

Abstract
Microbes survive in a variety of nutrient environments by modulating their intracellular metabolism. Balanced growth requires coordinated uptake of carbon and nitrogen, the primary substrates for biomass production. The mechanisms that balance carbon and nitrogen uptake are, however, poorly understood. We find in Escherichia coli that a sudden increase in nitrogen availability results in an almost immediate increase in glucose uptake. The concentrations of known glycolytic intermediates and regulators, however, remain homeostatic. Instead, we find that α-ketoglutarate, which accumulates in nitrogen limitation, directly blocks glucose uptake by inhibiting Enzyme I, the first step of the phosphotransferase system (PTS). This enables rapid modulation of glycolytic flux without marked concentration changes in glycolytic intermediates by simultaneously accelerating glucose import and consumption of the terminal glycolytic intermediate phosphoenolpyruvate. Quantitative modeling shows that this previously unidentified regulatory connection is in principle sufficient to coordinate carbon and nitrogen utilization.
Introduction
All organisms must produce biomass from multiple external nutrients in order to grow, and the availability of these nutrients is subject to environmental fluctuations. As growth can be limited by scarcity of any one nutrient, the rate at which each nutrient is assimilated must be sensitive not only to its own availability but also the availability of other nutrients. The problem of integrating information from multiple intra- and extracellular nutrient sensing pathways has gained significant attention recently with the identification of TOR as a highly conserved regulator of eukaryotic carbon and nitrogen signaling pathways and a major contributor to metabolic disregulation in cancer and diabetes1, 2. While TOR has been linked to a multitude of cellular processes related to the cell cycle, metabolism, stress response and ribosome activity, little understanding has been gained regarding the fundamental question of how cells quantitatively tailor carbon uptake to nitrogen availability and vice versa.
Bacteria are excellent models for studies of metabolic regulation, as they lack the extensive compartmentalization of eukaryotes while retaining much of the conserved core of central metabolism. The specific question of carbon and nitrogen coordination in bacteria is especially relevant, as biotechnology processes utilizing microbial cultures can take advantage of nitrogen-poor conditions to force carbon into pathways not needed for biomass production; nitrogen limitation has been used to induce microbial overproduction of organic solvents3, polyesters4, and hydrogen gas5-7. In such contexts, the microbes’ evolved mechanisms of nutrient balancing run contrary to the metabolic engineer’s goal of maximizing product yield. Understanding the means by which microbes coordinate the utilization of carbon and nitrogen therefore has value both as a model for more complex cases relevant to eukaryotic biology (human disease, food crops) and as a guide to the efficient reprogramming of microbial metabolism.
Regulation of nitrogen utilization pathways by carbon availability, and vice versa, at the level of gene expression has been studied extensively in E. coli, Bacillus subtilis, and Salmonella typhimurium8, 9. The well-studied PII family of proteins has been identified as a site of integration of carbon and nitrogen signals for the purpose of regulating nitrogen assimilation10, 11; no interaction has been demonstrated, however, between the PII regulatory system and carbon uptake pathways. In fact there is no known regulatory interaction which communicates information about nitrogen status to the enzymes involved in glycolysis or glucose transport on the short time scales required for adaptation to a rapidly changing nutrient environment.
Metabolomics, utilizing liquid chromatography coupled to tandem mass spectrometry to quantify the intracellular concentrations of metabolites throughout the metabolic network, has emerged as a powerful tool for generating the quantitative data needed to probe and model fast-acting regulatory interactions between small molecules and enzymes. In this work we investigate how E. coli glucose utilization is regulated in response to a perturbation in nitrogen, beginning from a rich metabolomics dataset which indicates that neither the known regulators of glycolysis nor intermediates of the pathway are responsive to nitrogen status. Instead we show that the carbon substrate of ammonia assimilation, α-ketoglutarate, which accumulates in nitrogen limitation, reduces glucose uptake in vivo and directly inhibits the first step of glucose transport, Enzyme I (EI) of the sugar:phosphoenolpyruvate phosphotransferase system. A simple quantitative model confirms that inhibition of EI by α-ketoglutarate is sufficient to match glucose consumption to the nitrogen-controlled growth rate without perturbing glycolytic intermediates, providing an elegant regulatory link between central carbon and nitrogen metabolism.
Results
Glycolytic flux but not intermediates respond to nitrogen
We previously monitored the concentrations of ~60 metabolites in wild-type NCM 3722 (hereafter simply wild-type) E. coli cultures experiencing a sudden 13-fold increase in external ammonia concentration and a concomitant 2.5-fold increase in growth rate12. Compounds in the nitrogen assimilation pathway and its immediate metabolic neighbors, including amino acids and tricarboxylic acid (TCA) cycle intermediates, exhibited large and rapid concentration changes upon nitrogen upshift; in contrast, the concentrations of intermediates in glycolysis, as well as glycolytic regulators such as adenosine 5’-triphosphate (ATP) and adenosine 5’-diphosphate (ADP), remained homeostatic (Fig. 1). The latter result is unexpected, as enzyme activity is dependent on the concentrations of substrates, products, and allosteric effectors; the result therefore implies either an uncoupling of glucose consumption and growth rate during nitrogen limitation or the existence of an unknown mechanism which controls glycolytic flux in response to nitrogen status without substantially altering the levels of glycolytic intermediates. To eliminate the former possibility we examined the glucose uptake response of nitrogen-upshifted E. coli.
Figure 1. Glycolytic intermediates are homeostatic during nitrogen upshift.
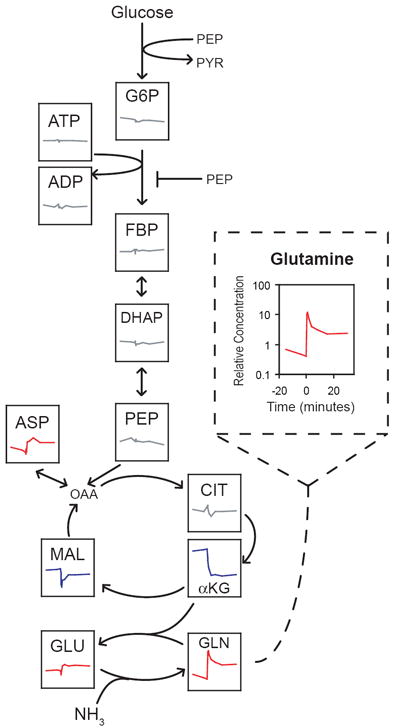
Wild-type E. coli cultures were subjected to a sudden 13-fold increase in extracellular ammonia at t = 0, inducing a 2.5-fold increase in growth rate. Each box shows the time-dependent concentration change of the indicated metabolite during the perturbation; concentration data were reported previously12. Concentrations are relative to the pool size in cells grown on ample nitrogen. All plots are on the same scale as the enlarged plot for glutamine (dashed box). Compounds are colored according to the change in steady-state concentration following the perturbation: red for increases of at least 50%, blue for decreases of at least 50%, gray otherwise. Abbreviations: G6P, glucose-6-phosphate and its isomer fructose-6-phosphate; FBP, fructose-1,6-bisphosphate; DHAP, dihydroxyacetone-phosphate; PEP, phosphoenolpyruvate; PYR, pyruvate; ADP/ATP, adenosine di-/tri-phosphate; OAA, oxaloacetate; CIT, citrate; αKG, α-ketoglutarate; MAL, malate; GLU, glutamate; GLN, glutamine; ASP, aspartate. See Supplementary Methods for experimental procedure.
Cultures were grown in minimal media batch culture with glucose as carbon source and L-arginine as nitrogen source to mid-logarithmic phase, at which point the culture was subjected to nitrogen upshift by addition of 10 mM ammonium chloride. We monitored cell growth and extracellular glucose concentration in the nitrogen-upshifted culture and in an unperturbed, nitrogen-limited control over the course of two hours. From these time courses we calculated growth rates and specific glucose uptake rates (Table 1, rows 1-2). Nitrogen upshift induced a 2.7-fold increase in growth rate and a 1.6-fold increase in specific glucose uptake rate relative to nitrogen limited cells. The increase in glucose uptake was nearly instantaneous, as the concentration of glucose consumed diverged appreciably within 5 minutes of the perturbation (Fig. 2a). Addition of L-glutamine, a nitrogen source of intermediate strength, also resulted in an immediate increase in glucose consumption, ruling out any ammonia-specific effect (Fig. 2b). Differences in scale between Figures 2a and 2b result from variation in the cell density at t = 0 between experiments; the estimated specific uptake rates for N-limited cells, obtained by dividing the slope of the line by the cell density, are comparable (6.3 mmol/gCDW/hr in 2a, 5.6 mmol/gCDW/hr in 2b).
Table 1.
Growth & sugar uptake rates following nitrogen upshift.
| Strain | C Source | N source | NH4Cl | Growth rate (hr-1) | Sugar uptake rate (mmol gCDW-1 hr-1) |
|---|---|---|---|---|---|
| NCM 3722 | Glucose | Arg | - | 0.341 ± 0.009 | 7.29 ± 0.90 |
| + | 0.919 ± 0.018 | 11.7 ± 0.76 | |||
| Glucose* | Arg | - | 0.346 ± 0.008 | 6.47 ± 1.09 | |
| + | 0.918 ± 0.012 | 11.1 ± 0.94 | |||
| Mannitol | Arg | - | 0.297 ± 0.014 | 5.75 ± 0.63 | |
| + | 0.685 ± 0.045 | 8.78 ± 1.24 | |||
| NCM 3722 Δppc | Glucose* | Arg | - | 0.370 ± 0.009 | 5.42 ± 1.00 |
| + | 0.805 ± 0.022 | 9.89 ± 1.20 | |||
| W3110 | Glucose | Asp | - | 0.280 ± 0.008 | 6.81 ± 0.76 |
| + | 0.561 ± 0.017 | 9.27 ± 0.73 | |||
| VH33# | Glucose | Asp | - | 0.215 ± 0.004 | 4.04 ± 0.34 |
| + | 0.347 ± 0.007 | 4.21 ± 0.39 |
E. coli were grown to mid-logarithmic phase in media containing the carbon (C) and nitrogen (N) sources specified. An asterisk (*) indicates a supplement of 1 g/L succinate, however sugar uptake rate refers only to the compound listed under C source. The cultures were perturbed by addition of either 10 mM NH4Cl or an equivalent volume of water. The values shown for growth rate are the mean ± standard error of three independent experiments. The sugar uptake rates and standard errors were calculated using data from all replicates as described in Methods.
W3110 derivative strain VH33 lacks the ptsH, ptsI, and crr genes, and imports glucose via overexpression of the galactose permease gene galP and the endogenous glucokinase activity
Figure 2. Glucose uptake increases rapidly upon nitrogen upshift, independent of glutamine concentration.
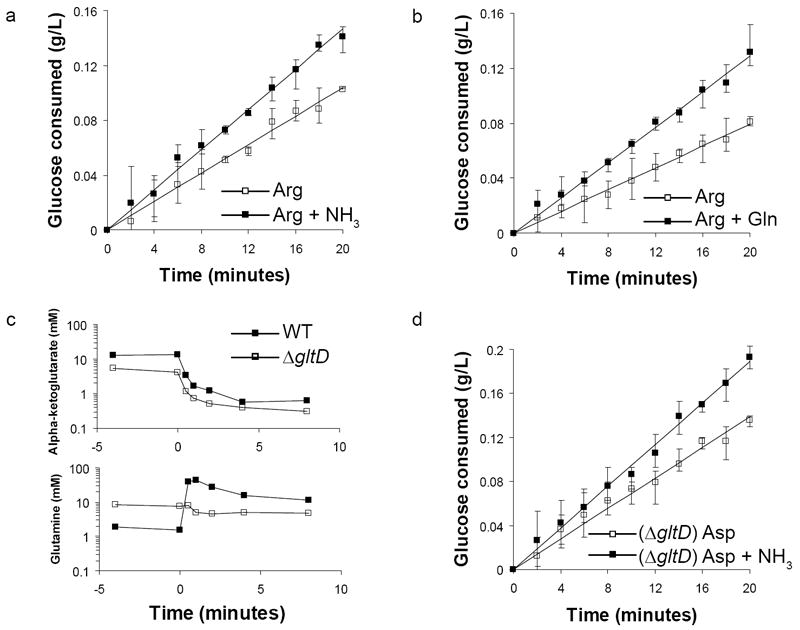
Batch cultures of (a,b) wild-type or (d) glutamate synthase deficient (ΔgltD) E. coli were grown to mid-logarithmic phase in media containing 2 g/L glucose and (a,b) 2.5 mM arginine or (d) 10 mM aspartate. At t = 0 the cultures were perturbed by addition of (a,d) 10 mM ammonia or (b) 2 mM glutamine. Plotted values are the glucose concentration (mean and range of three technical replicates) subtracted from the concentration at t = 0; lines are linear regression fits. (c) Intracellular α-ketoglutarate and glutamine concentrations during a nitrogen upshift of wild-type NCM 3722 and ΔgltD cultures12.
Anaplerosis and glutamine do not control glucose uptake
The rapidity of the glucose response to nitrogen upshift suggests a mechanism that bypasses regulation at the level of enzyme concentrations. The increased consumption of glucose upon nitrogen upshift could be explained by the increased demand for biosynthetic carbon; as the growth rate increases, more carbon will be routed into amino acid and nucleotide production. In particular, oxaloacetate and α-ketoglutarate from the TCA cycle will be consumed in the production of aspartate and glutamate, respectively. To balance this increased drain of TCA carbon, flux through phosphoenolpyruvate carboxylase (PPC), the anaplerotic reaction converting phosphoenolpyruvate (PEP) to oxaloacetate, must also increase, thereby creating a demand for PEP which could suffice to increase flux through glycolysis, as the reactions between fructose-1,6-bisphosphate and PEP are close to equilibrium and can therefore achieve substantial fold-changes in flux without proportionally large concentration changes13.
To test this hypothesis we compared the glucose uptake behavior of nitrogen limited wild-type and PPC deficient (Δppc) E. coli with and without nitrogen upshift, with a supplement of 1 g/L succinate to provide anaplerotic carbon. We found no substantial difference in growth or glucose consumption rate between wild-type and Δppc cultures, nor did the addition of succinate alter the glucose consumption of the wild-type (Table 1, rows 3-4 and 7-8). This result indicates that the rate of glucose consumption is uncoupled from the need for anaplerotic carbon. Moreover, even when anaplerosis is genetically eliminated, glucose uptake rate and nitrogen availability remain linked.
The observation that the increase in glucose consumption is not dependent on the direct routing of carbon from glycolysis to the reactions involved in nitrogen assimilation suggests that some small molecule signal might communicate nitrogen status to one or more glycolytic enzymes. The canonical signals of nitrogen limitation in enteric bacteria are decreased glutamine and increased α-ketoglutarate concentrations. In E. coli lacking glutamate synthase (ΔgltD), the glutamine response to nitrogen upshift is reversed, i.e. glutamine concentration is elevated during nitrogen limitation and falls rapidly upon nitrogen upshift; however the α-ketoglutarate response of ΔgltD is nearly identical to wild-type, with α-ketoglutarate falling by an order of magnitude within 1-2 minutes of nitrogen upshift (Fig. 2c) 12, 14. Therefore, if glutamine were to positively regulate glycolysis, a ΔgltD strain should fail to exhibit increased glucose uptake upon nitrogen upshift. ΔgltD is effectively unable to grow on arginine, a severely limiting nitrogen source, but exhibits only a minor growth defect on aspartate (Supplementary Results, Supplementary Table 1). Following nitrogen upshift from aspartate to ammonia, glucose uptake by the ΔgltD strain increased on a comparable time scale to the wild-type, indicating glutamine is not the physiological regulator of increased glucose uptake in response to nitrogen upshift (Fig. 2d).
α-ketoglutarate inhibits glucose uptake via Enzyme I
To test whether α-ketoglutarate negatively regulates glucose uptake, we grew wild-type E. coli in media with ample ammonia and monitored glucose uptake following addition of α-ketoglutarate. Addition of 20 mM α-ketoglutarate had little effect on wild-type (Table 2, rows 1-2). While a constitutively expressed α-ketoglutarate transporter exists, it is unclear whether the rate of transport would support accumulation of α-ketoglutarate in the cytoplasm15. Therefore we performed the identical experiment with a strain lacking α-ketoglutarate dehydrogenase (ΔsucA). ΔsucA grows on glucose minimal media when supplemented with a small amount of succinate to drive production of the succinyl-CoA required for the biosynthesis of methionine, lysine and heme. In ΔsucA the specific growth and glucose uptake rates each decreased by approximately 30% upon addition of 20 mM α-ketoglutarate, suggesting that α-ketoglutarate inhibits glucose consumption in vivo and induces glucose-limited growth under these conditions (Table 2, rows 7-8). Furthermore, addition of 20 mM dimethyl-ketoglutarate, a membrane-permeable ester which is cleaved by intracellular esterases to form α-ketoglutarate (Supplementary Fig. 1), induced a large decrease in glucose uptake and growth in wild-type (Table 2, rows 3-4), providing additional evidence in support of α-ketoglutarate as an inhibitor of glucose consumption.
Table 2.
Growth & glucose uptake rates following addition of α-ketoglutarate.
| Strain | C source | Perturbation | Growth rate (hr-1) | Sugar uptake rate (mmol gCDW-1 hr-1) |
|---|---|---|---|---|
| NCM 3722 | Glucose* | - | 0.999 ± 0.038 | 14.9 ± 1.84 |
| αKG | 0.990 ± 0.029 | 12.5 ± 1.41 | ||
| Glucose | - | 0.971 ± 0.049 | 13.1 ± 0.79 | |
| dmKG | 0.581 ± 0.049 | 6.70 ± 0.85 | ||
| Mannitol | - | 0.663 ± 0.044 | 8.16 ± 0.94 | |
| dmKG | 0.389 ± 0.037 | 3.49 ± 0.99 | ||
| NCM 3722 ΔsucA | Glucose* | - | 0.783 ± 0.024 | 12.65 ± 0.91 |
| αKG | 0.503 ± 0.023 | 8.82 ± 1.15 | ||
| W3110 | Glucose | - | 0.669 ± 0.027 | 11.13 ± 1.02 |
| dmKG | 0.457 ± 0.018 | 6.77 ± 0.78 | ||
| VH33# | Glucose | - | 0.388 ± 0.009 | 4.10 ± 0.59 |
| dmKG | 0.321 ± 0.013 | 3.43 ± 0.49 |
E. coli were grown to mid-logarithmic phase in media containing 4 g/L of the specified carbon (C) source and 10 mM NH4Cl. An asterisk (*) indicates a supplement of 1 g/L succinate, however sugar uptake rate refers only to the compound listed under C source. The cultures were perturbed by addition of either 20 mM α-ketoglutarate (αKG), 20 mM dimethyl-ketoglutarate (dmKG), or an equivalent volume of water in the control. The values shown for growth rate are the mean ± standard error of three independent experiments. The glucose uptake rates and standard errors were calculated using data from all replicates as described in Methods.
W3110 derivative strain VH33 lacks the ptsH, ptsI, and crr genes, and imports glucose via overexpression of the galactose permease gene galP and the endogenous glucokinase activity.
The main route of glucose transport in E. coli, as in many bacteria, is the sugar:phosphoenolpyruvate phosphotransferase system (PTS)16. The phosphate donor for phosphorylation of glucose is PEP, rather than ATP as in eukaryotes; the phosphate is transferred from PEP to histidine residues on a series of cytoplasmic proteins, Enzyme I (EI), HPr, EIIAGlc, and eventually to the membrane transporter EIIBCGlc, which simultaneously transports and phosphorylates glucose (Fig. 3a). PEP is thus a substrate for glucose transport as well as a product of glucose catabolism. Based on this network structure, we hypothesized the following strategy for varying glycolytic flux without perturbing the concentrations of glycolytic intermediates: inhibition of EI by α-ketoglutarate would have the effect of simultaneously reducing inflow of carbon at the top of glycolysis (less production of glucose-6-phosphate) and the outflow of carbon at the bottom (less consumption of PEP). Since many of the intermediate reactions are reversible and near equilibrium, the net flux may be modulated without substantial alteration to the concentration of intermediate compounds.
Figure 3. α-ketoglutarate inhibits Enzyme I in vitro.
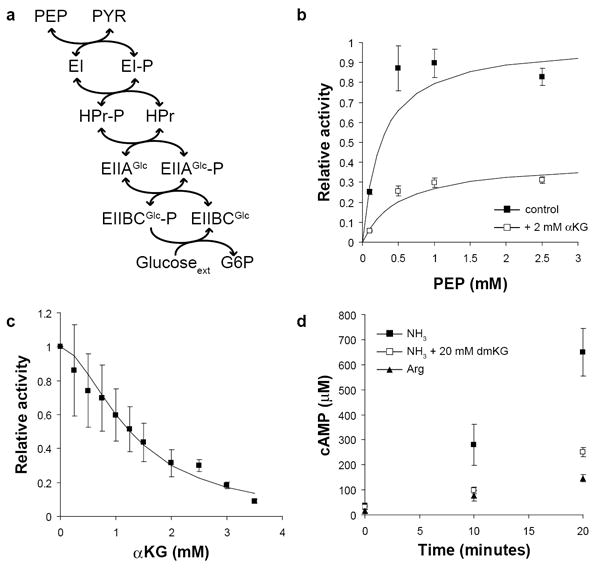
(a) Phosphoenolpyruvate (PEP) donates phosphate, which is transferred in turn to the cytosolic proteins EI, HPr, and EIIAGlc, the membrane transporter EIIBCGlc, and finally glucose, which is simultaneously transported and phosphorylated. All reactions except the final phosphorylation of glucose are reversible. (b) The activity of purified Enzyme I (EI) was determined in the presence (white squares) or absence (black squares) of 2 mM α-ketoglutarate. Plotted values are the estimated enzyme velocities ± standard error, and solid lines are the best fit Michaelis-Menten equation. Values are normalized to the estimated Vmax in the absence of α-ketoglutarate. (c) EI activity was determined in the presence of 1 mM PEP and varying concentrations of α-ketoglutarate. The solid line is the best fit Hill equation for non-competitive inhibition. Values are normalized to the activity with no α-ketoglutarate added. (d) Cyclic-AMP (cAMP) concentration was monitored following a carbon downshift from glucose to glycerol at t = 0. Cells were grown on either 10 mM ammonia or 2.5 mM arginine; where indicated, 20 mM dimethyl-ketoglutarate was present in the glycerol media following downshift. Plotted values are the mean and standard error of three biological replicates.
To directly test whether α-ketoglutarate inhibits EI, we purified overexpressed EI and determined its activity in vitro in the presence of varying concentrations of α-ketoglutarate. So that we could examine EI in isolation rather than reconstituting the entire PTS in vitro, which would be necessary for regeneration of dephosphorylated EI and net conversion of PEP to pyruvate, we assayed EI activity via its ability to equilibrate isotopically labeled carbon between PEP and pyruvate. After a brief pre-incubation of EI with 12C PEP and pyruvate, 13C pyruvate was added and the appearance of 13C PEP was monitored for the following 20 minutes.
Addition of 2 mM α-ketoglutarate inhibited EI activity in a non-competitive fashion (Fig. 3b). Glutamate, the transamination product of α-ketoglutarate, exhibited no ability to inhibit EI at 20 mM (Supplementary Fig. 2c). Surprisingly, the inhibition of EI by α-ketoglutarate was nearly complete at 4 mM, and the response to increasing inhibitor concentration was markedly cooperative (Fig. 3c); the Hill coefficient estimated by least squares regression is 1.8 ± 0.2, with a Ki of 1.3 ± 0.1 mM. Interestingly, α-ketoglutarate also non-competitively inhibits phosphoenolpyruvate synthetase (PPS)17. EI and PPS catalyze similar chemical reactions, and sequence alignment indicated that they share several important residues, including the active-site histidine which is phosphorylated, the catalytic cysteine, and four arginines which interact with the phosphate (Supplementary Fig. 3)18.
Further consequences of EI inhibition by α-ketoglutarate
The PTS catalyzes import of many sugars other than glucose, which should be similarly subject to regulation by α-ketoglutarate inhibition. We examined the rate of D-mannitol uptake by wild-type E. coli growing on arginine with and without addition of ammonia, and found that the mannitol uptake rate increased with nitrogen upshift (Table 1, rows 5-6). α-ketoglutarate concentration accumulated to a similar extent during nitrogen limitation on mannitol and glucose (Supplementary Table 2). We also observed that addition of 20 mM dimethyl-ketoglutarate reduced mannitol uptake by more than two-fold (Table 2, rows 5-6). Thus α-ketoglutarate inhibition of sugar uptake likely generalizes to all PTS sugars.
If regulation of PTS-catalyzed glucose import by α-ketoglutarate is the mechanism by which carbon and nitrogen uptake are coordinated, as we suggest, then genetically eliminating the PTS should uncouple glucose consumption from nitrogen status. Strain VH33 is a derivative of wild-type E. coli strain W3110 lacking the ptsHI and crr genes, and possessing a chromosomal promoter replacement upstream of galP driving overexpression of galactose permease, which allows glucose to enter the cell via an alternate route. The endogenous glucokinase activity of the W3110 background is sufficient to allow growth of VH33 on glucose, albeit with a slight growth defect. We examined the response of W3110 and VH33 to nitrogen upshift. Because of the known pyrimidine biosynthesis defect of W3110 growing on minimal media, arginine is too poor a nitrogen source to allow effective growth of these strains; accordingly we chose aspartate as the limiting nitrogen source (Supplementary Table 1). While W3110 increases its glucose uptake following a nitrogen upshift from aspartate to ammonia, VH33 does not, despite an increase in growth rate (Table 1, rows 9-12). Furthermore, dimethyl-ketoglutarate addition to VH33 causes little or no decrease in glucose uptake (Table 2, rows 9-12). These results confirm that the site of α-ketoglutarate action in vivo is the PTS, and that inhibition of the PTS is necessary and sufficient for carbon-nitrogen coordination.
Inhibition of EI has potential physiological consequences beyond regulation of PTS sugar uptake. Phosphorylated EIIAGlc activates adenylate cyclase. When glucose is present, EIIAGlc is 95% dephosphorylated, but upon glucose removal the fraction of phosphorylated EIIAGlc increases dramatically and adenylate cyclase is activated, producing cAMP19. We hypothesized that inhibition of EI by α-ketoglutarate would lead to a reduced level of phosphorylated EIIAGlc (assuming a basal rate of spontaneous hydrolysis of the histidine-phosphate bond) and thus a reduced rate of cAMP production. We therefore monitored cAMP concentration in NCM 3722 E. coli following a switch from glucose to glycerol media and found that cAMP production was indeed substantially slower when arginine is the nitrogen source compared to ammonia; furthermore, cAMP production by cells growing on ammonia was reduced in the presence of dimethyl-ketoglutarate (Fig. 3d). ATP concentration was not markedly perturbed in any of these conditions, and was consistently greater than 6 mM, far above the Km of adenylate cyclase, 0.21 mM (Supplementary Fig. 4)20. Accordingly, the effect of α-ketoglutarate on cAMP production is likely due to a difference in EIIAGlc phosphorylation rather than a difference in substrate availability. The observation that both nitrogen limitation and dimethyl-ketoglutarate addition inhibit cAMP production is consistent with our finding that α-ketoglutarate acts on the PTS at EI, a site upstream of EIIAGlc. It is further consistent with an earlier report that addition of α-ketoglutarate to cultures growing on glycerol results in decreased cAMP synthesis and inhibition of catabolite-repressed gene expression, and that this effect is dependent on the presence of EIIAGlc 21.
Mathematical modeling of carbon and nitrogen coordination
A simplified differential equation model of central carbon and nitrogen metabolism was designed to test whether the hypothesized regulatory strategy outlined in the previous section could account for the observed metabolite concentration changes. The metabolites and reactions represented in the model are depicted in Figure 4a: Glucose is transported and phosphorylated to glucose-6-phosphate with concomitant conversion of PEP to pyruvate. Glycolysis is simplified to the reversible interconversion of glucose-6-phosphate and two molecules of PEP. PEP and pyruvate condense to form α-ketoglutarate (PEP providing the oxaloacetate, and pyruvate the acetyl-CoA, required for synthesis of the α-ketoglutarate precursor citrate). α-ketoglutarate provides the carbon substrate for the glutamine synthetase/glutamate synthase cycle which assimilates ammonia, producing glutamate. Glutamate is consumed in producing biomass at a rate determined by the concentrations of α-ketoglutarate and glutamine (representing carbon and nitrogen availability respectively). As α-ketoglutarate is known to competitively inhibit citrate synthase, and glutamine synthetase is inhibited through a covalent modification cascade activated by glutamine, these metabolites each provide feedback inhibition on the respective reactions which form them in our model10, 22. Based on our observation that α-ketoglutarate inhibits EI, we also include feedback inhibition of the glucose uptake reaction by α-ketoglutarate. Parameters were manually fit to reproduce the observed steady-state growth rates and pool sizes of glutamine and α-ketoglutarate before and after the nitrogen upshift. The full specifications of the model are provided in Supplementary Methods.
Figure 4. Simulated pool size changes in response to varying nitrogen availability.
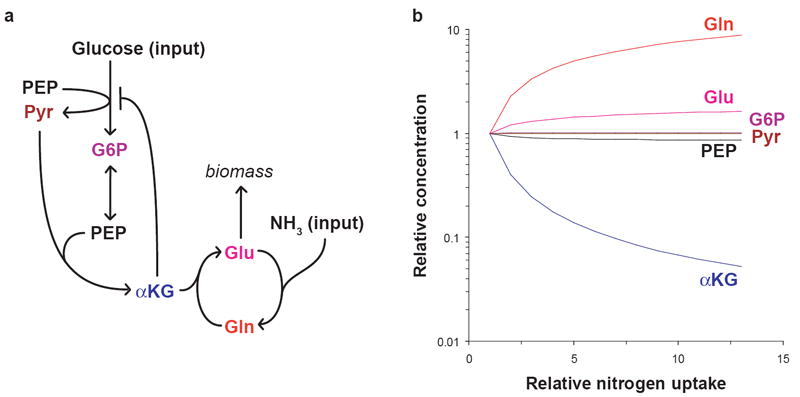
(a) The differential equation model consists of five metabolic reactions: glucose transport and phosphorylation with concomitant conversion of PEP to pyruvate; reversible glycolytic conversion of glucose-6-phosphate to two molecules of PEP; condensation of PEP and pyruvate to α-ketoglutarate; nitrogen transfer from glutamine to α-ketoglutarate producing two molecules of glutamate; and glutamine synthesis from glutamate and ammonia. Glutamate is consumed in the outflux to biomass, which is controlled by glutamine and α-ketoglutarate concentrations. Glucose and ammonia availability are the inputs to the model. α-ketoglutarate inhibits its own production as well as glucose transport, and glutamine inhibits glutamine synthesis. Abbreviations are as in the legend to Figure 1. (b) The model was used to simulate the steady-state pool sizes of each metabolite as external nitrogen availability (parameter SN in the model) was varied over a 13-fold range (reflecting the 13-fold increase in external ammonia in the experiment depicted in Figure 1) with carbon availability kept constant. The equations were simulated with all variables in the same unitless concentration scale; here, the fold changes in pool size, relative to the condition where nitrogen is most scarce (SN = 0.35), are plotted on a logarithmic scale. Abbreviations are as in the legend to Figure 1. The equations and parameter values are provided in Supplementary Methods.
We used the model to determine the steady-state concentration of each metabolite as nitrogen availability was varied over an order of magnitude with fixed carbon availability (Fig. 4b). The pool sizes of the glycolytic intermediates glucose-6-phosphate, pyruvate, and PEP remained nearly constant over this range, varying by less than 15% despite a 13-fold variation in the nitrogen-controlled growth rate. In contrast, consistent with the experimental results, glutamine and α-ketoglutarate were sensitive to nitrogen availability, accumulating and depleting respectively. Glutamate also increased with nitrogen availability but to a lesser extent than glutamine, consistent with previous observations (Fig. 1). Homeostasis of glycolytic intermediates was dependent on similar α-ketoglutarate binding constants for inhibition of glucose transport and of α-ketoglutarate production; this similarity is necessary for α-ketoglutarate levels to vary in parallel with the need to shutoff glucose uptake. Consistent with this constraint predicted by the model, our measured Ki for α-ketoglutarate acting on EI (1.3 mM) was comparable to the previously determined Ki for α-ketoglutarate acting on citrate synthase (0.7 mM)22. Otherwise, the trend in pool sizes for all simulated metabolites was generally robust to parameter perturbations (Supplementary Fig. 5; see Supplementary Methods for details of the sensitivity analysis). Thus, α-ketoglutarate inhibition of EI is sufficient to coordinate carbon and nitrogen uptake in the model without perturbation of glycolytic intermediates, as observed in the initial experiment.
Discussion
To our knowledge this work represents the first demonstration of a direct biochemical mechanism by which a bacterial cell regulates the transport of one elemental nutrient in response to the availability of another. PTS inhibition by α-ketoglutarate amounts to feedback inhibition on excess carbon uptake. Such feedback has been identified as a straightforward strategy for homeostasis maintenance and maximization of nutrient utilization efficiency when multiple nutrients are required for growth23. As the carbon substrate of nitrogen assimilation, α-ketoglutarate accumulation is a reliable indicator of nitrogen limitation. In contrast, depletion of glutamine, the intracellular proxy for external nitrogen in enteric bacteria, can be disrupted, as when glutamate synthase activity is lost24. The accumulation of α-ketoglutarate during nitrogen limitation is also observed in the eukaryotic microbe Saccharomyces cerevisiae, which like E. coli assimilates ammonia via condensation with α-ketoglutarate to form glutamate25. Cultures of S. cerevisiae grown in nitrogen-, phosphorus-, or sulfur-limited chemostats consume glucose at a reduced rate, whereas glucose is exhaustively consumed by auxotrophic strains in leucine- or uracil-limited chemostats (i.e. unnatural limitations)26. This finding indicates that the carbon uptake rate is tailored not just to growth rate but more specifically to the availability of other nutrients, and implies that specific mechanisms have evolved to achieve this coordination of nutrient uptake.
Increased flux through a pathway requires increased flux through each individual enzyme. When an enzymatic reaction operates close to equilibrium, as is the case for the majority of glycolytic reactions in E. coli growing on glucose, small changes in the ratio of reactants and products are sufficient to achieve increased flux; for reactions far from equilibrium, i.e. glucose import and phosphofructokinase, a combination of small changes in allosteric control and substrate/product occupancy of the active site may collectively produce a substantial flux change. We propose this is achieved for glucose import primarily by relief of allosteric α-ketoglutarate inhibition, and that the simultaneous increase in flux at either end of the linear pathway results in slight concentration adjustments (i.e. within the measurement error afforded by mass spectrometry-based metabolomics) driving increased net flux through the reversible reactions in between. Phosphofructokinase is subject to allosteric control by PEP and nucleotide diphosphates, and slight adjustments in these, along with the substrate fructose-6-phosphate and product fructose-1,6-bisphosphate, could together increase enzyme activity in proportion to the total pathway flux increase27. It is also possible, however, that some previously unidentified regulator of phosphofructokinase, perhaps α-ketoglutarate, plays a role in controlling its activity when nitrogen availability varies.
In combination with the well-characterized regulation of the PII nitrogen assimilation signaling network by α-ketoglutarate, our findings indicate a dominant role for α-ketoglutarate concentration in controlling central metabolism in response to external nitrogen availability. α-ketoglutarate is a promiscuous enzymatic regulator, competitively inhibiting citrate synthase and 3-phosphoglycerate dehydrogenase, the first step of serine biosynthesis22,28, and controlling the rate of aspartate production during nitrogen limitation by product inhibition of aspartate transaminase12. While both EI and PPS are non-competitively inhibited by α-ketoglutarate, PtsP (the EI homolog in the “nitrogen PTS”) is insensitive to α-ketoglutarate; in addition to an EI-like domain, PtsP contains an additional N-terminal domain of 127 residues, the presence of which may preclude interaction with α-ketoglutarate29.
Our model relies on EI activity being able to control overall PTS flux. Although exogenous expression of EI only slightly increases glucose uptake in wild-type E. coli30, this reflects feedback inhibition of endogenous EI transcription by the transcriptional repressor Mlc. In the absence of Mlc, exogenous EI expression increases the glucose uptake rate, indicating that EI has a substantial flux control coefficient31. Here we show that EI is a direct site of α-ketoglutarate inhibition and that α-ketoglutarate is a physiological inhibitor of overall PTS flux.
Consistent with α-ketoglutarate inhibition of the PTS being the primary means of coordinating carbon and nitrogen metabolism in E. coli, we find that glucose uptake is minimally sensitive to nitrogen availability in the PTS-deficient VH33 strain that has been engineered to import glucose via galactose permease. Interestingly, strains similar to VH33 are commonly used in industrial metabolic engineering. While such strains were initially designed for their higher PEP availability, an additional advantage in bioengineering may be their lack of carbon-nitrogen coordination: such strains will continue to actively metabolize carbon even when nitrogen is scarce. For applications where PEP accumulation is not desired, e.g., in biofuel production, a PTS-containing strain whose carbon metabolism is not impaired by nitrogen limitation might be preferred. Such a strain should be achievable by mutation of EI to render it α-ketoglutarate resistant.
In summary, metabolomic analysis of the transition from limiting to ample nitrogen yielded a rich data set which enabled us to confirm the physiological significance of previous biochemical observations regarding nitrogen assimilation, as well as to identify fundamental gaps in current knowledge, in this case regarding integration of carbon and nitrogen metabolism. While the inhibition of the PTS protein EI by α-ketoglutarate plays a central role in such regulation, the regulation of downstream carbon metabolic reactions, e.g., those controlling the pentose phosphate pathway flux and anaplerosis, still needs to be addressed. The general approach of using metabolomics-driven kinetic modeling to assess the adequacy of current regulatory understanding and to hypothesize new regulatory interactions, which can then be tested by targeted genetic and biochemical experiments, holds the potential to deliver a comprehensive understanding of metabolic regulation.
Methods
Strains and media
All strains were constructed from prototrophic E. coli NCM 372232, with the exceptions of W3110 and VH33, which were kindly provided by Dr. Guillermo Gosset. Strain FG 1195 (ΔgltD) was the generous gift of Dr. Dalai Yan. Strains CD001 (Δppc) and CD006 (ΔsucA) were constructed by P1 phage transduction of the kanamycin marker from the Keio collection strains JW3928 and JW0715, respectively, into the NCM 3722 background33. Strain VH33 is a derivative of previously reported W3110 derivative VH32 obtained by excision of the kanamycin marker34, 35. All media formulations contained Gutnick minimal salts36, D-glucose or D-mannitol at 2 or 4 g/L as specified, and the specified nitrogen source at 10 mM, or in the case of L-arginine, 2.5 mM to maintain an equal concentration of elemental nitrogen. For W3110 and VH33 cultures, thiamine was added to a final concentration of 50 ng/mL.
Glucose & mannitol concentration measurement and uptake rate calculation
1 mL samples were taken from liquid batch cultures and passed through a 0.2 micron filter to remove cells. For glucose media, the residual glucose concentration was measured by a YSI 7100 Select Biochemistry Analyzer (YSI Incorporated) set to recalibrate after every 5 samples. Each concentration was determined as the median of three technical replicates. For the fine time resolution assays shown in Figure 2, single experiments are displayed and each data point represents the mean and range of the technical replicates. The first timepoint is corrected for the change in culture volume upon perturbation.
For mannitol media, residual mannitol was determined by 1H-NMR spectroscopy. After dilution in deionized water, combination in a 9:1 ratio with D2O containing 5 mM sodium 2,2-dimethyl-2-silapentane-5-sulfonate (DSS), and thorough mixing, 600 uL of sample was pipetted into a 5 mm NMR test tube (Wako Pure Chemical Industries, Ltd). All NMR experiments were run on a Bruker Avance-III 500 MHz instrument (Bruker) under control by TopSpin 2.1/ICON automation using a BACS-120 autosampler. We calibrated the 90 degree pulse width on each sample using Bruker’s “pulsecal” routine prior to acquiring data with water suppression37. Water suppression was done using the excitation sculpting method using an in-house modified version of the Bruker program library38. Spectra were analyzed using MNova version 7.0.0 (Mestrelab Research S.L). In each sample the integral peak area ratio of mannitol to DSS was compared to a standard curve to obtain the mannitol concentration in g/L.
To calculate the sugar uptake rates following a media perturbation shown in Tables 1 & 2, three independent experiments were conducted in which the sugar concentration was measured every 30 minutes for two hours following the perturbation. The specific sugar uptake rate q was estimated by a simultaneous linear regression of the data from all experiments:
| (1) |
where S is the median extracellular sugar concentration in grams per liter, C is biomass density in grams cell dry weight (gCDW) per liter, and μ is the growth rate (mean of three experiments). An OD650 value of 0.6 was taken to correspond to 0.274 gCDW per liter39; no substantial difference was found in the OD/CDW ratio between wild-type cells grown in ammonia and arginine.
Enzyme I assay
PtsI (Enzyme I) containing an N-terminal His tag was purified from ASKA library strain b241640. The protocol for the determination of PtsI activity was modified from the previously reported 14C-exchange procedure41. In all reactions, PtsI was pre-incubated for 5 minutes at 37 °C with a 1:4 mixture of sodium phosphoenolpyruvate (Sigma-Aldrich, ≥97%) and 12C sodium pyruvate (Sigma-Aldrich, ≥99%), in a buffer containing 25 mM sodium phosphate adjusted to pH 7 and 2.5 mM MgCl2. The reaction was initiated by addition of an amount of 13C-labeled sodium pyruvate (Campbridge Isotope Laboratories, ≥98%) equivalent to the amount included in the pre-incubation mixture, yielding a final PEP:pyruvate ratio of 1:8. Approximately 1 μg of purified protein was used in each reaction and the total reaction volume was 300 μL. Putative inhibitors were included in the pre-incubation mixture. At each time point of interest, 20 μL samples were simultaneously quenched and diluted by pipetting into 980 μL of 80% v/v methanol in water at -70 °C. The fraction of 13C-labeled PEP in the total PEP pool was determined by negative-mode LC/MS-MS using ion-pairing reversed phase chromatography42. Supplementary Figures 2a & 2b show the PEP labeling time courses of the experiments represented in Figure 3a. Each time course (the mean and standard error of three experimental replicates) was fitted by weighted non-linear least squares regression in R to the following equation:
| (2) |
where L is the 13C-labeled fraction of the PEP pool at time t, and k is the fitted parameter representing the rate constant of the labeling reaction; 0.45 is the 13C-labeled fraction of total carbon. From the estimated k, the reaction velocity v was calculated as v = k[PEP], where [PEP] is the concentration of PEP in the reaction mixture. These velocities are plotted in Figure 3a, normalized to the estimated Vmax in the absence of α-ketoglutarate. The values plotted in Figure 3b, collected on two different days, are normalized to the velocity of the uninhibited reaction performed on the corresponding day, with the appropriately propagated standard errors.
Supplementary Material
Acknowledgments
The authors thank Dr. Jie Yuan, whose experiments laid the groundwork for the current study; Dr. Dalai Yan for providing the GOGAT- strain; and Dr. Guillermo Gosset for providing the W3110 and VH33 strains. This research was funded by NSF CAREER award MCB-0643859, Joint DOE-AFOSR Award DOE DE-SC0002077 - AFOSR FA9550-09-1-0580, and the NIH Center for Quantitative Biology Award P50 GM071508.
Footnotes
Author Contributions C.D.D. designed and performed experiments, designed the model and wrote the paper; D.J.S. designed the model and performed the model analysis; N.S.W. designed the model; J.D.R. designed experiments, designed the model and wrote the paper.
Competing Financial Interests Statement The authors declare no competing financial interests.
References
- 1.Guertin DA, Sabatini DM. Defining the Role of mTOR in Cancer. Cancer Cell. 2007;12:9–22. doi: 10.1016/j.ccr.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 2.Kapahi P, et al. With TOR, Less Is More: A Key Role for the Conserved Nutrient-Sensing TOR Pathway in Aging. Cell Metab. 2010;11:453–465. doi: 10.1016/j.cmet.2010.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Monot F, Engasser JM. Production of acetone and butanol by batch and continuous culture of Clostridium acetobutylicum under nitrogen limitation. Biotechnol Lett. 1983;5:213–218. [Google Scholar]
- 4.Kessler B, Weusthuis R, Witholt B, Eggink G. Production of microbial polyesters: fermentation and downstream processes. Adv Biochem Eng Biotechnol. 2001;71:159–182. doi: 10.1007/3-540-40021-4_5. [DOI] [PubMed] [Google Scholar]
- 5.Aoyama K, Uemura I, Miyake J, Asada Y. Fermentative metabolism to produce hydrogen gas and organic compounds in a cyanobacterium, Spirulina platensis. J Ferment Bioeng. 1997;83:17–20. [Google Scholar]
- 6.Kumazawa S, Mitsui A. Characterization and optimization of hydrogen photoproduction by a saltwater blue-green alga, Oscillatoria sp. Miami BG7. I. Enhancement through limiting the supply of nitrogen nutrients. Int J Hydrogen Energy. 1981;6:339–348. [Google Scholar]
- 7.Troshina O, Serebryakova L, Sheremetieva M, Lindblad P. Production of H2 by the unicellular cyanobacterium Gloeocapsa alpicola CALU 743 during fermentation. Int J Hydrogen Energy. 2002;27:1283–1289. [Google Scholar]
- 8.Commichau FM, Forchhammer K, Stulke J. Regulatory links between carbon and nitrogen metabolism. Curr Opin Microbiol. 2006;9:167–172. doi: 10.1016/j.mib.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 9.Wacker I, et al. The regulatory link between carbon and nitrogen metabolism in Bacillus subtilis: regulation of the gltAB operon by the catabolite control protein CcpA. Microbiology. 2003;149:3001–3009. doi: 10.1099/mic.0.26479-0. [DOI] [PubMed] [Google Scholar]
- 10.Jiang P, Ninfa AJ. Escherichia coli PII signal transduction protein controlling nitrogen assimilation acts as a sensor of adenylate energy charge in vitro. Biochemistry. 2007;46:12979–12996. doi: 10.1021/bi701062t. [DOI] [PubMed] [Google Scholar]
- 11.Ninfa AJ, Jiang P. PII signal transduction proteins: sensors of alpha-ketoglutarate that regulate nitrogen metabolism. Curr Opin Microbiol. 2005;8:168–173. doi: 10.1016/j.mib.2005.02.011. [DOI] [PubMed] [Google Scholar]
- 12.Yuan J, et al. Metabolomics-driven quantitative analysis of ammonia assimilation in E. coli. Mol Syst Biol. 2009;5:302. doi: 10.1038/msb.2009.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bennett BD, et al. Absolute metabolite concentrations and implied enzyme active site occupancy in Escherichia coli. Nat Chem Biol. 2009;5:593–599. doi: 10.1038/nchembio.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yan D. Protection of the glutamate pool concentration in enteric bacteria. Proc Natl Acad Sci U S A. 2007;104:9475–9480. doi: 10.1073/pnas.0703360104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Seol W, Shatkin AJ. Escherichia coli alpha-ketoglutarate permease is a constitutively expressed proton symporter. J Biol Chem. 1992;267:6409–6413. [PubMed] [Google Scholar]
- 16.Meadow ND, Fox DK, Roseman S. The bacterial phosphoenolpyruvate: glycose phosphotransferase system. Annu Rev Biochem. 1990;59:497–542. doi: 10.1146/annurev.bi.59.070190.002433. [DOI] [PubMed] [Google Scholar]
- 17.Chulavatnatol M, Atkinson DE. Phosphoenolpyruvate Synthetase from Escherichia coli. J Biol Chem. 1973;248:2712–2715. [PubMed] [Google Scholar]
- 18.Teplyakov A, et al. Structure of phosphorylated enzyme I, the phosphoenolpyruvate:sugar phosphotransferase system sugar translocation signal protein. Proc Natl Acad Sci U S A. 2006;103:16218–16223. doi: 10.1073/pnas.0607587103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bettenbrock K, et al. Correlation between growth rates, EIIACrr phosphorylation, and intracellular cyclic AMP levels in Escherichia coli K-12. J Bacteriol. 2007;189:6891–6900. doi: 10.1128/JB.00819-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Amin N, Peterkofsky A. Requirement for GLY-60 of Escherichia coli adenylyl cyclase for ATP binding and catalytic activity. Biochem Biophys Res Commun. 1992;182:1218–1225. doi: 10.1016/0006-291x(92)91861-j. [DOI] [PubMed] [Google Scholar]
- 21.Daniel J, Danchin A. 2-Ketoglutarate as a possible regulatory metabolite involved in cyclic AMP-dependent catabolite repression in Escherichia coli K12. Biochimie. 1986;68:303–310. doi: 10.1016/s0300-9084(86)80027-x. [DOI] [PubMed] [Google Scholar]
- 22.Pereira DS, Donald LJ, Hosfield DJ, Duckworth HW. Active site mutants of Escherichia coli citrate synthase. Effects of mutations on catalytic and allosteric properties. J Biol Chem. 1994;269:412–417. [PubMed] [Google Scholar]
- 23.Goyal S, Yuan J, Chen T, Rabinowitz JD, Wingreen NS. Achieving optimal growth through product feedback inhibition in metabolism. PLoS Comput Biol. 2010;6:e1000802. doi: 10.1371/journal.pcbi.1000802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ikeda TP, Shauger AE, Kustu S. Salmonella typhimurium apparently perceives external nitrogen limitation as internal glutamine limitation. J Mol Biol. 1996;259:589–607. doi: 10.1006/jmbi.1996.0342. [DOI] [PubMed] [Google Scholar]
- 25.Boer VM, Crutchfield CA, Bradley PH, Botstein D, Rabinowitz JD. Growth-limiting Intracellular Metabolites in Yeast Growing under Diverse Nutrient Limitations. Mol Biol Cell. 2010;21:198–211. doi: 10.1091/mbc.E09-07-0597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brauer MJ, et al. Coordination of growth rate, cell cycle, stress response, and metabolic activity in yeast. Mol Biol Cell. 2008;19:352–367. doi: 10.1091/mbc.E07-08-0779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Blangy D, Buc H, Monod J. Kinetics of the allosteric interactions of phosphofructokinase from Escherichia coli. J Mol Biol. 1968;31:13–35. doi: 10.1016/0022-2836(68)90051-x. [DOI] [PubMed] [Google Scholar]
- 28.Zhao G, Winkler ME. A novel alpha-ketoglutarate reductase activity of the serA-encoded 3-phosphoglycerate dehydrogenase of Escherichia coli K-12 and its possible implications for human 2-hydroxyglutaric aciduria. J Bacteriol. 1996;178:232–239. doi: 10.1128/jb.178.1.232-239.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rabus R, Reizer J, Paulsen I, Saier MH. Enzyme INtr from Escherichia coli. J Biol Chem. 1999;274:26185–26191. doi: 10.1074/jbc.274.37.26185. [DOI] [PubMed] [Google Scholar]
- 30.Rohwer JM, Meadow ND, Roseman S, Westerhoff HV, Postma PW. Understanding glucose transport by the bacterial phosphoenolpyruvate:glycose phosphotransferase system on the basis of kinetic measurements in vitro. J Biol Chem. 2000;275:34909–34921. doi: 10.1074/jbc.M002461200. [DOI] [PubMed] [Google Scholar]
- 31.Nishio Y, Usuda Y, Matsui K, Kurata H. Computer-aided rational design of the phosphotransferase system for enhanced glucose uptake in Escherichia coli. Mol Syst Biol. 2008;4:160. doi: 10.1038/msb4100201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Soupene E, et al. Physiological studies of Escherichia coli strain MG1655: growth defects and apparent cross-regulation of gene expression. J Bacteriol. 2003;185:5611–5626. doi: 10.1128/JB.185.18.5611-5626.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Baba T, et al. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol. 2006;2 doi: 10.1038/msb4100050. 2006.0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.De Anda R, et al. Replacement of the glucose phosphotransferase transport system by galactose permease reduces acetate accumulation and improves process performance of Escherichia coli for recombinant protein production without impairment of growth rate. Metab Eng. 2006;8:281–290. doi: 10.1016/j.ymben.2006.01.002. [DOI] [PubMed] [Google Scholar]
- 36.Gutnick D, Calvo JM, Klopotowski T, Ames BN. Compounds which serve as the sole source of carbon or nitrogen for Salmonella typhimurium LT-2. J Bacteriol. 1969;100:215–219. doi: 10.1128/jb.100.1.215-219.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu PS, Otting G. Rapid pulse length determination in high-resolution NMR. J Magn Reson. 2005;176:115–119. doi: 10.1016/j.jmr.2005.05.018. [DOI] [PubMed] [Google Scholar]
- 38.Hwang TL, Shaka AJ. Water Suppression That Works. Excitation Sculpting Using Arbitrary Wave-Forms and Pulsed-Field Gradients. J Magn Res. 1995;112:275–279. [Google Scholar]
- 39.Lu W, Kwon YK, Rabinowitz JD. Isotope ratio-based profiling of microbial folates. J Am Soc Mass Spectrom. 2007;18:898–909. doi: 10.1016/j.jasms.2007.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kitagawa M, et al. Complete set of ORF clones of Escherichia coli ASKA library (A Complete Set of E. coli K-12 ORF Archive): Unique Resources for Biological Research. DNA Res. 2005;12:291–299. doi: 10.1093/dnares/dsi012. [DOI] [PubMed] [Google Scholar]
- 41.Hoving H, Lolkema JS, Robillard GT. Escherichia coli phosphoenolpyruvate-dependent phosphotransferase system: equilibrium kinetics and mechanism of Enzyme I phosphorylation. Biochemistry. 1981;20:87–93. doi: 10.1021/bi00504a015. [DOI] [PubMed] [Google Scholar]
- 42.Lu W, Bennett BD, Rabinowitz JD. Analytical strategies for LC-MS-based targeted metabolomics. J Chromatogr, B; Anal Technol Biomed Life Sci. 2008;871:236–242. doi: 10.1016/j.jchromb.2008.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.