzelmann K. The cystic fibrosis transmembrane conductance regulator and its function in epithelial transport. Rev Physiol Biochem Pharmacol. 1999;137:1–70. doi: 10.1007/3-540-65362-7_4. [DOI] [PubMed] [Google Scholar]
- Schwiebert EM, Benos DJ, Egan ME, Stutts MJ, Guggino WB. CFTR is a conductance regulator as well as a chloride channel. Physiol Rev. 1999;79:S145–166. doi: 10.1152/physrev.1999.79.1.S145. [DOI] [PubMed] [Google Scholar]
- Bezprozvanny I, Maximov A. PDZ domains: more than just a glue. Proc Natl Acad Sci USA. 2001;98:787–789. doi: 10.1073/pnas.98.3.787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karthikeyan S, Leung T, Ladias JAA. Structural basis of the NHERF PDZ1 interaction with the carboxyl-terminal region of the cystic fibrosis transmembrane conductance regulator. J Biol Chem. 2001;as 20101074/jbc.C100154200 doi: 10.1074/jbc.C100154200. [DOI] [PubMed] [Google Scholar]
- Jiang Q, Mak D, Devidas S, Schwiebert EM, Bragin A, Zhang Y, Skach WR, Guggino WB, Foskett JK, Engelhardt JF. Cystic fibrosis transmembrane conductance regulator-associated ATP release is controlled by a chloride sensor. J Cell Biol. 1998;143:645–657. doi: 10.1083/jcb.143.3.645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwiebert EM, Morales MM, Devidas S, Egan ME, Guggino WB. Chloride channel and chloride conductance regulator domains of CFTR, the cystic fibrosis transmembrane conductance regulator. Proc Natl Acad Sci U S A. 1998;95:2674–2679. doi: 10.1073/pnas.95.5.2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagan KA, Mahey R, Cooper DM. Functional co-localization of transfected Ca2+-stimulable adenylyl cyclases with capacitative Ca2+ entry sites. J Biol Chem. 1996;271:12438–12444. doi: 10.1074/jbc.271.21.12438. [DOI] [PubMed] [Google Scholar]
- Gu C, Cooper DM. Ca2+, Sr2+, and Ba2+ identify distinct regulatory sites on adenylyl cyclase (AC) types VI and VIII and consolidate the apposition of capacitative cation entry channels and Ca2+-sensitive ACs. J Biol Chem. 2000;275:6980–6986. doi: 10.1074/jbc.275.10.6980. [DOI] [PubMed] [Google Scholar]
- Lin S, Fagan KA, Li KX, Shaul PW, Cooper DM, Rodman DM. Sustained endothelial nitric-oxide synthase activation requires capacitative Ca2+ entry. J Biol Chem. 2000;275:17979–17985. doi: 10.1074/jbc.275.24.17979. [DOI] [PubMed] [Google Scholar]
- Hofmann F, Ammendola A, Schlossmann J. Rising behind NO: cGMP-dependent protein kinases. J Cell Sci. 2000;113:1671–1676. doi: 10.1242/jcs.113.10.1671. [DOI] [PubMed] [Google Scholar]
- Vaandrager AB, Smolenski A, Tilly BC, Houtsmuller AB, Ehlert EM, Bot AG, Edixhoven M, Boomaars WE, Lohmann SM, de Jonge HR. Membrane targeting of cGMP-dependent protein kinase is required for cystic fibrosis transmembrane conductance regulator Cl- channel activation. Proc Natl Acad Sci U S A. 1998;95:1466–1471. doi: 10.1073/pnas.95.4.1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philipp S, Flockerzi V. Molecular characterization of a novel human PDZ domain protein with homology to INAD from Drosophila melanogaster. FEBS Lett. 1997;413:243–248. doi: 10.1016/s0014-5793(97)00877-6. [DOI] [PubMed] [Google Scholar]
- Suh SH, Vennekens R, Manolopoulos VG, Freichel M, Schweig U, Prenen J, Flockerzi V, Droogmans G, Nilius B. Characterisation of explanted endothelial cells from mouse aorta: electrophysiology and Ca2+ signalling. Pflüg Arch Eur J Physiol. 1999;438:612–620. doi: 10.1007/s004249900085. [DOI] [PubMed] [Google Scholar]
- Wei L, Vankeerberghen A, Jaspers M, Cassiman J, Nilius B, Cuppens H. Suppressive interactions between mutations located in the two nucleotide binding domains of CFTR. FEBS Lett. 2000;473:149–153. doi: 10.1016/s0014-5793(00)01519-2. [DOI] [PubMed] [Google Scholar]
- Wei L, Vankeerberghen A, Cuppens H, Eggermont J, Cassiman JJ, Droogmans G, Nilius B. Interaction between calcium-activated chloride channels and the cystic fibrosis transmembrane conductance regulator. Pflügers Arch Eur J Physiol. 1999;438:635–641. doi: 10.1007/s004249900108. [DOI] [PubMed] [Google Scholar]