

Abstract
In human disease induced by Salmonella enterica serovar Typhimurium (S. Typhimurium), transepithelial migration of neutrophils rapidly follows attachment of the bacteria to the epithelial apical membrane. We have previously shown that during S. Typhimurium infection the multidrug resistance associated protein 2 (MRP2) is highly expressed at the apical surface of the intestinal epithelia, and that it functions as an efflux pump for the potent neutrophil chemoattractant hepoxilin A3. However, the molecular mechanisms regulating its apical localization during active states of inflammation remain unknown. Thus, our objective was to determine the mechanistic basis for the translocation of MRP2 to the apical surface of intestinal epithelial cells during S. Typhimurium infection. We show that suppression of ezrin, through either RNAi or truncation of the C-terminus, results not only in a decrease in S. Typhimurium-induced neutrophil transmigration but also significantly attenuates the apical membrane expression of MRP2 during Salmonella infection. In addition, we determined that S. Typhimurium induces the activation of ezrin via a PKC-α dependent pathway and that ezrin activation is coupled to apical localization of MRP2. Based on these results we propose that activation of ezrin is required for the apical localization of MRP2 during S. Typhimurium infection.
Introduction
Salmonella enterica serovar Typhimurium (S. Typhimurium) is a Gram-negative, facultative intracellular pathogen that causes a variety of diseases in humans and animals ranging from mild/severe gastroenteritis (intestinal mucosal inflammation) to systemic infection. The pathogen is typically transmitted through contaminated food or water, and upon ingestion, S. Typhimurium colonizes and invades the intestinal mucosa, where it induces disease. The gastroenteritis induced by S. Typhimurium is usually self-limiting, causing such symptoms as fever, nausea, abdominal cramps, and diarrhea, which can present with mucus and/or blood.
S. Typhimurium evokes a potent inflammatory response in the host, the hallmark of which is the migration of polymorphonuclear leukocytes (PMN) across the intestinal epithelium to the luminal surface (Gewirtz et al., 1999). This process can be separated into three distinct phases: (a) extravasation of circulating PMN from the microvasculature; (b) passage of PMN across the lamina propria to a subepithelial space; (c) paracellular movement of PMN across the epithelial monolayer. During S. Typhimurium infection PMN recruitment is coordinated in part by the epithelial release of an array of proinflammatory cytokines, among which are two potent PMN chemoattractants, interleukin 8 (IL-8) and hepoxilin A3 (HXA3). IL-8, which is secreted primarily at the basolateral surface, establishes a gradient across the lamina propria that guides PMN to the basal aspect of enterocytes. HXA3, which is released apically, has been shown to direct PMN paracellular transit across the epithelial monolayer to the luminal surface (“PMN transepithelial migration”) (McCormick et al., 1998; Mrsny et al., 2004). HXA3 is a derivative of arachidonic acid formed from the enzymatic action of the 12-lipoxygenase (12-LOX) pathway, and is a member of the eicosanoid class of lipids, which include inflammatory mediators such as leukotrienes and prostaglandins.
The S. Typhimurium effector protein, SipA, has been found to be both necessary and sufficient for induction of PMN transepithelial migration across model intestinal epithelia (Lee et al., 2000). More recently, we determined that SipA mediates PMN transepithelial migration by directly facilitating the release of HXA3 via an increase in the protein expression of the ATP binding cassette (ABC) transporter, multidrug resistance associated protein 2 (MRP2). Thus, MRP2 functions as an efflux pump for the vectorial release of HXA3 to the apical surface. Secreted HXA3 then establishes a chemotactic gradient through the tight junction complex that is used by PMNs to target the lumen of intestinal mucosal tissues at sites of inflammation (Mrsny et al., 2004). Therefore, in order to perform its function as an efflux transporter for HXA3, MRP2 is translocated to the apical membrane surface. At present, the molecular mechanism by which the apical expression of MRP2 is regulated during active states of intestinal inflammation induced by S. Typhimurium has yet to be determined.
Members of the ERM (for ezrin, radixin, moesin) protein family are critical regulators of cytoskeletal-plasma membrane interactions, especially in polarized cells (Bretscher et al., 2000). ERM proteins share two conserved domains; the NH2-terminal domain, which directly binds to plasma membrane proteins such as ICAM-1, -2, CD43 and CD44, and the –COOH terminal domain, which directly binds to F-actin (reviewed in Bretscher et al., 2000). ERM proteins also interact indirectly with other membrane proteins and transporters including Na+/H+ exchanger (NHE3), cystic fibrosis transmembrane regulator (CFTR), and platelet derived growth factor receptor (PDGFR). The linkage of these proteins with ERM members is thought to facilitate their proper localization within the cell membrane (ERM or the aforementioned bound proteins). In the inactive state, the NH2-terminal domain of ERM proteins directly interacts with the –COOH terminal domain and renders the protein “dormant.” Phosphorylation of ezrin at Thr-567, releases this interaction and activates the protein, enabling it to interact with specific membrane proteins.
In mice, loss of radixin gene expression results in a decrease of MRP2 in the bile canalicular membrane without altering the overall expression levels of the protein, suggesting radixin plays a role in the apical membrane localization of MRP2 in the bile canalicular membrane (Kikuchi et al., 2002). Furthermore, both ezrin and radixin have been reported to regulate the apical membrane localization of MRP2 in the human intestinal cell line Caco-2 (Yang et al., 2007). It has also recently been determined that there is a correlation between the activation status of ezrin and the apical localization of MRP2 in the rat intestine (Nakano et al., 2009). Of the three ERM family members, ezrin is highly enriched on the apical surface of intestinal epithelial cells. Therefore, the goal of the present study was to test the hypothesis that during infection, S. Typhimurium triggers the activation of ezrin as a means to regulate the localization of MRP2 to the apical surface, a function critical for release of HXA3, and hence the induction of PMN transepithelial migration.
Results
S. Typhimurium induced PMN transmigration is ezrin dependent
Because ezrin has been implicated in the trafficking and cellular localization of MRP2 (Yang et al., 2007; Nakano et al., 2009), we sought to determine whether ezrin is involved in the signal transduction cascade required for the ability of S. Typhimurium to induce PMN transepithelial migration. We employed a genetic siRNA approach to knock down the cellular expression levels of ezrin using ezrin-specific siRNA. Western blotting was performed to confirm and quantify the loss of ezrin by siRNA with densitometric analysis by ImageJ (Rasband, 1997-2000). We found a ~90% reduction in the amount of ezrin in uninfected and infected HCT8 monolayers expressing ezrin siRNA, in comparison to the control monolayers expressing nonspecific siRNA (Figure 1A). Our specific cell surface biotinylation experiments revealed that siRNA against ezrin, but not the vector control, decreased the apical membrane surface expression of MRP2 (Figure 1B, left panel) during S. Typhimurium infection. Ezrin specific siRNA had no effects on membrane expression of MRP2 in uninfected cells (Figure 1B, right panel) or whole cell MRP2 expression (data not shown).
Figure 1.
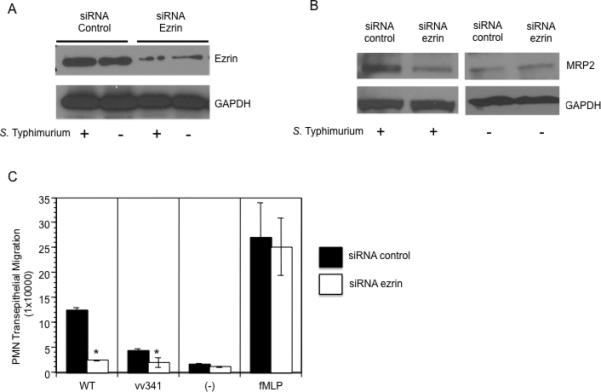
Knockdown of ezrin decreases membrane expression of MRP2 and S. Typhimurium induced PMN transmigration: (A). HCT8 cells were stably transfected with 4 μg of ezrin-specific siRNA or control siRNA, whole cell lysates were generated, separated by SDS-PAGE and Western blotted for ezrin. Ezrin knockdown cells show decreased ezrin protein expression compared to siRNA control transfected cells. (B) Membrane fractions of cells transfected with either siRNA control or ezrin-specific siRNA were extracted and Western blotted for MRP2. Cells transfected with ezrin-specific siRNA show a decreased membrane expression of MRP2 during infection with wild-type S. Typhimurium (left panel). Ezrin-specific siRNA had no effect on the levels of MRP2 in uninfected cells (right panel). GAPDH was used as loading control (C) PMN transepithelial migration: Cells transfected with control or ezrin siRNA were either infected with wild-type S. Typhimurium (WT), the hilA mutant (vv341) or treated with fMLP (positive control) or HBSS+ (negative control). Ezrin knockdown cells show a significant decrease in PMN transmigration compared to siRNA control cells following infection with wild-type S. Typhimurium. Ezrin knockdown did not have any effect on fMLP. (* =P<0.05 Student t-test). Data are from a single experiment and representative of at least three experiments that showed an identical pattern of results.
We next investigated the pathophysiological importance of ezrin during S. Typhimurium-induced PMN migration. Wild type S. Typhimurium infection-induced PMN transepithelial migration was significantly reduced across HCT8 monolayers expressing siRNA against ezrin mRNA in comparison to monolayers expressing nonspecific siRNA (Figure 1C). However, treatment of ezrin knock-down cells with imposed gradients of the PMN chemoattractant, fMLP, did not have any effect on PMN transmigration, suggesting that the decreased PMN transmigration observed in ezrin knock down cells is specific to Salmonella infection. These results demonstrate that ezrin is necessary and sufficient for both cell surface localization of MRP2 and PMN transmigration during S. Typhimurium infection.
Ezrin is required for the apical cell surface localization of MRP2 during S. Typhimurium infection and is dependent on the effector protein SipA
We have previously determined that the S. Typhimurium effector protein, SipA, mediates PMN transepithelial migration by directly affecting the release of HXA3 via an increase in MRP2 protein expression (Pazos et al., 2008). Since ezrin plays a role in anchoring many transport proteins to the apical plasma membrane, including MRP2, we sought to determine whether the effector protein SipA also governs the involvement of ezrin in facilitating the translocation of MRP2 to the apical surface during S. Typhimurium infection.
Polarized HCT8 monolayers expressing either full length or C- terminal truncated ezrin (kindly provided by Dr. Jerrold Turner, University of Chicago) were infected with either wild-type S. Typhimurium or the isogenic SipA mutant (EE633), and subsequently examined for the apical surface localization of MRP2. As shown in Figure 2A, left panel, infection of cells with wild-type S. Typhimurium induced a significant increase in the apical localization of MRP2. In contrast, cells infected with the isogenic SipA mutant strain failed to promote the recruitment of MRP2 to the apical membrane domain in cells expressing full type ezrin (Figure 2A). As a control, infection of cells expressing the truncated form of ezrin with either wild-type or SipA mutant S. Typhimurium failed to translocate MRP2. The protein expression of MRP2 was quantified by densitometry (Figure 2A, right panel). We also demonstrated that truncation of ezrin resulted in a significant decrease in PMN transmigration compared to HCT8 cells expressing full-length ezrin (supplementary figure 1).
Figure 2.
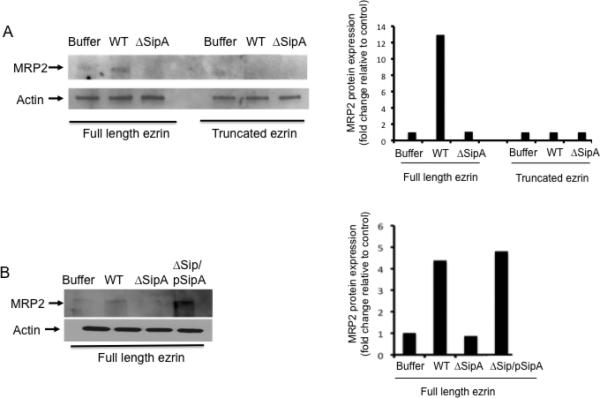
(A). Ezrin is required for apical surface localization of MRP2 during S. Typhimurium infection. Polarized HCT8 cells expressing full-length ezrin or truncated ezrin (C-terminal truncated) were infected with either wild-type S. Typhimurium (WT), SipA mutant S. Typhimurium (ΔSipA) or buffer control for one hour and the apical cell surfaces were biotinylated. Cell extracts were generated, pulled-down with streptavidin, and Western blotted for MRP2 (left panel). β-actin was used as loading control. Only cells expressing full-length ezrin and infected with WT S. Typhimurium showed increased apical localization of MRP2. The levels of MRP2 were quantified by densitometry to show the difference (right panel). Densitometry was performed using the KODAK 1D™ image analysis program and results are expressed as fold change relative to control (B) Polarized HCT8 cells expressing full length ezrin were infected with either wild-type (WT), SipA mutant (ΔSipA) or SipA complement (ΔSipA/pSipA) S. Typhimurium for 1hr and the apical cell surfaces were biotinylated, pulled-down with streptavidin, and Western blotted for MRP2. Results show that only in WT and the ΔSipA/pSipA infected cells (cells expressing full length ezrin) did we observe increased apical expression of MRP2 (left panel). MRP2 quantification was done by densitometry (right panel). Densitometry was performed using the KODAK 1D™ image analysis program. Results are expressed as fold change relative to control. Data are from a single experiment and representative of at least three experiments that showed an identical pattern of results.
As these results suggest that SipA might be inducing the activation of ezrin, we confirmed that the loss of the apical expression of MRP2 in cells infected with the SipA mutant strain could be complemented. Polarized HCT8 monolayers expressing wild-type ezrin were infected with a SipA- complemented strain (AJK63) (Lee et al., 2000) and examined for the ability to induce the translocation of MRP2. As shown in Figure 2B (left and right panels), while cells infected with the SipA mutant failed to localize MRP2 to the apical surface, cells infected with the SipA complemented strain induced the apical localization of MRP2 to the same extent as cells infected with wild-type S. Typhimurium. These results indicate that the S. Typhimurium effector, SipA, is necessary for the apical translocation of MRP2 during S. Typhimurium infection.
S. Typhimurium infection results in the activation of ezrin
Based on our observations thus far, we hypothesize that SipA is involved in the apical trafficking of MRP2 by activating ezrin. Ezrin consists of two protein interaction domains; the N-terminal domain targets the cell membrane and binds cargo proteins, whereas the C-terminal domain binds F-actin (Bretscher et al., 2000). In the inactive form, these two ezrin domains interact with each other masking the membrane and the F-actin binding sites, which keeps ezrin in its monomeric conformation (Bretscher et al., 2000). However, phosphorylation of ezrin at Thr-567 activates ezrin by releasing these interactions. As a result, ezrin is free to redistribute and interacts with the cellular cytoskeleton, most notably with F-actin.
To examine whether S. Typhimurium infection results in the activation of ezrin, polarized monolayers of HCT8 cells were infected with wild-type S. Typhimurium for up to 60 minutes, and thereafter the cells were lysed with cytoskeletal stabilization buffer (csk buffer). The detergent soluble and insoluble (cytoskeletal) fractions were collected and Western blotting was performed to assess the cellular distribution of ezrin (i.e. ezrin activation) during infection of Salmonella. As shown in figure 3A (upper panel), a marked increase in the association of ezrin with the detergent -insoluble fraction was found 30 mins after infection with wild-type S. Typhimurium. Ezrin levels stayed the same 1 hour after infection with wild-type S. Typhimurium. Infection with wild-type S. Typhimurium was also accompanied by a corresponding decrease of ezrin in the detergent -soluble fraction compared to the corresponding -insoluble fraction (Figure 3A, lower panel). We next determined whether the movement of ezrin from the detergent-soluble to the detergent–insoluble fraction was a consequence of phosphorylation at Thr-567. To do this, polarized monolayers of HCT8 cells were infected with either wild-type S. Typhimurium or the SipA mutatnt (EE633), whole cell lysates were extracted and western blotted for phospho-ezrin Thr-567. As shown in Figure 3B, infection with wild-type S. Typhimurium resulted in a profound increase in the phosphorylation of ezrin at Thr-567 as early as 30 minutes after infection. Such increase in ezrin Thr-567 phosphorylation was lost when cells were infected with the SipA mutant S. Typhimurium strain (Figure 3B). Collectively, these results demonstrate that S. Typhimurium infection of polarized intestinal cell monolayers not only promotes the activation of ezrin, but also, such activation appears to be dependent on SipA.
Figure 3.
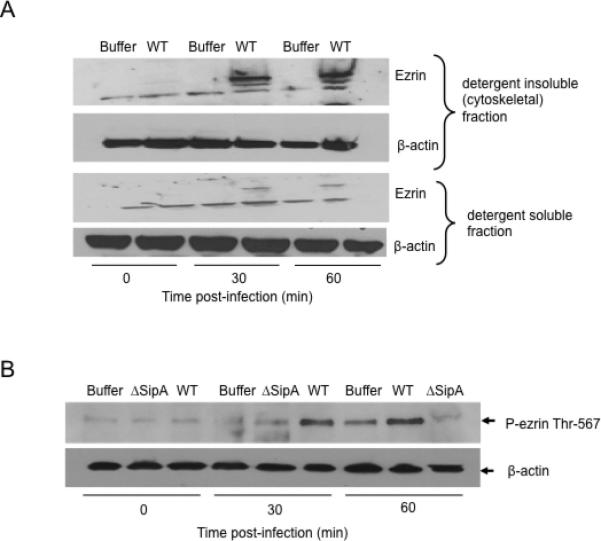
Ezrin activation during S. Typhimurium infection (A). HCT8 cells expressing full-length ezrin were infected with wild-type S. Typhimurium (WT) or left uninfected (Buffer) for indicated time points. The membrane -soluble and -insoluble (cytoskeletal) fractions were generated by lysing cells with csk buffer and analyzed by Western blotting for ezrin to examine ezrin movement from the detergent -soluble to the detergent –insoluble fraction. Results show increased movement (activation) of ezrin from the detergent -soluble to the -insoluble (cytoskeletal) fractions upon infection of cells with WT S. Typhimurium (upper panel) accompanied by a decrease in the membrane –soluble fraction (lower panel). β-actin was used as loading control. (B). HCT8 cells expressing full-length ezrin were infected with either wild-type (WT), SipA mutant (ΔSipA) S. Typhimurium or buffer control for indicated time points. Whole cell lysates were generated, normalized for protein concentration, and Western blotted for phospho-ezrin Thr-567 and β-actin. 30 mins and 1 hr infection with WT S. Typhimurium resulted in a significant increase in ezrin Thr-567 phosphorylation compared to buffer control and ΔSipA. β-actin was used as loading control. Data are from a single experiment and representative of at least three experiments that showed an identical pattern of results.
Ezrin activation and MRP2 apical localization is mediated by Protein kinase C (PKC) during S. Typhimurium infection
We have previously described evidence linking the activation of PKC-α induced by the S. Typhimurium effector protein SipA to signaling pathways that lead to PMN migration (Silva et al., 2004). In addition, a recent report further suggests that PKC-α regulates MRP2 transport activity (Ito et al., 2005). Therefore, we performed experiments to determine the extent to which PKC could be involved in both ezrin activation and the apical localization of MRP2. Polarized monolayers of HCT8 cells were treated with the pan-PKC inhibitor chelerythrine chloride (CCL) prior to infection with either the wild-type or the isogenic SipA mutant S. Typhimurium strains, and the detergent soluble and insoluble fractions were harvested and Western blotted for ezrin and β-actin. We found that inhibition of PKC resulted in a significant decrease in the association of ezrin within the detergent -insoluble fraction as compared to the untreated controls, suggesting that PKC may be required for ezrin activation and its movement (Figure 4A). The protein expression was quantified by densitometry (Figure 4A, lower panel).
Figure 4.
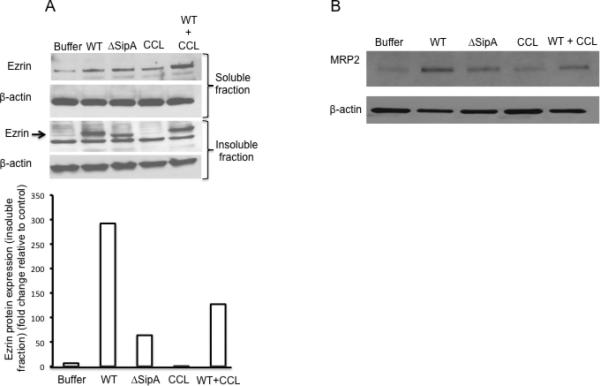
Role of PKC in ezrin activation and apical localization of MRP2 during S. Typhimurium infection. (A). Polarized HCT8 cells expressing full length ezrin were either treated for 1h with 5 μM chelerythrine chloride (CCL; a pan PKC inhibitor) or left untreated prior to infection with indicated S. Typhimurium strains (WT = wild-type, ΔSipA = SipA mutant, buffer = control) for 1hr. Ezrin activation was analyzed by lysing cells with csk buffer and examined for the movement of ezrin from the detergent –soluble to the detergent –insoluble fractions. Treatment of cells with CCL resulted in decreased movement of ezrin to the detergent –insoluble fraction (middle panel). Levels of activated ezrin were quantified by densitometry and presented on the bar chart (lower panel). Densitometry was performed using the KODAK 1D™ image analysis program and results are expressed as fold change relative to control (B). Polarized HCT8 cells expressing full-length ezrin were either treated with 5 μM CCL or left untreated for 1 hour before infection with indicated S. Typhimurium strains (WT = wild type, ΔSipA = SipA mutant, CCL alone or CCL + WT S. Typhimurium) for 1hr before apical cell surfaces were biotinylated. Cell lysates were generated, pulled-down with streptavidin, and Western blotted for MRP2. Treatment of cells with CCL (WT+CCL) resulted in loss of apical localization of MRP2 during S. Typhimurium infection. β-actin was used as loading control. Data are from a single experiment and representative of at least three experiments that showed an identical pattern of results.
We next examined whether the inhibition of PKC adversely affects the apical localization of MRP2 during S. Typhimurium infection. As described above, polarized monolayers of HCT8 cells were treated with CCL prior to infection with S. Typhimurium and the apical cell surfaces were biotinylated to examine the localization of MRP2. As shown in Figure 4B, we found that inhibition of PKC prior to S. Typhimurium infection decreased the apical expression/localization of MRP2 (Figure 4B). This result is consistent with the notion that PKC is involved in ezrin activation and the apical expression/localization of MRP2 during S. Typhimurium infection.
Ezrin Thr-567 phosphorylation during S. Typhimurium infection is PKC-α dependent
The PKC protein family consists of approximately 13 isoforms, classified into three groups based on the biochemical requirements for activation (all require phosphatidyserine for their activation; reviewed in Breitkreutz et al., 2007). Classical PKCs (cPKC) (including PKC-α, βI, βII and γ) are calcium sensitive and require diacylglycerol (DAG) for their activation; novel or nPKCs (PKC-δ, η, ε and θ) are calcium independent but require DAG for their activity, while the atypical or aPKCs (PKC-ζ, λ) are also calcium independent but phosphatidylserine alone is sufficient for their maximal activity (reviewed in Breitkreutz et al., 2007). We have previously described the involvement of PKC-α in Salmonella induced PMN transmigration (Silva et al., 2004), thus we sought to investigate whether ezrin activation could also be mediated by PKCα. Prior to infection, HCT8 cells were treated with the PKC-α inhibitors Gö6976 (5μM) and HBDDE (5μM), or the PKC-δ inhibitor, Rottlerin (10 μM), as a non-specific control. Following infection with wild-type S. Typhimurium or the SipA mutant, whole cell lysates were harvested and Western blotted for phospho-ezrin Thr-567. As shown in Figure 5A and B, pre-treatment of cells with either Gö6976 or HBDDE decreased the phosphorylation of ezrin at Thr-567 following infection with wild-type S. Typhimurium when compared to untreated cells or cells treated with the non-specific PKC-δ control, rottlerin (Figure 5C). These results indicate that ezrin phosphorylation at Thr-567 and its activation during S. Typhimurium infection is PKC-α dependent.
Figure 5.
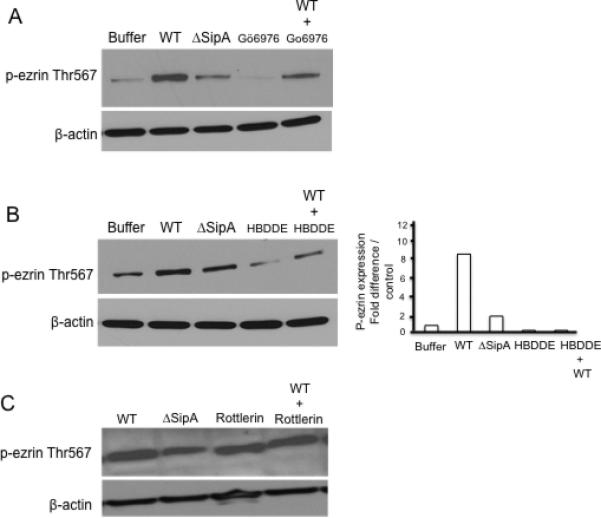
PKC-α decreases ezrin Thr-567 phosphorylation during S. Typhimurium infection. Polarized HCT8 cells were treated for 1hr with the PKC-α inhibitors (A). Gö6976, (B). HBDDE and (C). Rottlerin (PKC-δ inhibitor) prior to infection with S. Typhimurium (WT = wild-type, ΔSipA = SipA mutant, buffer = control) for 1hr. Whole cell lysates were generated, protein normalized and analyzed by Western blotting for p-ezrin Thr-567. Results demonstrate that treatment of cells with the PKC-α inhibitors Gö6976 and HBDDE resulted in a significant decrease in p-ezrin Thr-567 compared to control. Treatment of cells with a PKC-δ inhibitor (Rottelrin) did not show any change in p-ezrin Thr569 expression. β-actin was used as loading control. Densitometry analysis of p-ezrin Thr-567 Western blots following treatment of cells with the PKC-α inhibitor HBDDE. Densitometry was performed using the KODAK 1D™ image analysis program. Results are expressed as fold change relative to control. Data are from a single experiment and representative of at least three experiments that showed an identical pattern of results.
Thus far, we have shown that treatment of cells with the pan PKC inhibitor (CCL) results in a significant decrease in apical membrane translocation of MRP2 during S. Typhimurium infection (Figure 4B). We sought to determine whether apical translocation and/or expression of MRP2 during S. Typhimurium infection are specific to PKC-α (as is ezrin phosphorylation). To address this question, we carried out apical cell surface biotinylation. Prior to infection, HCT8 cells were treated with the PKC-α specific inhibitors Gö6976 (5μM) or HBDDE (5μM). Following infection with wild-type S. Typhimurium, the cells were washed and the apical cell surface was biotinylated (see methods). As shown in Figure 6A and 6B, treatment of cells with either PKC-α specific inhibitor Gö6976 or HBDDE, respectively, resulted in a significant decrease in apical translocation of MRP2 during S. Typhimurium infection. These results demonstrate that PKC-α is not only required for ezrin phosphorylation/activation but also required for apical translocation of MRP2 during S. Typhimurium infection.
Figure 6.
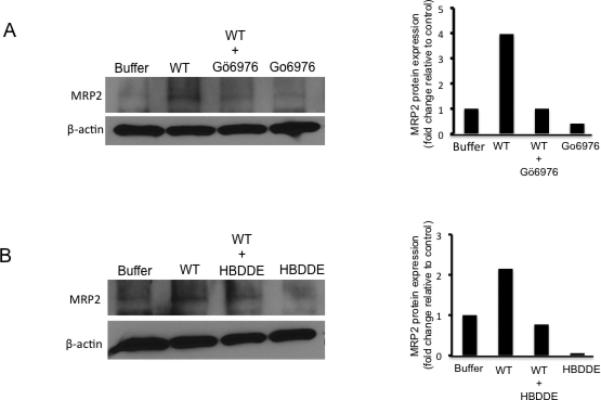
PKC-α treatment decreases apical translocation of MRP2 during S. Typhimurium infection. Polarized HCT8 cells were either treated with (A) 5 μM Gö6976 or (B) 5 μM HBDDE for 1 hour before infection with wild-type S. Typhimurium (WT = wild type) for 1hr before apical cell surfaces were biotinylated. Cell lysates were generated, pulled-down with streptavidin, and Western blotted for MRP2. Treatment of cells with the PKC-α specific inhibitors (A) Gö6976 or (B) HBBDE resulted in a significant loss of apical localization of MRP2 during S. Typhimurium infection. Bar charts represent densitometry analysis and are provided on the right side of each western blot. β-actin was used as loading control. Data are from a single experiment and repetitive of at least three experiments that showed an identical pattern of results.
S. Typhimurium infection induces ezrin/MRP2 co-localization and interaction
The results described thus far point to a role of ezrin in the apical translocation/localization of MRP2 during S. Typhimurium infection. However, the question of how ezrin may be involved in the apical transport of MRP2 remains to be determined. One possibility is that during Salmonella infection ezrin and MRP2 interact with each other to facilitate apical translocation of MRP2. To test this hypothesis, we used two different but complementary approaches. First, we used fluorescence microscopy combined with confocal microscopy to examine the extent to which infection of cells with wild-type Salmonella resulted in co-localization of ezrin and MRP2. Polarized monolayers of HCT8 cells maintained on membrane filters were infected with wild-type S. Typhimurium, the SipA mutant, or incubated with the HBSS+ control prior to co-immunostaining for both ezrin and MRP2. We found that infection of wild-type S. Typhimurium resulted in a significant increase in ezrin/MRP2 co-localization on the apical cell membrane (Figure 7A, middle panel), while no such co-localization was observed in cells infected with the isogenic SipA mutant (Figure 7A, bottom panel) or the uninfected control cells (Figure 7A, upper panel). These results were confirmed by confocal microscopy (Figure 7B).
Figure 7.
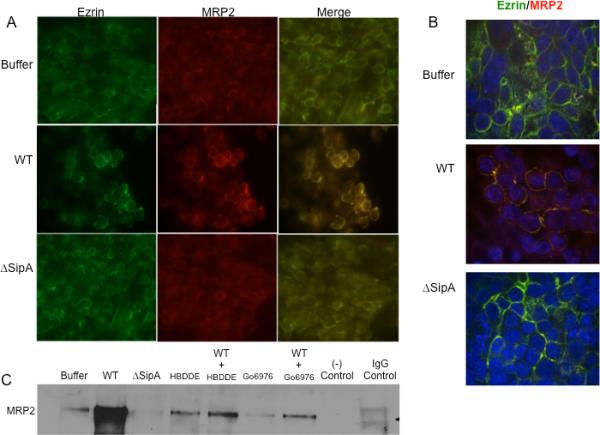
S. Typhimurium infection induces ezrin/MRP2 co-localization. (A) Immunoflourescence analysis of ezrin and MRP2 following infection with wild-type (WT), SipA mutant (ΔSipA) S. Typhimurium or buffer control for 1hr and co-immunostained for ezrin (green) and MRP2 (red) or merged for co-localization as indicated. Infection of cells with WT S. Typhimurium resulted in a significant increase in ezrin:MRP (yellow) co-localization (right, middle panel) compared to buffer control and ΔSipA infected cells. (B). Cells were treated as above and subjected to confocal microscopy. As above cells infected with WT S. Typhimurium showed a significant increase in ezrin/MRP co-localization at the apical surface (yellow, middle panel). (C). Polarized HCT8 cells were either treated with 5 μM of HBDDE, 5 μM Gö6976 or buffer control as indicated for 1 hr prior to infection with either wild-type (WT) or SipA mutant (ΔSipA) S. Typhimurium or buffer control for 1hr. Whole cell lysates were generated, and the proteins were normalized and immunoprecipitated with rabbit anti-ezrin antibody (1:100) prior to Western blotting for anti-goat MRP2. Goat IgG was used as matched control. Infection of cells with WT S. Typhimurium resulted in increased ezrin/MRP2 interaction, which was decreased upon treatment of cells with PKC-α inhibitors (HBDDE and Gö6976). Data are from a single experiment and representative of at least three experiments that showed an identical pattern of results.
Second, we used a biochemical “pull-down” approach in which polarized monolayers of HCT8 cells were infected in the absence or presence of either wild-type S. Typhimurium or the isogenic SipA mutant. Following infection, whole cell lysates were immunoprecipitated with anti-ezrin antibody, and then Western blotted for MRP2. Infection of cells with wild-type S. Typhimurium increased ezrin:MRP2 interactions as shown by the intense MRP2 band in the Western blot, as compared to cells infected with either the SipA mutant strain, or the uninfected control (Figure 7C). These results demonstrate that S. Typhimurium infection increases ezrin:MRP2 interactions and that the SipA effector protein is required to instigate this interaction. To further examine whether this interaction is PKC-α dependent, we used a similar approach. As shown in Figure 7C, treatment of cells with both PKC-α inhibitors (HBDDE and Gö6976) decreased the interaction of ezrin and MRP2 during S. Typhimurium infection demonstrating that PKC-α phosphorylation of ezrin is required for its interaction with MRP2 and that this interaction is PKC-α dependent.
Discussion
In the present study, we show that suppression of ezrin adversely influences the ability of S. Typhimurium to induce PMN transmigration because the apical surface expression of MRP2 also becomes compromised. As MRP2 facilitates the apical release of the PMN chemoattractant, HXA3, suppression of this efflux mechanism directly correlates with a decrease in S. Typhimurium-induced PMN migration. Thus, our study tested the hypothesis that during infection S. Typhimurium triggers the activation of ezrin as a means to regulate the localization of MRP2 to the apical surface, a function critical for release of HXA3, and hence the induction of PMN transepithelial migration. In support of this hypothesis we reveal three key findings.
First, we determined a link between the activation of ezrin and the ability of S. Typhimurium (in a SipA dependent manner) to not only induce PMN migration, but also to translocate MRP2 to the apical membrane surface. Our observations are consistent with recent reports documenting the involvement of ERM proteins in the apical localization of MRP2. As an example, the predominant ERM protein expressed in hepatocytes and bile canalicular is radixin, and this protein was shown to be necessary for the apical localization of MRP2 (Kikuchi et al., 2002). Mutations in the human MRP2 gene results in conjugated hyperbilirubinemia, defective excretion of organic anions, and leads to a disease known as Dubin-Johnson syndrome (Wakusawa et al., 2003). Remarkably, radixin deficiency in mice results in a similar phenotype with loss of MRP2 from the apical canalicular membranes, emphasizing the critical role played by radixin in directing the apical localization of MRP2 in the bile canalicular membrane (Kikuchi et al., 2002). ERM proteins have also been reported to play a critical role in the modulation of membrane expression of P-glycoprotein (another member of the ABC family) in cells of human lymphoid origin (Luciani et al., 2002). However, whether ERM regulation of apical membrane protein expression of members of the ABC family is a general phenomenon remains to be determined.
Another study has described the requirement of radixin in maintaining the structure, and function of the apical canalicular membrane in rat hepatocytes (Wang et al., 2006). In keeping with this observation, in the developing mouse intestine, ezrin was shown to be essential for the establishment and/or maintenance of epithelial cell polarity, as well as villus morphogenesis (Saotome et al., 2004). The same study also determined that ezrin regulates the localization and/or function of some apical membrane proteins required for normal intestinal functions, including MRP2. These studies are consistent with our findings where we demonstrate the ERM protein, ezrin, plays a critical role in regulating the apical surface localization of MRP2 during S. Typhimurium infection.
Second, we have determined that infection of cells with wild-type S. Typhimurium but not the SipA mutant strain resulted in increased ezrin:MRP2 interactions. These results suggest that ezrin is functioning as a scaffold where MRP2 binds and is transported to the apical cell surface, and offer an explanation of how ezrin may induce the apical transport of MRP2 during S. Typhimurium infection. In rat intestinal epithelial cells and Caco-2 cells ezrin:MRP2 interactions have also been shown to be associated with apical surface localization of MRP2 (Yang et al., 2007; Nakano et al., 2009), supporting our observations described above. However, whether ezrin and MRP2 interact directly or indirectly through another adaptor protein remains to be determined.
Speculatively, a potential candidate protein that may function as a scaffold, mediating ezrin-MRP2 interaction in intestinal epithelial cells is NHERF-1 (Na+/H+ exchanger regulatory factor 1). NHERF-1 binds to both MRP2 and ezrin through its PDZ domain located on its C- and N-termini (Reczek et al., 1997; Weinman et al., 1995). Moreover, the PDZ domain of NHERF-1 has been reported to bind to many transport and membrane proteins including the CFTR, the β2-adrenegic receptor, and the Na+/H+ exchanger-3 (NHE3) where it facilitates their cross linking to F-actin cytoskeleton (Bretscher et al., 2000; Cao et al., 1999; Reczek et al., 1997). In the liver, NHERF-1 was also shown to directly bind to MRP2, an interaction that promoted the apical localization of MRP2 in hepatocytes (Li et al., 2010). As NHERF-1 is also expressed in intestinal epithelial cells, its potential involvement in apical localization of MRP2 or ezrin-MRP2 interaction during S. Typhimurium infection is an obvious extension of our current study.
Our third key finding revealed that S. Typhimurium infection resulted in the activation of ezrin via phosphorylation at Thr-567, and this occurred in a SipA dependent manner. As PKC is known to play a significant role in the signaling cascades leading to ezrin activation, we first addressed whether PKC was involved in activating ezrin during S. Typhimurium infection, and if so, which isoform was involved. We found that inhibitors specific to PKC-α decreased the apical localization of MRP2, attenuated ezrin-MRP2 interactions, and significantly reduced the phosphorylation of ezrin at Thr-567, suggesting that PKC-α activates ezrin during S. Typhimurium infection.
Several protein kinases including the p38 MAPK, protein kinase B (Akt2), phosphatidyl inositides and members of the PKC family have been implicated in ezrin activation/phosphorylation in normal physiology and upon stimulation (Zhao et al., 2004; Shiue et al., 2005; Rasmussen et al., 2008; Nakano et al., 2007). While PKC iota (PKC-ι) has been shown to be involved in ezrin activation during normal intestinal epithelial differentiation, in response to initiation of Na+-glucose co-transport (Wald et al., 2007), there is a rapid increase in the apical membrane association of NHE3 and in its cytoskeletal association with ezrin, which parallels ezrin Thr-567 phosphorylation (Zhao et al., 2004). In this study, the p38 MAP kinase was identified as the kinase involved in ezrin Thr-567 phosphorylation.
Another study reported a correlation between the apical localization of MRP2 in rat intestines with ezrin activation upon conventional PKC activation (Nakano et al., 2009). However, we have previously described a critical role played by PKC-α in PMN transmigration whereby inhibition of PKC-α resulted in a decrease in PMN transepithelial migration during S. Typhimurium infection (Silva et al., 2004) consistent with the role of PKC-α in ezrin activation, apical translocation of MRP2, and HXA3 secretion. Thus, based on these results, we propose that S. Typhimurium infection, through a SipA dependent mechanism, induces the activation of PKC-α (Silva et al., 2004), which in turn phosphorylates ezrin at Thr-567. Phosphorylated ezrin then modulates apical surface localization of MRP2 in intestinal epithelial cells (Figure 8). Taken together these results reiterate the central role played by PKC-α in Salmonella induced enteritis suggesting PKC-α can be used as an important therapeutic target during S. Typhimurium infection.
Figure 8. Working model by which S. Typhimurium induces the apical expression of MRP2.
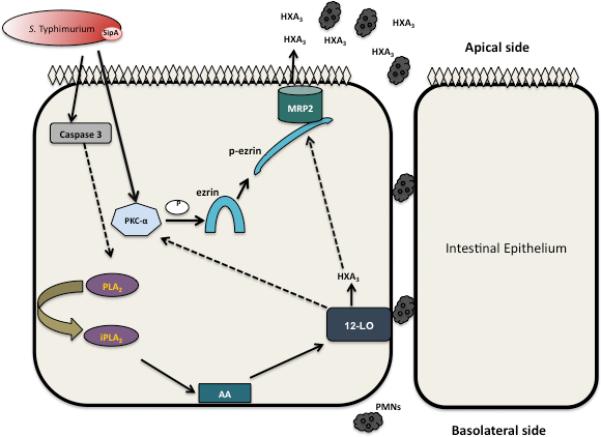
S. Typhimurium infection induces activation of PKC-α (Silva et al., 2004) and caspase-3 (Srikanth et al., 2010). Once activated, PKC-α activates ezrin via its phosphorylation at Thr-567. p-ezrin then associates with the cellular cytoskeleton, binding and facilitating transport of MRP2 to the apical cell membrane of intestinal epithelial cells and efflux of HXA3. Also, inhibition of 12-LOX has been reported to decrease apical expression of MRP2 in mice infected with S. Typhimurium, and PMN migration (Pazos et al., 2008). Dotted arrows indicate unknown mechanisms that require further examination. Abbreviations: AA = Arachidonic acid, PLA2 = phospholipase A2, 12-LOX = 12-Lipoxygenase, PKC-α = protein kinase C alpha, HXA3 = Hepoxilin A3, PMN = polymorphonuclear leukocytes.
In conclusion, we have shown that S. Typhimurium infection, in a process mediated by SipA; induces ezrin activation via a PKC-α dependent mechanism and that ezrin activation is coupled to apical localization of MRP2. We also provide evidence that there is a direct correlation between apical membrane localization of MRP2 in intestinal epithelial cells and the ability to induce S. Typhimurium-induced PMN transmigration. These results indicate that during S. Typhimurium infection ezrin plays a critical role in the pathogenesis of S. Typhimurium induced enteritis. Whether other inducers of intestinal inflammation (i.e. autoimmune or idiopathic colitis such as Crohn's disease and ulcerative colitis) share a similar mechanism of inflammation remains to be determined.
Experimental Procedures
Reagents and antibodies
Protein kinase C inhibitors; Chelerythrine chloride (CCL), HBDDE, Gö6976, and Rottlerin were purchased from Enzo (Plymouth Meeting, PA), polyclonal goat anti-MRP2 antibody, mouse anti-MRP2 antibody, donkey anti-goat rhodamine, sheep anti-rabbit FITC were obtained from Santa Cruz (Santa Cruz, CA), rabbit anti-ezrin and rabbit anti-phosphoezrin Thr-567 were purchased from Cell Signaling (Danvers, MA), Sulfo-NHS-LC biotin and Sulfo-NHS-LC-acetate were purchased from Thermo-Scientific (Rockford, IL), and mouse anti-β-actin antibody and N-formylmethionyl-leucyl-phenylalanine (fMLP) were purchased from Sigma (St. Loius, MO). All secondary antibodies used for Western blotting were purchased from Santa Cruz.
Cell culture
HCT8 intestinal epithelial cells (passage number 40-56) expressing ezrin or with truncated ezrin (C-terminal truncated) were grown in RPMI 1640 (Invitrogen, Carlsbad, CA) supplemented with 10% FBS and 100 units/ml Penicillin/Streptomycin and maintained on tissue culture treated flasks (Costar, Cambridge, MA 3151) using the protocol described by McCormick et al. (McCormick et al., 1993). Briefly, monolayers were grown on polycarbonate filters (Costar 3412) and used 6-8 days after plating. Inverted monolayers (Costar 3421) were used for PMN transmigration assays. For biotinylation and immunoprecipitation, cells were plated on transwells in 6 well plates (Costar 3412) or on transwells in 100 mm tissue culture dishes (Costar 3419) using RPMI 1640 (Invitrogen) supplemented with 10% FBS and 100 units/ml Penicillin/Streptomycin. HCT8 cells stably overexpressing full length and C-terminal truncated ezrin (kind gift from Dr. Jerrold Turner, University of Chicago) were maintained as described above for normal HCT8 cells.
Bacterial strains and growth conditions
The S. Typhimurium strains used include EE633 (sipA::lazZY4), VV341 (hilA::kan-339) and AJK63 (EE633/pAK68C, SipA complement). All of these strains are isogenic mutants of S. Typhimurium SL1344 (Lee et al., 2000). S. Typhimurium cultures were grown in Luria Bertani broth (LB; Becton Dickinson, Sparks, MD) aerobically with agitation at 37°C for ~8 h to reach stationary phase of growth. Once stationary phase was reached, bacteria were diluted 1:1000 into fresh LB and incubated overnight (~18 h) at 37°C without agitation. Bacteria from these cultures were in the late logarithmic phase of growth and correlated to 5-7 × 108 bacteria ml-1.
Generation of siRNA
Plamids used to generate small interfering RNA (siRNA) against ezrin were constructed using the pSUPER vector (Oligoengine, Seattle, WA) using the method described by Mumy et al. (Mumy et al., 2008). Briefly, oligonucleotides were designed by incorporating a 19-nucleotide sequence (shown below in italics) from the targeted human ezrin (EZRIN; GeneBank accession number, NM_003379) transcript and its reverse complement (also shown in italics) separated by a short spacer region along with Bg1II and HindIII restriction sites. The Ezrin oligonucleotide sequenceusedis: 5’-GATCCCC GAGAAGAAAAGGAGAGAAATTCAAGAGATTTCTCTCCTTTTCTTCTC TTTTTGAAA-3’ and 5’AGCTTTTCCAAAAAGAGAAGAAAAGGAGAGAAATCTCTTGAATTTCTCTCCTTTTCTTCTCGGG-3’. Random oligonulceotides were also generated to use as controls. The oligonucelotides were annealed, yielding double-stranded DNAs with overhanging restriction sites and ligated into digested pSUPER. Constructs were transformed into competent E. coli DH5α by standard methods and plated on LB with 50 μg/ml Ampicillin. Plasmids were extracted using the Qiaprep spin miniprep kit (Qiagen, Valencia, CA) and sequenced for confirmation prior to use.
Transfection of cells with siRNA
The HCT8 intestinal cell line (a kind gift of Cheleste Thorpe, Tufts University School of Medicine), a transformed polarizing human intestinal epithelial with high transepithelial electrical resistance (>1200 Ohms cm2) was used for this study because of its high transfection efficiency and ease of genetic manipulation. The cells were transfected with the modified pSUPER (4μg) using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) transfection reagent according to manufacturer's instructions. The next day cells were passaged into fresh media with a selection agent (neomycin-G418, 1mg/ml; Sigma-Aldrich, St. Loiuis, MO). Cells underwent two more selection cycles before use for experiments.
PMN transepithelial migration assay
The PMN (polymorphonuclear leukocytes) transepithelial migration assay was performed as previously described with some modifications (Parkos et al., 1992). Briefly, inverted polarized HCT8 cell monolayers seeded on 0.33-cm2 filters were apically infected with S. Typhimurium at a multiplicity of infection (MOI) of 100:1 at 37°C for one hour. Following infection, cells were washed and transferred to a new 24-well plate containing 1 ml HBSS+ in the bottom chamber (apical side). One hundred μl HBSS+ was added on the basolateral surface of the monolayers followed by 20 μl of prepared PMNs (1 × 106 PMNs). The monolayers were incubated at 37°C for 2 hours, after which the monolayers and non-migrating PMNs were gently removed leaving only those PMNs that had migrated through the monolayer. As a positive control for PMN transmigration, imposed gradients of the potent chemoattractant, N-formylmethionyl-leucyl-phenylalanine (fMLP, 1 μM Sigma, St. Louis, MO) were generated. Transmigration was quantified by assaying for the PMN azurophilic granule marker myeloperoxidase (MPO) as previously described (Parkos et al., 1992).
Treatment of cells with PKC inhibitors
Cells maintained on transwells were washed with Hank's balanced salt solution (HBSS+, plus calcium chloride) and treated for one hour with indicated concentrations of the PKC inhibitors. After treatment, cells were infected with either the wildtype or the SipA mutant S. Typhimurium for 1 hour. The inhibitor(s) was administered throughout the infection period. Chelerythrine chloride (CCL), HBDDE and Gö6976 were used at 5 μM concentrations while Rottlerin was used at 10 μM concentration. These concentrations have previously been reported to inhibit PKCs (Silva et al., 2004).
Cell surface biotinylation
Apical cell surface biotinylation was performed using the protocol described by Strohmeier et al. with some modifications (Strohmeier et al., 1997). Briefly, polarized HCT8 cell monolayers seeded on 7.5-cm2 filters were apically infected with either wild-type S. Typhimurium (SL1344), the isogenic SipA mutant strain (EE633) or uninfected (HBSS+) for 1 hour at a MOI of approximately 100:1. After infection, non-adhering bacteria were removed by washing with HBSS+ and the cells were incubated for an additional 60 min. Briefly, after two biotinylation steps (20 minutes each at pH 9.0 and pH 8.5 respectively) with 0.5 mg/ml Sulfo-NHS biotin (apical) and 0.5 mg/ml Sulfo-NHS acetate (basolateral; Pierce), the cells were incubated with quenching buffer (50 mM NH4Cl in HBSS+) for 20 min at 4°C. Proteins were extracted following a 30 min incubation at 4°C with lysis buffer containing 150 mM NaCl, 50 mM Tris-HCl (pH 8), 5 mM EDTA, 1% Triton X-100, 5 mM Na3VO4, 20 mM NaF, 0.8 mM PMSF and complete protease inhibitor cocktail tablets. The extracted proteins were centrifuged at 12,000 rpm for 5 min at 4°C, protein normalized and equal protein of in the supernatant (input) exposed to streptavidin beads (Sigma) that had been pre-washed with high salt buffer (500 mM NaCl, 5 mM EDTA, 50 mM Tris-HCl at pH 7.5 and 0.1 % Triton X-100) and lysis buffer. Following an overnight incubation at 4°C with slight agitation, the beads were collected by centrifugation, boiled 5 min in tricine sample buffer (Bio-Rad), and Western blot analysis was performed, immunoblotting for MRP2. The input was also separated by SDS-PAGE and immunoblotted for β-actin.
Protein extraction for Western blotting
For generation of whole cell lysates, cells were lysed in accordance with Kohler et al., (Kohler et al., 2007). Once lysed, the cells were passed through a 25-gauge needle 5 times prior to centrifuging at 12,000 rpm for 5 minutes. Analysis of the detergent-soluble and –insoluble fractions was performed according to Zhou et al. (Zhou et al., 2004). Following cell lysis, the proteins were normalized, separated by SDS-PAGE gels and immunoblotted accordingly.
Co-immunoprecipitation
Immunoprecipitation was carried out as described in Cummins et al. (Cummins et al., 2006). Cells grown and maintained on Costar transwells™ in 100 mm tissue culture dishes were apically infected with either wild-type S. Typhimurium, the isogenic SipA mutant strain or the uninfected control (HBSS+) for one hour. Following this incubation the cells were washed and whole cell lysates were generated. Protein normalized samples were then pre-cleared with AG agarose beads (Santa Cruz), and the supernatant was incubated with anti-ezrin antibody (1:50). The resulting samples were resolved on a 10% SDS-poly-acrylamide gel, transferred to nitrocellulose membranes, blocked with 5% non-fat milk and incubated overnight with mouse-anti MRP2 (1:500). Membranes were washed and incubated with the anti-mouse IgG horseradish peroxidase labelled secondary antibody. Bands were identified using enhanced chemiluminescence (Thermo-Scientific).
Immunoflourscence and confocal microscopy
Cells maintained on Costar transwells™ were infected with either wild-type S. Typhimurium, the isogenic SipA mutant or left uninfected for one hour. Cells were washed with HBSS+ and incubated at 37°C for another one hour before immunoflourescence studies were carried out. Briefly, cells were washed twice with PBS and fixed with 4% paraformadehyde/PBS for one hour. Following two washes in PBS, cells were incubated with 50mM ammonium chloride/PBS for 15 minutes (quenching) and permeabilized with 0.1% Triton-X 100 in PBS for 7 minutes. The cells were then washed twice with PBS before the filter membranes were carefully cut and placed onto a humid chamber (cells facing upwards) where 50μl of blocking buffer (3% BSA/PBS) was added for 30 minutes followed by the addition of the primary antibodies prepared in 3% BSA/PBS (anti-rabbit ezrin (1:20), anti-goat MRP2 (1:20)) and incubated overnight at 4°C. After two washes a 1:100 dilution (diluted in 3% BSA/PBS) of the appropriate species-labelled secondary antibody (FITC for anti-rabbit or rhodamine for anti-goat) was added at room temperature for one hour. Next, the filter membranes were washed twice with PBS, rinsed in distilled water and mounted on ethanol cleaned glass slides using Vectashield mounting medium with DAPI (Vector Lab Inc., Burlingame, CA) and left overnight. Slides were examined and images acquired on an upright fluorescent microscope (Nikon Eclipse E800) and an integrated Canon DS126131 Camera. Confocal images were acquired using an MVI Eclipse TE2000E Confocal microscpe using metamorph software and the integrated technologies and image analysis was done using the Image J program (NIH).
Supplementary Material
Acknowledgements
The research was supported by grants from the Crohn's and Colitis Foundation of America and the National Institutes of Health (DK56754) to B.A.M. The use of human volunteers in this study was in accordance with appropriate guidelines and was approved by the University of Massachusetts Medical School Review Board for the Protection of Human Subjects (approval #13006). Dr. Jerrold R. Turner, University of Chicago School of Medicine, is thanked for generous gift of the ezrin mutant constructs.
References
- Breitkreutz D, Braiman-Wiksman L, Daum N, Denning MF, Tennenbaum T. Protein kinase C family: on the crossroads of cell signaling in skin and tumor epithelium. J Cancer Res Clin Oncol. 2007;133:793–808. doi: 10.1007/s00432-007-0280-3. [DOI] [PubMed] [Google Scholar]
- Bretscher A, Chambers D, Nguyen R, Reczek D. ERM-Merlin and EBP50 protein families in plasma membrane organization and function. Annu Rev Cell Dev Biol. 2000;16:113–43. doi: 10.1146/annurev.cellbio.16.1.113. [DOI] [PubMed] [Google Scholar]
- Cao TT, Deacon HW, Reczek D, Bretscher A, von Zastrow M. A kinase-regulated PDZ-domain interaction controls endocytic sorting of the beta2-adrenergic receptor. Nature. 1999;401:286–90. doi: 10.1038/45816. [DOI] [PubMed] [Google Scholar]
- Cummins EP, Berra E, Comerford KM, Ginouves A, Fitzgerald KT, Seeballuck F, et al. Prolyl hydroxylase-1 negatively regulates IkappaB kinase-beta, giving insight into hypoxia-induced NFkappaB activity. Proc Natl Acad Sci U S A. 2006;103:18154–9. doi: 10.1073/pnas.0602235103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gewirtz AT, Siber AM, Madara JL, McCormick BA. Orchestration of neutrophil movement by intestinal epithelial cells in response to Salmonella typhimurium can be uncoupled from bacterial internalization. Infect Immun. 1999;67:608–17. doi: 10.1128/iai.67.2.608-617.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, Wakabayashi T, Horie T. Mrp2/Abcc2 transport activity is stimulated by protein kinase Calpha in a baculo virus co-expression system. Life Sci. 2005;77:539–50. doi: 10.1016/j.lfs.2004.10.071. [DOI] [PubMed] [Google Scholar]
- Kikuchi S, Hata M, Fukumoto K, Yamane Y, Matsui T, Tamura A, et al. Radixin deficiency causes conjugated hyperbilirubinemia with loss of Mrp2 from bile canalicular membranes. Nat Genet. 2002;31:320–5. doi: 10.1038/ng905. [DOI] [PubMed] [Google Scholar]
- Kohler H, Sakaguchi T, Hurley BP, Kase BA, Reinecker HC, McCormick BA. Salmonella enterica serovar Typhimurium regulates intercellular junction proteins and facilitates transepithelial neutrophil and bacterial passage. Am J Physiol Gastrointest Liver Physiol. 2007;293:G178–87. doi: 10.1152/ajpgi.00535.2006. [DOI] [PubMed] [Google Scholar]
- Lee CA, Silva M, Siber AM, Kelly AJ, Galyov E, McCormick BA. A secreted Salmonella protein induces a proinflammatory response in epithelial cells, which promotes neutrophil migration. Proc Natl Acad Sci U S A. 2000;97:12283–8. doi: 10.1073/pnas.97.22.12283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Wang W, Soroka CJ, Mennone A, Harry K, Weinman EJ, et al. NHERF-1 binds to Mrp2 and regulates hepatic Mrp2 expression and function. J Biol Chem. 2010;285:19299–307. doi: 10.1074/jbc.M109.096081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luciani F, Molinari A, Lozupone F, Calcabrini A, Lugini L, Stringaro A, Puddu P, Arancia G, Cianfriglia M, Fais S. P-Glycoprotein-Actin Association through Erm Family Proteins: A Role in P-Glycoprotein Function in Human Cells of Lymphoid Origin. Blood. 2002;99:641–8. doi: 10.1182/blood.v99.2.641. [DOI] [PubMed] [Google Scholar]
- McCormick BA, Colgan SP, Delp-Archer C, Miller SI, Madara JL. Salmonella typhimurium attachment to human intestinal epithelial monolayers: transcellular signalling to subepithelial neutrophils. J Cell Biol. 1993;123:895–907. doi: 10.1083/jcb.123.4.895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick BA, Parkos CA, Colgan SP, Carnes DK, Madara JL. Apical secretion of a pathogen-elicited epithelial chemoattractant activity in response to surface colonization of intestinal epithelia by Salmonella typhimurium. J Immunol. 1998;160:455–66. [PubMed] [Google Scholar]
- Mrsny RJ, Gewirtz AT, Siccardi D, Savidge T, Hurley BP, Madara JL, et al. Identification of hepoxilin A3 in inflammatory events: a required role in neutrophil migration across intestinal epithelia. Proc Natl Acad Sci U S A. 2004;101:7421–6. doi: 10.1073/pnas.0400832101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mumy KL, Bien JD, Pazos MA, Gronert K, Hurley BP, McCormick BA. Distinct isoforms of phospholipase A2 mediate the ability of Salmonella enterica serotype typhimurium and Shigella flexneri to induce the transepithelial migration of neutrophils. Infect Immun. 2008;76:3614–27. doi: 10.1128/IAI.00407-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano T, Sekine S, Ito K, Horie T. Correlation between apical localization of Abcc2/Mrp2 and phosphorylation status of ezrin in rat intestine. Drug Metab Dispos. 2009;37:1521–7. doi: 10.1124/dmd.108.024836. [DOI] [PubMed] [Google Scholar]
- Parkos CA, Colgan SP, Delp C, Arnaout MA, Madara JL. Neutrophil migration across a cultured epithelial monolayer elicits a biphasic resistance response representing sequential effects on transcellular and paracellular pathways. J Cell Biol. 1992;117:757–64. doi: 10.1083/jcb.117.4.757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pazos M, Siccardi D, Mumy KL, Bien JD, Louie S, Shi HN, et al. Multidrug resistance-associated transporter 2 regulates mucosal inflammation by facilitating the synthesis of hepoxilin A3. J Immunol. 2008;181:8044–52. doi: 10.4049/jimmunol.181.11.8044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen M, Alexander RT, Darborg BV, Mobjerg N, Hoffmann EK, Kapus A, Pedersen SF. Osmotic Cell Shrinkage Activates Ezrin/Radixin/Moesin (Erm) Proteins: Activation Mechanisms and Physiological Implications. Am J Physiol Cell Physiol. 2008;294:197–212. doi: 10.1152/ajpcell.00268.2007. [DOI] [PubMed] [Google Scholar]
- Reczek D, Berryman M, Bretscher A. Identification of EBP50: A PDZ-containing phosphoprotein that associates with members of the ezrin-radixin-moesin family. J Cell Biol. 1997;139:169–79. doi: 10.1083/jcb.139.1.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saotome I, Curto M, McClatchey AI. Ezrin is essential for epithelial organization and villus morphogenesis in the developing intestine. Dev Cell. 2004;6:855–64. doi: 10.1016/j.devcel.2004.05.007. [DOI] [PubMed] [Google Scholar]
- Shiue H, Musch MW, Wang Y, Chang EB, Turner JR. Akt2 phosphorylates ezrin to trigger NHE3 translocation and activation. J Biol Chem. 2005;280:1688–95. doi: 10.1074/jbc.M409471200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siccardi D, Mumy KL, Wall DM, Bien JD, McCormick BA. Salmonella Enterica Serovar Typhimurium Modulates P-Glycoprotein in the Intestinal Epithelium. Am J Physiol Gastrointest Liver Physiol. 2008;294:1392–400. doi: 10.1152/ajpgi.00599.2007. [DOI] [PubMed] [Google Scholar]
- Silva M, Song C, Nadeau WJ, Matthews JB, McCormick BA. Salmonella typhimurium SipA-induced neutrophil transepithelial migration: involvement of a PKC-alpha-dependent signal transduction pathway. Am J Physiol Gastrointest Liver Physiol. 2004;286:G1024–31. doi: 10.1152/ajpgi.00299.2003. [DOI] [PubMed] [Google Scholar]
- Srikanth CV, Wall DM, Maldonado-Contreras A, Shi HN, Zhou D, Demma Z, Mumy KL, McCormick BA. Salmonella Pathogenesis and Processing of Secreted Effectors by Caspase-3. Science. 2010;330:390–3. doi: 10.1126/science.1194598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strohmeier GR, Lencer WI, Patapoff TW, Thompson LF, Carlson SL, Moe SJ, et al. Surface expression, polarization, and functional significance of CD73 in human intestinal epithelia. J Clin Invest. 1997;99:2588–601. doi: 10.1172/JCI119447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakusawa S, Machida I, Suzuki S, Hayashi H, Yano M, Yoshioka K. Identification of a novel 2026G-->C mutation of the MRP2 gene in a Japanese patient with Dubin-Johnson syndrome. J Hum Genet. 2003;48:425–9. doi: 10.1007/s10038-003-0052-0. [DOI] [PubMed] [Google Scholar]
- Wald FA, Oriolo AS, Mashukova A, Fregien NL, Langshaw AH, Salas PJ. Atypical protein kinase C (iota) activates ezrin in the apical domain of intestinal epithelial cells. J Cell Sci. 2008;121:644–54. doi: 10.1242/jcs.016246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Soroka CJ, Mennone A, Rahner C, Harry K, Pypaert M, et al. Radixin is required to maintain apical canalicular membrane structure and function in rat hepatocytes. Gastroenterology. 2006;131:878–84. doi: 10.1053/j.gastro.2006.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinman EJ, Steplock D, Wang Y, Shenolikar S. Characterization of a protein cofactor that mediates protein kinase A regulation of the renal brush border membrane Na(+)-H+ exchanger. J Clin Invest. 1995;95:2143–9. doi: 10.1172/JCI117903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Q, Onuki R, Nakai C, Sugiyama Y. Ezrin and radixin both regulate the apical membrane localization of ABCC2 (MRP2) in human intestinal epithelial Caco-2 cells. Exp Cell Res. 2007;313:3517–25. doi: 10.1016/j.yexcr.2007.07.033. [DOI] [PubMed] [Google Scholar]
- Zhao H, Shiue H, Palkon S, Wang Y, Cullinan P, Burkhardt JK, et al. Ezrin regulates NHE3 translocation and activation after Na+-glucose cotransport. Proc Natl Acad Sci U S A. 2004;101:9485–90. doi: 10.1073/pnas.0308400101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.