

Abstract
Structurally well-defined IgG-Fc glycoforms are highly demanded for understanding the effects of glycosylation on antibody’s effector functions. We report in this paper chemoenzymatic synthesis and Fcγ receptor binding of an array of homogeneous IgG-Fc glycoforms. The chemoenzymatic approach consists of the chemical synthesis of defined N-glycan oxazolines as donor substratess, the expression of the Fc domain in a CHO cell line in the presence of an α-mannosidase inhibitor kifunensine, and an endoglycosidase-catalyzed glycosylation of the deglycosylated Fc domain (GlcNAc-Fc homodimer) with the synthetic glycan oxazolines. The enzyme from Arthrobacter protophormiae (Endo-A) was found to be remarkably efficient to take various modified N-glycan core oxazolines, including the bisecting sugar-containing derivatives, for Fc glycosylation remodeling, resulting in the formation of the corresponding homogeneous Fc glycoforms. Nevertheless, neither Endo-A, nor the Mucor hiemalis endoglycosidase mutants (EndoM-N175A and EndoM-N175Q), was able to transfer full-length complex-type N-glycan to the Fc domain, implicating the limitations of these two enzymes in Fc glycosylation remodeling. SPR binding studies with the synthetic IgG-Fc glycoforms unambiguously proved that the presence of a bisecting GlcNAc moiety could significantly enhance the binding of Fc to FcγRIIIa, the activating Fcγ receptor, independent of Fc core-fucosylation. Interestingly, the Fc glycoforms carrying an unusual bisecting sugar moiety such as a mannose or a LacNAc moiety also demonstrated enhanced affinity to FcγRIIIa. On the orther hand, the presence of a bisecting GlcNAc or core fucosylation had little effect on the affinity of Fc to the inhibitory Fcγ receptor, FcγRIIb. Our experimental data also showed that the α-linked mannose residues in the pentasaccharide Man3GlcNAc2 core was essential to maintain a high-affinity of Fc to both FcγRIIIa and FcγRIIb. The synthetic homogeneous Fc glycoforms thus provide a useful tool for elucidating how a fine Fc N-glycan structure precisely affects the function of the Fc domain.
Keywords: chemoenzymatic synthesis, Transglycosylation, glycoprotein, IgG-Fc glycosylation, ADCC, endoglycosidase, bisecting GlcNAc
INTRODUCTION
Monoclonal antibodies (MAbs) are a class of therapeutic glycoproteins used for the treatment of various human diseases including cancer and inflammatory disorders.1–3 Almost all the therapeutic monoclonal antibodies currently used for disease treatment are of the immunoglobulin G (IgG) type, which are composed of two light chains and two heavy chains that are associated to form three distinct protein domains linked by a flexible hinge region. The two identical Fab domains are specific for antigen-binding, while the Fc domain, a homodimer consisting of the CH2 and CH3 subdomains, is engaged in the “downstream” effector functions of antibodies including antibody-dependent cellular cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC).2,4 ADCC and CDC effector functions are mediated by the interactions of the Fc domain with respective Fcγ receptors (such as FcγRIIIa and FcγRIIb) on effector cells and the C1q component of the complement cascade, respectively.4 The Fc homodimer carries two N-glycans at each of the conserved N-glycosylation sites (Asn-297) of the two CH2 domains. Structural analysis has indicated that human IgG-Fc N-glycans are typical bi-antennary complex type N-glycans with considerable structural heterogeneity.5–7 More than 30 different Fc oligosaccharides were characterized, in which the core Asn-linked heptasaccharide GlcNAc2Man3GlcNAc2-Asn can be differentially decorated with core fucosylation, bisecting GlcNAc attachment, and varied terminal galactosylation and sialylation (Figure 1).
Figure 1.

Schematic presentations of the natural and synthetic human IgG1-Fc glycoforms. A) natural heterogeneous Fc glycforms; B) synthetic homogeneous Fc glycoforms. The IgG1-Fc structure was modeled on the basis of the crystal structure of an anti-HIV antibody b12 (PDB code, 1hzh) ( E. O. Saphire et al, Science, 2001, 293, 1155). GlcNAc, N-acetylglucosamine; Man, mannose; Gal, galactose; Glc, glucose; Fuc, L-fucose; Sia, sialic acid. The dash lines represent variable decorations.
Recent advances in glycobiology and immunology have suggested that antibody Fc glycosylation can significantly impact the structure and biological function of antibodies.2,8 It has been demonstrated that aglycosylated or de-glycosylated IgG antibodies are almost completely devoid of Fc-mediated effector functions as a result of reduced or ablated binding to Fcγ receptors or those proteins in the complement system.7,9 Crystallographic studies and NMR analysis have shown that the Fc N-glycans have multiple non-covalent interactions with the Fc protein domain, implicating an important role of the N-glycan in maintaining an appropriate Fc domain conformations required for the specific interactions between Fc and Fc receptors for antibody’s effector functions.6,9–16 It has been further demonstrated that the distinct fine structures of Fc N-glycans can define whether the antibody goes for activation (such as ADCC) or inhibitory (such as anti-inflammatory) effector functions.2,17 For example, core fucosylation has been shown to significantly decrease the affinity of Fc to FcγIIIa receptor that correlates with the antibody’s ADCC function and anti-cancer efficacy in vivo.18–22 On the other hand, specific terminal α-2,6-sialylation of IgG-Fc N-glycan, a minor population (about 5%) of the intravenous immunoglobulin (IVIG), has recently demonstrated to be responsible for the anti-inflammatory activity of IVIG in the treatment of auto-immune diseases.4,23–25 As to the role of the bisecting GlcNAc residue in the Fc N-glycans, studies have shown that increasing the population of the bisecting GlcNAc contents leads to enhanced affinity to FcγIIIa receptor and thus enhanced ADCC function of the antibodies.21,26–28 However, another study has suggested that the lack of the core fucose, rather than the presence of a bisecting GlcNAc, plays the key role in enhancing the ADCC, as pre-addition of the bisecting GlcNAc residue to the core-β-mannose inhibits core fucosylation in the biosynthetic pathway, resulting in an increased population of non-fucosylated glycoforms that demonstrate enhanced ADCC.19 It should be pointed out that many of the structure-activity relationship studies so far reported have used mixtures of Fc glycoforms, although some of the specific glycoforms were enriched by lectin affinity purification and/or enzymatic trimming. This situation sometimes makes a conclusive interpretation of the experimental results difficult.
The urgent need of various glycosylation-defined pure IgG-Fc glycoforms, which are difficult to isolate from natural source, for structure-activity relationship studies and for biomedical applications has stimulated an intense interest in developing methods to control Fc glycosylation. These include glycan biosynthetic pathway engineering in plant, mammalian, and yeast expression systems to produce non-fucosylated IgG-Fc glycoforms with improved ADCC activity;26,29–33 sequential enzymatic trimming of Fc N-glycans to produce truncated IgG-Fc glycoforms for structural analysis and Fcγ receptor binding studies;13,28,34 and chemoselective Fc glycosylation by site-directed mutagenesis at the glycosylation site (N297C) followed by chemoselective ligation with a glycan.35 We have previously described a convergent chemoenzymatic method for IgG-Fc glycosylation remodeling that takes advantage of the transglycosylation activity of the Arthrobacter endoglycosidase (Endo-A) and the highly active N-glycan oxazoline as the donor substrates.36 In this approach, an IgG-Fc was first expressed in yeast to give the IgG-Fc carrying yeast N-glycans at the Fc domain. Then the heterogeneous N-glycans were cleaved to leave only the innermost GlcNAc attached at the glycosylation site (Asn-297). Finally a defined core N-glycan was transferred to the GlcNAc moiety by Endo-A to provide a homogeneous IgG-Fc glycoform. This enzymatic transglycosylation approach preserves the natural N-glycan core structure in the Fc domain, which appears essential for the effector functions of antibodies.2 Our initial studies have suggested that the enzymatic transglycosylation was feasible for glycosylation remodeling of recombinant IgG-Fc dimers under mild conditions without the need of denaturing the Fc protein domain.36 Despite this initial success, it is still to be demonstrated whether different types of N-glycans can be introduced at the Fc domain by this method. Glycosylation remodeling of the IgG-Fc homodimer could be particularly challenging as the two Fc N-glycans at the glycosylation sites (Asn-297) are sandwitched between the two Fc domains, which might be less accessible for enzymatic reactions.9–15 Our initial success in the chemoenzymatic glycosylation remodeling of IgG-Fc,36 together with recent advances in the method development,37–48 prompted us to expand the chemoenzymatic approach to the synthesis of various homogeneous IgG-Fc glycoforms while examining the scope and limitations of the endoglycosidase-catalyzed transglycosylation for IgG-Fc glycosylation remodeling. We report in this paper the chemeonzymatic synthesis of an array of specific, homogeneous IgG-Fc glycoforms (Figure 1, glycoforms Fc-1 to Fc-6), with a focus on elucidating the roles of individual sugar residues within and/or flanking the Fc N-glycan core in the binding to Fcγ receptors FcγRIIIa and FcγRIIb. In particular, several bisecting sugar containing Fc glycoforms, including the unusual bisecting LacNAc Fc glycoform that was recently discovered as a minor IgG-Fc glycoform with unknown function,49 were synthesized. Our experimental data indicate that Endo-A is remarkably efficient to take various modified N-glycan core oxazolines, including the bisecting sugar-containing derivatives, for Fc glycosylation remodeling. Nevertheless, neither Endo-A, nor the Mucor hiemalis endoglycosidase mutant (EndoM-N175A and EndoM-N175Q), was able to transfer full-length complex-type N-glycan oxazoline to the Fc domain, implicating the limitations of these two enzymes in Fc glycosylation remodeling. Our SPR binding studies with well-defined synthetic IgG-Fc glycoforms provide unambiguous evidence indicating that the presence of a bisecting GlcNAc moiety could directly enhance the interaction between Fc to FcγRIIIa, independent of core-fucosylation, whereas the presence of a bisecting GlcNAc has little effect on the affinity of Fc to FcγRIIb. Our experimental data also showed that the two α-linked mannose residues in the pentasaccharide Man3GlcNAc2 core is essential to maintain a high affinity of Fc to the FcγRIIIa, while the core β-mannose residue could be changed to a β-glucose moiety without significantly affecting the binding to FcγRIIIa.
RESULTS AND DISCUSSION
Synthesis of bisecting GlcNAc-containing N-glycan oxazoline (1) and bisecting LacNAc-containing N-glycan oxazoline (2)
Chemoenzymatic synthesis of the homogeneous Fc glycoforms by the endoglycosidase-catalyzed transglycosylation requires the preparation of the corresponding N-glycan oxazolines as donor substrates. The structures of sugar oxazolines synthesized and used for the present study were listed in Figure 2. These glycan oxazolines were designed for the preparation of the well-defined homogeneous Fc glycoforms that will allow a detailed probing of the effects of varied glycan structures on the binding of Fc to respective Fcγ receptors that are essential for antibody effector functions. Chemical synthesis of bisecting GlcNAc-containing N-glycans of varied size has been reported by several research groups.50–55 These studies suggested that the 4-hydroxyl group of the core β-mannose was sterically hindered and difficult to glycosylate when the 3- and 6-positions of the core β-mannose were pre-occupied with bulky sugar residues. Thus, for the synthesis of the bisecting GlcNAc-containing sugar oxazoline (1), we adopted a synthetic strategy of introducing the bisecting GlcNAc residue prior to the glycosylation at the 3- and 6-positions of the β-mannose moiety (Scheme 1).
Figure 2.

Structures of synthetic sugar oxazolines
Scheme 1.

Synthesis of bisecting GlcNAc- and LacNAc-containing N-glycan oxazolines a
aReagents and conditions: (a) BSP, TTBP, Tf2O, CH2Cl2, 56%. (b) 80% aq. AcOH, 80%. (c) TBDMSCl, pyridine, 95%. (d) BF3·OEt2, CH2Cl2, 76%. (e) TFA, CH2Cl2, 88%. (f) TMSOTf, CH2Cl2, 54%. (g) 1) NH2NH2 monohydrate, EtOH, H2O; 2) Ac2O, pyridine, 77% (2 steps). (h) 1) Pd(OH)2-C, H2, CH2Cl2, MeOH; 2) Ac2O, pyridine, 89% (2 steps). (i) TMSBr, BF3·OEt2, 2,4,6-collidine, CH2Cl2, 48%. (j) MeONa, MeOH, quantitative. (k) MeONa, MeOH, quantitative. (l) DMC, Et3N, H2O, quantitative. (m) UDP-Gal, β-1,4-galactosyltransferase, buffer, quantitative. (n) DMC, Et3N, H2O, quantitative.
Glycosylation of monosaccharide 7 56 with thioglycoside donor 8 57 under the promotion of 1-benzenesulfinyl piperidine (BSP)/2,4,6-tri-tert-butylpyrimidine (TTBP)/trifluoromethanesulfonic anhydride (Tf2O)56 gave the corresponding disaccharide product (β/α = 6:1), from which the desired β-isomer 9 was isolated in 56% yield. Removal of the benzylidene group by mild acidic hydrolysis provided diol 10 in 80% yield. The primary hydroxyl group was then selectively protected with TBDMSCl in pyridine to give disaccharide 11 in excellent yield. BF3 · Et2O-catalyzed glycosylation of the 4-OH with glucosamine building block 12 58 gave the trisaccharide 13 in 76% yield, in which the bisecting glucosamine moiety was introduced with a complete β-selectivity. The TBDMS and PMB groups in trisaccharide 13 were simultaneously removed by treatment with TFA in CH2Cl2 to give diol 14 in 88% yield. Di-glycosylation of 14 was achieved with an excess of glycosyl donor 15 under the catalysis of TMSOTf, giving pentasaccharide 16 in 54% yield. The phthalimido groups were then changed to the acetamido groups in two steps by treatment with hydrazine hydrate followed by acetylation with Ac2O/pyridine to afford 17, which was subject to catalytic hydrogenation and subsequent O-acetylation to give the peracetate (18) in 89% yield. Initial attempt to form the sugar oxazoline by the Lewis acid catalyzed reaction using TMSBr/BF3·Et2O/2,4,6-collidine as the promoter59,60 gave only a moderate yield (48%) of 19, mainly due to decomposition of the oligosaccharide under the acidic reaction conditions. De-O-acetylation of 19 with catalytic amount of MeONa in MeOH provided pentasaccharide oxazoline 1. Recently, a method for one-step conversion of unprotected N-acetyl-2-amino sugars to sugar oxazolines using a chloroformamidinium reagent in water was reported,61 and we have also shown that this method was equally efficient for making even very large complex N-glycan oxazolines from free N-glycans.41 Thus, we tested this method for preparing oxazoline 1 from free pentasaccharide 20, which was obtained by de-O-acetylation of 18. Treatment of 20 with an excess of 2-chloro-1,3-dimethylimidazolinium chloride (DMC) and Et3N in water afforded the desired sugar oxazoline 1 in quantitative yield, which was readily purified by gel filtration on a Sephadex G-10 column. These results clearly indicate that the one-step conversion of free glycan to the sugar oxazoline in an aqueous solution is much more efficient than the Lewis acid-catalyzed oxazoline ring formation from the O-acetylated derivative. The synthesis of the bisecting LacNAc containing sugar oxazoline (2) started with enzymatic extension of the sugar chain in tetrasaccharide 20. We found that addition of a β-1,4-linked galactose to the bisecting GlcNAc could be readily achieved by incubation of 20 with UDP-Gal and a β-1,4-galactosyltransferase from bovine milk in a phosphate buffer, giving hexasaccharide 21 in quantitative yield. Compound 21 was then converted into hexasaccharide oxazoline 2 in a single step upon treatment with an excess of DMC and Et3N (Scheme 1). The bisecting mannose-containing oxazoline, Man4GlcNAc-oxazoline (3), was synthesized following the previously reported procedure.44
Synthesis of core-glucose-containing glycan oxazoline (4)
Another sugar oxazoline that would be interesting for structure-activity relationship studies related to Fc glycosylation is the tetrasaccharide oxazoline (4), in which the core β-mannose moiety was replaced with a glucose moiety. Oxazoline 4 was previously synthesized by Fairbanks and co-workers and was shown to be a substrate for Endo-A.46 We describe here an alternative synthesis of 4 (Scheme 2). Briefly, coupling of thioglycoside 22 62 and acceptor 23 56 gave the β-linked disaccharide 24. the PMB group was selectively removed by DDQ to provide 25, which was then converted to diol 26 in 94% yield by regioselective reductive ring opening of the benzylidene group using Et3SiH/PhBCl2 in CH2Cl2. Glycosylation of 26 with glycosyl donor 27 under the catalysis of TMSOTf, with simultaneous introduction of two mannose residues, afforded tetrasaccharide 28 in 95% yield. A one-step transformation of the azido group to an acetamido group was carried out by treatment of 28 with AcSH in pyridine to give 29 (78% yield), which was then converted into the per-acetate derivative (30) by sequential de-O-acylation, catalytic hydrogenation, and O-acetylation. Finally, compound 30 was de-O-acetylated and the resulting free oligosaccharide was converted to sugar oxazoline 4 in excellent yield through the single-step, DMC-promoted oxazoline-forming reaction (Scheme 2). The synthesis of Man3GlcNAc-oxazoline (5) and ManGlcNAc-oxazoline (6) was previously reported.58
Scheme 2.

Synthesis of a glucose-containing N-glycan oxazoline a
a Reagents and conditions: (a) NIS, TfOH, dichloroethane, 63%. (b) DDQ, CH2Cl2, H2O, 87%. (c) Et3SiH, PhBCl2, CH2Cl2, 94%. (d) TMSOTf, CH2Cl2, 95%. (e) AcSH, pyridine, CHCl3, 78%. (f) 1) MeONa, MeOH, then Dowex (H+); 2) Pd(OH)2/C, H2, MeOH; 3) Ac2O, pyridine, 92% (three steps). (g) 1) MeONa, MeOH, then Dowex (H+); 2) DMC, Et3N, H2O, 95% (two steps).
Expression of human IgG1-Fc homodimer carrying a homogeneous Man9GlcNAc2 glycan at each of the Fc glycosylation site (Asn-297)
We have previously expressed human IgG1-Fc in yeast Pichia pastoris to obtain a recombinant IgG1-Fc carrying yeast N-glycans.36 The N-glycans were a mixture of yeast high-mannose type glycans ranging from Man9GlcNAc2 to Man13GlcNAc2. To obtain a homogeneous, human-like Man9GlcNAc2 glycoform of the Fc domain, we expressed human IgG1-Fc in Chinese hamster ovary (CHO) cell lines in the presence of kifunensine, a potent class I α-mannosidase inhibitor that blocks further processing of the Man9GlcNAc2 N-glycan during glycoprotein biosynthesis.63–65 It has been previously reported that expression of a monoclonal antibody in CHO or human HEK293 cell lines in the presence of kifunensine could efficiently control the N-glycosylation at the high-mannose type stage.11,66 Recently we have successfully overproduced Man9GlcNAc2-glycoform of Fc-fused HIV-1 V3 domain using kifunensine as the α-mannosidase inhibitor.67 Following this protocol, we overproduced human IgG1-Fc in CHO cell lines in the presence of kifunensine (2 μg/mL). The recombinant Fc was purified by protein A affinity chromatography. SDS-PAGE analysis indicated that the recombinant Fc expressed in the presence of kifunensine appeared as a single band of a size of ca. 65 kDa and ca. 33 kDa under non-reducing and reducing conditions, respectively (Figure 3A, lanes 1 and 2). This result suggests that the recombinant IgG1-Fc was produced as a homodimer. Treatment of the IgG1-Fc with PNGase F, which would completely de-glycosylate the glycoprotein by hydrolyzing the glycosylamide linkage between N-glycan and the Asn residue, gave a new single band that was about 2 kDa less than the glycosylated monomeric IgG1-Fc (Figure 3A, lane 3, shown under a reducing SDS-PAGE condition). These results implicate the attachment of two N-glycans in the HM-Fc homodimer. The recombinant HM-Fc was also characterized by direct MALDI-TOF MS analysis of the Fc homodimer, which gave a species with an average m/z of 65447 (Figure 3B). The experimental data was in good agreement with the theoretical molecular weight (M = 65444 Da) of Fc homodimer carrying two high-mannose type (Man9GlcNAc2) N-glycans, which was calculated on the basis of the amino acid sequence of the recombinant Fc plus two Man9GlcNAc2 glycans (Figure S1, supporting information). The N-glycans in the recombinant IgG1-Fc was further verified by MALDI-TOF MS analysis. The MS spectrum of the PNGase F-released N-glycans indicated that more than 90% of the N-glycans attached were Man9GlcNAc2, with Man8GlcNAc2 being a minor fraction (Figure 3C). In contrast, the N-glycans released from the wild type recombinant IgG1-Fc produced in CHO cell lines in the absence of kifunensine, designated as CT-Fc, were a mixture of bi-antennary complex type N-glycans that were fucosylated and contained 0, 1, and 2 terminal galactose residues, respectively (designated as G0F, G1F, and G2F glycoforms) (Figure 3D).
Figure 3.

SDS-PAGE and MALDI-TOF MS analysis of recombinant IgG1-Fc. A, SDS-PAGE: Lane M, protein marker; lane 1, HM-Fc (non-reducing condition); lane 2, HM-Fc, reducing condition; lane 3, PNGase F-treatment of HM-Fc; B, MALDI-TOF MS of HM-Fc; C, MALDI-TOF MS of N-glycans released from HM-Fc; D, MALDI-TOF MS of N-glycans released from CHO-expressed Fc (CT-Fc).
Synthesis of IgG-Fc homogeneous glycoforms through chemoenzymatic glycosylation remodeling
Our preliminary studies have shown that the Endo-A catalyzed transglycosylation with Man3GlcNAc oxazoline (5) could be efficiently applied to the seemingly hindered GlcNAc residues of the Fc homodimer (GlcNAc-Fc) to make a homogeneous glycoform of IgG-Fc under a very mild condition, without the need of denaturing the Fc domain. Thus the native structure of IgG1-Fc homodimer was kept intact during the enzymatic de-glycosylation and subsequent enzymatic transglycosylation.36 In the present work, we used the high-mannose type IgG-Fc homodimer (HM-Fc) that was expressed in CHO cell line in the presence of kifunensine as the starting material for glycosylation remodeling to prepare a panel of new homogeneous IgG-Fc glycoforms, as shown in Scheme 3. Treatment of HM-Fc with Endo-H under mild conditions (phosphate buffer, pH 6.5, 23°C) led to an efficient conversion of HM-Fc (Figure 4A lane 1) to the GlcNAc-Fc (Figure 4A, lane 2). The identity of the GlcNAc-Fc homodimer was confirmed by its MALDI-TOF MS (Calculated, M = 62119 Da; found (m/z), 62122) (Figure 4B). We first examined the Endo-A catalyzed reaction of bisecting GlcNAc oxazoline 1 and the GlcNAc-Fc homodimer (donor/acceptor, 20:1, molar ratio; 10 molar equivalent to each GlcNAc moiety of the homodimer) in a phosphate buffer (pH 7.0), and monitored the reaction by SDS-PAGE. We found that the bisecting GlcNAc-containing oxazoline (1) was an excellent substrate for Endo-A and the transglycosylation reaction went smoothly to give a product, which appeared as a new band that was about 1 kDa larger than the GlcNAc-Fc under reducing SDS-PAGE conditions (Figure 4A, lane 3 vs. lane 2). After 3 h, all the GlcNAc-Fc was converted to the new product. The single band for the glycosylated IgG-Fc under the reducing conditions suggests that both the glycosylation sites in the Fc homodimer were glycosylated (The Fc homodimer would be reduced to monomeric Fc on SDS-PAGE in the presence of 2-mercaptoethanol. If any one of the two glycosylation sites was not transglycosylated, it would show up as the monomeric GlcNAc-Fc in SDS-PAGE). The product, Fc-1, was readily purified by affinity chromatography on a protein A column. Treatment of glycoform Fc-1 with PNGase F led to the removal of the N-glycans, giving a single band that was about 1 kDa less in size when compared with the Fc-1 under reducing conditions (Figure 4A, lane 4 vs. lane 3). Since PNGase F specifically hydrolyzes the amide linkage between the N-glycan and the Asn residue in the protein, the result suggests that the transferred glycans were specifically attached to the GlcNAc moieties in the GlcNAc-Fc homodimer to form an intact N-glycan in the product, rather than on any other amino acid residues. MALDI-TOF MS spectrum (Figure 4C) of the Fc-1 showed a single species (m/z) at 63909, which matched well with the expected molecular mass (63905 Da) of the Fc homodimer carrying two bisecting-GlcNAc-Man3GlcNAc2 glycans. Further characterization of the product was performed by analysis of the attached N-glycans. The N-glycans were first released by PNGase F and were then analyzed by MALDI-TOF MS: calculated for GlcNAcMan3GlcNAc2, M = 1113.41 Da; Found (m/z), 1136.80 [M + Na]+ (Figure 4D). It should be noted that we have previously demonstrated that terminal GlcNAc in an N-glycan was itself eligible to serve as an acceptor for enzymatic transglycosylation.42,68 In the present case, we did not observe cross-transglycosylation to the bisecting GlcNAc moiety under the transglycosylation conditions used (5 mM sugar oxazoline, 0.5 mM GlcNAc-Fc). This result suggests that the bisecting GlcNAc moiety might be relatively hindered in comparison with the terminal GlcNAc on the α1,3 and α1,6 arms of the N-glycan core.42 Nevertheless, we did observe self-condensation of the bisecting GlcNAc-containing Man3GlcNAc-oxazoline (1) when a much higher concentration (50 mM) of the sugar oxazoline (1) alone was incubated with a larger amount of Endo-A, leading to the formation of a series of novel oligomers (data not shown). A more detailed study on this self-condensation reaction of the sugar oxazoline will be described elsewhere.
Scheme 3.

Chemoenzymatic synthesis of homogeneous glycoforms of human IgG1-Fc a
a Reagents and conditions. The high-mannose IgG1-Fc glycoform was produced in CHO cells in the presence of kifunensine. Then the high-mannose N-glycan was removed by Endo-H to give the GlcNAc-containing IgG1-Fc, which served as an acceptor for the Endo-A catalyzed transglycosylation with respective synthetic sugar oxazolines (1 to 6) to afford the corresponding homogeneous IgG1-Fc glycoforms.
Figure 4.

SDS-PAGE and MALDI-TOF MS analysis of GlcNAc-Fc and transglycosylation product Fc-1. A, SDS-PAGE: Lane 1, HM-Fc; lane 2, GlcNAc-Fc; lane 3, transglycosylation product Fc-1; lane 4, PNGase F-treatment of Fc-1; B, MALDI-TOF MS of GlcNAc-Fc; C, MALDI-TOF MS of product Fc-1; D, MALDI-TOF MS of N-glycans released from Fc-1.
Transglycosylation of GlcNAc-Fc with the bisecting LacNAc-containing glycan oxazoline (2) was performed in the similar way as for the preparation of the bisecting GlcNAc glycoform (Fc-1). It was found that the bisecting LacNAc-containing glycan oxazoline (2) was also an excellent substrate for Endo-A, leading to the formation of another Fc glycoform, Fc-2. Again, when an excess of sugar oxazoline was used (10-fold molar excess for each GlcNAc acceptor in the GlcNAc-Fc homodimer), the transglycosylation could go to completion and the product (Fc-2) was readily purified by Protein A affinity chromatography. The identity of glycoform Fc-2 was characterized by MALDI-TOF MS analysis of both the intact glycoprotein and the N-glycan released by PNGase F treatment: MALDI-TOF MS of Fc-2, Calculated for Fc-2, M = 64229 Da; found (m/z), 64222. MALDI-TOF MS of the attached N-glycan, calculated for LacNAc-Man3GlcNAc2, M = 1275.46 Da; found (m/z), 1298.40 [M + Na]+. The Endo-A catalyzed transglycosylation of GlcNAc-Fc with the other sugar oxazolines, including Man4GlcNAc-oxazoline (3), Man2GlcGlcNAc-oxazoline (4), Man3GlcNAc-oxazoline (5),58 and ManGlcNAc-oxazoline (6),58 was performed in the similar way to give the corresponding glycoforms, Fc-3 to Fc-6, respectively (Scheme 3). Except for the enzymatic reaction with the disaccharide oxazoline (6), which was much slower than the reaction with sugar oxazoline 1, all the enzymatic transglycosylations went efficiently to give the corresponding products. In the case of disaccharide oxazoline 6, more sugar oxazoline (15-fold molar excess for each GlcNAc residue in the GlcNAc-Fc homodimer) was added to push the transglycosylation to completion. Again, the Fc glycosylation products were purified by protein A affinity chromatography and characterized by MALDI-TOF MS of the intact glycoprotein as well as the N-glycans released (see Experimental Section).
We also attempted to make a homogeneous Fc glycoform carrying a full-length biantennary complex-type N-glycan. Endo-A is an endoglycosidase specific for the hydrolysis and transglycosylation of high-mannose type N-glycans. But we have recently found that Endo-A also possesses a low activity on complex-type N-glycan oxazoline for transglycosylation without product hydrolysis when a large amount of Endo-A and a high concentration of substrates were used.41 Thus we tested the ability of Endo-A to glycosylate GlcNAc-Fc using a complex type glycan oxazoline as the donor substrate (see Figure S2, supporting information). SDS-PAGE analysis of the reaction mixture after 3 h incubation did not indicate the formation of transglycosylation product (Figure S2, lane 3). This result suggests that although Endo-A is efficient to make various Fc glycoforms carrying the core N-glycans, its activity is not sufficient to transfer the full-length complex-type N-glycan to GlcNAc-Fc domain. We then tested the mutants of the fungus enzyme, EndoM-N175A and EndoM-175Q. We have previously reported that the two glycosynthase mutants were able to transfer complex-type glycan oxazolines to GlcNAc-ribonuclease and GlcNAc-peptides, using the corresponding sugar oxazoline (CT-oxazoline) as the donor substrate.38,43,44,69 When the CT-oxazoline and GlcNAc-Fc were incubated with EndoM-N175A, SDS-PAGE analysis did not reveal the formation of any transglycosylation product (Figure S2, lane 4). Under the same reaction condition, the EndoM-N175Q mutant did not glycosylate the GlcNAc-Fc, either (data not shown). Surprisingly, Endo-M (either the wild-type or the N175A and N175Q mutants) also failed to transfer the simple Man3GlcNAc-oxazoline to GlcNAc-Fc. Given the fact that EndoM-N175A and EndoM-N175Q could efficiently transfer the complex-type sugar oxazoline and simple Man3GlcNAc-oxazoline to GlcNAc-peptides and even GlcNAc-containing ribonuclease B 38,43,44,69 the present results suggest that the active site of Endo-M enzyme might not be able to access to the GlcNAc moiety in acceptor GlcNAc-Fc, which was sandwiched by the two Fc domains and could be sterically hindered. As Endo-A could efficient transfer various core-modified N-glycan oxazolines to GlcNAc-Fc, the failure of Endo-A to transfer complex-type glycan oxazoline might simply reflect the intrinsic low activity of Endo-A on complex type N-glycans, rather than its recognition to acceptor GlcNAc-Fc. These results indicate the difference in acceptor substrate flexibility for the two endoglycosidases (Endo-A and Endo-M). We are currently working on site-directed mutagenesis of Endo-A and are also testing other endoglycosidases in order to find an enzyme or mutants capable of effectively attaching full-length complex type N-glycans to IgG-Fc to make homogeneous complex-type IgG-Fc glycoforms.
Binding of IgG-Fc glycoforms with the stimulatory Fcγreceptor (FcγRIIIa) and the inhibitory Fcγreceptor (FcγRIIb)
With the recombinant and synthetic Fc glycoforms in hands, we evaluated their affinity to Fcγ receptors FcγRIIIa and FcγRIIb using surface plasmon resonance (SPR) technology. In our previous study,36 we directly immobilized FcγRIIIa on the chips and flowed different IgG-Fc glycoforms at varied concentrations. While the direct “random” immobilization provided a quick comparison of the binding of different Fc glycoforms when the same chip was used, the affinity as measured by the apparent KD values (found at μM concentrations for the interaction between wild type IgG-Fc and FcγRIIIa) seemed to be significantly under-estimated, probably due to partial denaturing of the receptor structure during random immobilization. In the present study, we measured the affinity by site-specific immobilization of individual Fc glycoforms through their affinity capture with pre-immobilized protein A on the chips, as demonstrated in previous reports for site-specific immobilization of monoclonal antibodies.34,70 The Fcγ receptors at various concentrations were injected as analytes. A typical SPR profile for the interactions between Fc glycoforms and the FcγRIIIa was shown in Figure 5. The KD values for the binding of the various Fc glyocoforms to Fcγ receptors FcγRIIIa and FcγRIIb, which were obtained by fitting of the binding data with a 1:1 steady state binding model uisng BIAcore T100 evaluation software, were listed in Table 1. We found that the site-directed immobilization of the Fc glycoforms by affinity capture provided an accurate and highly reproducible measurement of the affinity of Fc glycoforms to Fcγ receptors, as it provides a consistent surface orientation of the immobilized proteins from experiment to experiment. The protein A chips could be easily re-generated and reused for capturing and measuring different Fc glycoforms. It should be pointed out that protein A binding (capture) would not interfere with the interaction between Fc domain and Fcγ receptors, as the protein A binds the CH2/CH3 interface of the Fc domain, while the Fcγ receptors’ binding sites locate presumably at the interface of the low hinge region and the up CH2 domain.71
Figure 5.

SPR sensorgrams of the binding of various Fc glycoforms to FcγIIIa receptor from a representative experiment. Fc glycoforms were immobilized by Protein A capture and the binding was analyzed by injecting the respective Fcγ receptors at varied concentrations.
Table 1.
Binding affinities of IgG-Fc glycoforms to Fcγ receptors FcγRIIIa and FcγRIIba)
| IgG-Fc glycoforms | FcγRIIIa | FcγRIIb | |||
|---|---|---|---|---|---|
| Name | N-glycan attached | KD (nM) | Relative Affinity b) | KD (μM) | Relative Affinity b) |
| CT-Fc | Complex type | 136.4±3.1 | 1 | 2.10±0.12 | 1 |
| HM-Fc |
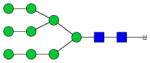
|
45.0±1.7 | 3.1 | 3.5±0.17 | 0.60 |
| Fc-1 |
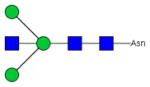
|
22.0±2.1 | 6.2 | 2.20±0.15 | 0.95 |
| Fc-2 |
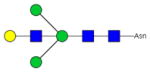
|
40.7±2.5 | 3.4 | N.D.c) | - |
| Fc-3 |
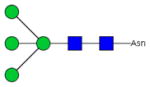
|
46.8±1.2 | 2.9 | 2.52±0.20 | 0.83 |
| Fc-4 |
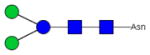
|
78.5±2.4 | 1.7 | N.D.c) | - |
| Fc-5 |
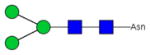
|
58.4±0.6 | 2.3 | 2.90±0.22 | 0.72 |
| Fc-6 |
|
354±11 | 0.4 | >10 | - |
| GlcNAc-Fc |
|
>3000 | <0.05 | >10 | - |
The affinity was measured on a BIAcore T100 and the KD values were calculated using an equilibrium binding model by fitting the SPR data.
The KD data (mean ± S.D.) were obtained from a set of two independent expreiments.
Relative affinity was calculated as KD (natural complex type Fc)/KD (variant Fc glycoforms).
Not determined.
Using the KD as an estimate of the relative affinity, we found that the simple GlcNAcMan3GlcNAc2-Fc glycoform (Fc-1) carrying a bisecting GlcNAc moiety had the highest affinity for FcγRIIIa (KD = 22 nM). This is about 6-fold as high as that of the wild type IgG-Fc (CT-Fc) carrying the fucosylated complex type N-glycans (KD = 136 nM), 2-fold as high as that of the Man9GlcNAc2 glycoform (HM-Fc) (KD = 45 nM), and about 3-fold as high as that of the Man3GlcNAc2-glycoform (Fc-5, KD = 58 nM) (Table 1). Interestingly, the Fc glycoforms with an unnatural bisecting LacNAc moiety (Fc-2) (KD = 41 nM) or a mannose moiety (Fc-3) (KD = 47 nM) also showed enhanced affinity in comparison with the Man3GlcNAc2 Fc-glycoform (Fc-5, KD = 58 nM), although the enhancement is less significant than the addition of the natural bisecting GlcNAc moiety. These results indicate that adding a bisecting sugar moiety to the core N-glycan could enhance the affinity of Fc domain to FcγIIIa receptor, but the enhancement depends on the nature of the sugar moiety inserted. Trimming the high-mannose structure from Man9GlcNAc2 down to the pentasaccharide core Man3GlcNAc2 only slightly decreased the affinity (Man9-Fc, KD = 45 nM; Fc-5, KD = 58 nM). However, removal of the two outer mannose residues from the Man3GlcNAc2 core to the ManGlcNAc2 glycoform (Fc-6, KD = 354 nM) resulted in about 6-fold drop of the Fc affinity to FcγRIIIa. Further trimming of the oligosaccharide down to the glycoform (GlcNAc-Fc, KD > 3000 nM), which carries only the innermost GlcNAc moiety, resulted in dramatic loss of the Fc affinity. These results clearly indicate the essential role of the pentasaccharide core at the Fc domain for its high affinity binding to FcγRIIIa. On the other hand, changing the core β-mannose moiety in the Man3GlcNAc2- glycoform (Fc-5, KD = 58 nM) to a glucose moiety as the Man2GlcGlcNAc2-glycoform (Fc-4, KD = 79 nM) did not have significant effect on the Fc affinity, suggesting that the C-2 configuration of the core β-mannose moiety in the Fc N-glycan is not critical for Fc’s recognition to FcγRIIIa. Taken together, our experimental data clearly indicate that the Fc domain affinity to FcγRIIIa could be precisely tuned by the fine structures of the Fc N-glycan. Our experimental data with the pure, homogeneous synthetic Fc glycoforms provided unambiguous evidence showing that the presence of a bisecting GlcNAc moiety on the Fc glycan core could directly enhance the binding of Fc to FcγRIIIa. This result clarifies a long-standing controversial problem in the field as to the role of bisecting GlcNAc.19,21,26–28. The bisecting GlcNAc moiety plays both a direct and an indirect role in enhancing antibody Fc’s affinity to FcγRIIIa. On one hand, the presence of a bisecting GlcNAc moiety can directly impact the affinity of Fc to FcγRIIIa, independent of core-fucosylation. On the other hand, pre-addition of a bisecting moiety to the N-glycan core can inhibit the enzymatic attachment of a core fucose moiety during biosynthesis, playing an indirect role in enhancing the affinity of Fc to FcγRIIIa resulting from reduced population of the non-fucosylated Fc glycoforms.
We also measured the affinity of selected Fc glycoforms to the inhibitory Fcγ receptor FcγRIIb. FcγRIIb is an important regulator of both adaptive and innate immunity.4 We found that the Fc glycoforms with or without the bisecting sugar moiety (comparing Fc-1, Fc-2, and Fc-3 to Fc-5) showed similar affinity to FcγRIIb (KD = 2–3 μM) (Table 1). Thus, in contrast to the observation that addition of a bisecting GlcNAc moiety significantly enhanced Fc’s affinity to the activating receptor (FcγRIIIa), the presence of a bisecting sugar moiety had little effect on the affinity to the inhibitory Fcγ receptor. The similar binding affinity of the Fc glycoform carrying the core-fucosylated N-glycan (CT-Fc) to those without a core fucose moiety (HM-Fc, Fc-1, Fc-2, Fc-3, and Fc-5) also indicated that the presence of a core fucose moiety in the N-glycan did not have much influence on the binding of Fc to FcγRIIb. However, when the Fc N-glycan was trimmed off to leave only the trisaccharide ManGlcNAc2 core or the innermost GlcNAc moiety (Fc-6 and GlcNAc-Fc), the binding of the Fc domain to FcγRIIb became very weak, for which we did not obtain an accurate KD value. This observation is consistent with the previously reported observation on FcγRIIb-binding of core-fucosylated and truncated IgG-Fc glycoforms.34 Thus, it seems that the pentasaccharide (Man3GlcNAc2) core of the Fc N-glycan is essential and sufficient for the binding of Fc domain to FcγRIIb, but further decoration such as addition of a bisecting GlcNAc moiety to the core has little effect on the affinity.
Identification of high-affinity FcγRIIIa-binding glycoforms with enhanced ADCC function is clinically significant to address the issue of Fcγ receptor polymorphism found in cancer patients who are less or not responsive to the treatment with common MAbs. In these patients, the FcγRIIIa-F158 allele has low affinity to therapeutic antibody such as rituximab in comparison with the high-affinity receptor FcγRIIIa-V158 allele.72–74 ADCC function was also reported to be an important mechanism for achieving protective immunity for HIV-neutralizing antibodies.75 Our synthetic and Fcγ receptor-binding studies of various pure Fc glycoforms demonstrate that addition of an appropriate bisecting sugar moiety at the N-glycan core could significantly enhance the affinity of Fc to the activating receptor (FcγRIIIa) while having little effects on affinity to the inhibitory receptor (FcγRIIb), pointing to a way to enhance the ADCC function of antibodies. It should be pointed out that previous structural and functional studies have suggested that the impact of Fc glycan structures on the affinity of Fc domain to Fcγ receptors are mainly due to their effects on the functional conformations of the Fc domain that are critical for the interactions between Fc domain and respective Fcγ receptors.9–15 Indeed, the Fc oligosaccharide can form multiple noncovalent interactions with the protein surface of the CH2 domain and, as shown in the crystal structures, a large portion of the glycans appears sandwiched between the two CH2 domains. Nevertheless, it is still not clear how the addition of the bisecting GlcNAc or other (unnatural) bisecting sugar moiety precisely affects the local and/or global conformations of the Fc domain to favor its binding to FcγRIIIa. The synthetic, homogeneous Fc glycoforms obtained in the present work should be valuable for NMR and X-ray crystallographic structural studies for deciphering the precise molecular mechanism by which fine Fc N-glycans modulate the interaction between Fc domain and Fcγ receptors.
CONCLUSION
A convergent chemoenzymatic synthesis of an array of homogeneous IgG-Fc glycoforms was achieved through the Endo-A catalyzed glycosylation remodeling approach. The results indicate that Endo-A is remarkably efficient to introduce various modified N-glycan core to Fc domain by transglycosylation with the N-glycan core oxazolines. However, either Endo-A or the Endo-M mutant (EndoM-N175A and EndoM-N175Q) was unable to efficiently transfer a full-length complex type N-glycan to the Fc domain, implicating the limitations of Endo-A and Endo-M for IgG-Fc glycosylation remodeling. The availability of the synthetic homogeneous Fc glycoforms allowed a clear assessment on how the individual sugar residues within or flanking the pentasaccharide Man3GlcNAc2 core affect the binding of Fc to Fcγ receptors, FcγRIIIa and FcγRIIb. Specifically, our SPR binding studies provide unambiguous evidence that the presence of a bisecting sugar moiety at the N-glycan core, such as a GlcNAc or a mannose moiety, could directly enhance the affinity of Fc to FcγIIIa receptor, independent of Fc fucosylation. In contrast, the presence of a bisecting sugar moiety shows no effect on the intercation between Fc and the inhibitory Fcγ receptor FcγRIIb. These results implicate a new way to enhance antibody’s ADCC function by introducing an appropriate bisecting sugar moiety at the Fc glycan. To further expand the scope of this chemoenzymatic method for IgG-Fc glycosylation remodeling, we are currently performing site-directed mutagenesis of Endo-A and are also testing other endoglycosidases in order to find an enzyme or mutants capable of effectively transferring full-length complex type N-glycans to IgG-Fc domain. We look forward to reporting new findings from these studies.
EXPERIMENTAL SECTION
Materials and Methods
Endo-β-N-acetylglucosaminidase from Arthrobacter protophormiae (Endo-A) was overproduced in E. coli following the reported procedure.76 Bovine milk β-1,4-galactosyltransferase was purchased from Sigma-Aldrich (St. Louis, MO). PNGase F was purchased from New England Biolabs (Ipswich, MA). UDP-Gal was purchased from EMD Chemicals Inc. (Gibbstown, NJ). All other reagents were purchased from Sigma-Aldrich (St. Louis, MO) without further purification. TLC was performed using Silica-gel on aluminum plates (Sigma-Aldrich). Flash column chromatography was performed on silica gel 60 (230–400 mesh). NMR spectra were recorded on JEOL ECX 400 MHz spectrometer. The chemical shifts were assigned in ppm. Analytical RP-HPLC was performed on a Waters 626 HPLC instrument with a Symmetry300™ C18 column (3.5 μm, 4.6 × 250 mm) at 40 °C. The column was eluted with a linear gradient 0–90% MeCN containing 0.1% TFA for 20 min at the flow rate of 1.0 mL/min. MALDI-TOF/MS spectra were recorded on an Autoflex MALDI-TOF mass spectrometer (Bruker Daltonics, Billerica, MA) by using 2,5-dihydroxybenzoic acid as a matrix under positive ion conditions. High-resolution mass spectra (HRMS) were measured on a MALDI-TOF/TOF 4800 spectrometer (Applied Biosystems) with 2,5-dihydroxybenzoic acid as matrix and Cal 4700 standard peptide mixture (Applied Biosystems) was used as the internal standard.
Synthesis of benzyl 2-O-benzyl-4,6-O-benzylidene-3-O-p-methoxybenzyl-β-D-mannopyranosyl-(1→4)-3,6-di-O-benzyl-2-deoxy-2-phthalimido-β-D-glucopyranoside (9)
A mixture of phenyl 2-O-benzyl-4,6-O-benzylidene-3-O-p-methoxybenzyl-1-thio-α-D-mannopyranoside 8 57 (140 mg, 0.25 mmol), BSP (56.7 mg, 0.27 mmol), TTBP (122 mg, 0.49 mmol), and activated 3 Å molecular sieves (588 mg) in CH2Cl2 (4.9 mL) was stirred for 20 min at −60 °C under an argon atmosphere. Then Tf2O (50 μL, 0.30 mmol) was added, followed by the addition of a solution of benzyl 3,6-di-O-benzyl-2-deoxy-2-phthalimido-β-D-glucopyranoside 7 (111 mg 0.19 mmol) in CH2Cl2 (4.8 mL). The mixture was stirred at −60 °C for 1 h and then warmed to room temperature over a period of 1 h. The mixture was filtered through a Celite pad. The filtrate was diluted with CH2Cl2, washed sequentially with saturated NaHCO3 and brine. The organic layer was dried over MgSO4, filtered, and concentrated. The residue was subjected to silica gel column chromatography (hexanes/EtOAc, 7:2) to afford 9 (112 mg, 56%) as a white amorphous powder. ESI-MS: calcd. for C63H61NO13, M = 1039.41; Found (m/z), 1062.43 [M+Na]+; 1H NMR (400 MHz, CDCl3, TMS) δ 7.77–6.79 (m, 33H), 5.51 (s, 1H), 5.11 (d, 1H, J = 8.3 Hz), 4.89–4.78 (m, 2H), 4.68–4.64 (m, 2H), 4.55–4.47 (m, 3H), 4.43–4.39 (m, 2H), 4.26–4.15 (m, 3H), 4.08–4.00 (m, 2H), 3.79 (s, 3H), 3.72 (m, 1H), 3.67 (dd, 1H, J = 11.3, 1.6 Hz), 3.59–3.53 (m, 2H), 3.47 (m, 1H), 3.41 (dd, 1H, J = 9.9, 3.0 Hz), 3.14 (dt, 1H, J = 9.6, 4.6 Hz); 13C NMR (100 MHz, CDCl3) δ 167.65, 159.08, 138.70, 138.59, 137.74, 137.61, 137.13, 133.53, 131.57, 130.54, 129.00, 128.75, 128.48, 128.23, 128.10, 127.90, 127.76, 127.70, 127.55, 127.51, 127.47, 126.82, 126.04, 123.11, 113.66, 101.92, 101.25, 97.32, 79.44, 78.60, 77.93, 76.95, 76.90, 74.90, 74.64, 74.55, 73.51, 72.21, 70.66, 68.50, 68.46, 67.27, 55.64, 55.20.
Synthesis of benzyl 2-O-benzyl-3-O-p-methoxybenzyl-β-D-mannopyranosyl-(1→4)-3,6-di-O-benzyl-2-deoxy-2-phthalimido-β-D-glucopyranoside (10)
A solution of 9 (50 mg, 48 μmol) in 80% aqueous solution of AcOH (5 mL) was stirred at 40 °C. After stirring for 6 h, the mixture was concentrated and purified by silica gel column chromatography (hexanes/EtOAc, 1:2) to afford 10 (36 mg, 80%) as a white amorphous powder. ESI-MS: calcd. for C56H57NO13, M = 951.38; Found (m/z), 973.52 [M+Na]+; 1H NMR (400 MHz, CDCl3, TMS) δ 7.87–6.82 (m, 28H), 5.14 (d, 1H, J = 7.8 Hz), 4.89–4.71 (m, 5H), 4.55–4.49 (m, 3H), 4.43–4.37 (m, 2H), 4.29–4.20 (m, 3H), 4.02 (dd, 1H, J = 9.6, 8.2 Hz), 3.81–3.69 (m, 8H), 3.60 (m, 1H), 3.45 (m, 1H), 3.17 (m, 1H), 3.11 (dd, 1H, J = 9.6, 2.8 Hz), 2.18 (d, 1H, J = 1.8 Hz), 1.98 (t, 1H, J = 6.6 Hz); 13C NMR (100 MHz, CDCl3) δ 167.66, 159.37, 138.57, 138.42, 137.82, 137.14, 133.61, 131.56, 129.72, 129.25, 128.53, 128.20, 128.11, 127.84, 127.58, 127.53, 127.50, 127.41, 127.00, 123.17, 113.92, 101.10, 97.33, 81.63, 78.91, 76.80, 75.67, 74.71, 74.44, 74.29, 74.18, 73.62, 70.93, 70.67, 68.69, 67.15, 62.75, 55.63, 55.24.
Synthesis of benzyl 2-O-benzyl-6-O-tert-butyldimethylsilyl-3-O-p-methoxybenzyl-β-D-mannopyranosyl-(1→4)-3,6-di-O-benzyl-2-deoxy-2-phthalimido-β-D-glucopyranoside (11)
To a solution of 10 (455 mg, 0.48 mmol) in pyridine (5 mL) was added tert-butyldimethylchlorosilane (144 mg, 0.96 mmol) at room temperature. After 2 h, the reaction solution was diluted with CH2Cl2, washed with saturated NaHCO3 and brine, and dried over MgSO4. The solvent was evaporated and the residue was purified by column chromatography on silica gel (hexanes/EtOAc, 3:1) to provide 11 (483 mg, 95%) as a white amorphous powder. ESI-MS: calcd. for C62H71NO13Si, M = 1066.31; Found (m/z), 1089.58 [M+Na]+; 1H NMR (400 MHz, CDCl3, TMS) δ 7.87–6.74 (m, 28H), 5.11 (m, 1H), 4.87–4.75 (m, 4H), 4.67 (d, 1H, J = 11.9 Hz), 4.53–4.38 (m, 6H), 4.22–4.18 (m, 2H), 4.04–3.94 (m, 2H), 3.81 (dd, 1H, J = 10.1, 4.6 Hz), 3.78 (s, 3H), 3.74–3.67 (m, 2H), 3.65–3.59 (m, 2H), 3.51 (m, 1H), 3.36 (s, 1H), 3.22–3.13 (m, 2H), 0.84 (s, 9H), 0.02, −0.01 (2s, 6H); 13C NMR (100 MHz, CDCl3) δ 167.67, 159.11, 138.89, 138.83, 137.86, 137.16, 133.48, 131.59, 130.45, 129.18, 128.46, 128.07, 128.05, 127.80, 127.75, 127.68, 127.60, 127.55, 127.49, 127.24, 126.67, 123.07, 113.73, 101.29, 97.32, 81.23, 78.98, 76.79, 75.07, 74.74, 74.32, 74.20, 74.11, 73.48, 71.53, 70.65, 68.57, 65.38, 55.65, 55.20, 25.74, 18.02, −5.65, −5.70.
Synthesis of benzyl 3,4,6-tri-O-acetyl-2-deoxy-2-phthalimido-β-D-glucopyranosyl-(1→4)-2-O-benzyl-6-O-tert-butyldimethylsilyl-3-O-p-methoxybenzyl-β-D-mannopyranosyl-(1→4)-3,6-di-O-benzyl-2-deoxy-2-phthalimido-β-D-glucopyranoside (13)
A solution of 11 (23 mg, 22 μmol) and 2,3,4-tri-O-acetyl-2-deoxy-2-phthalimido-β-D-glucopyranosyl tricholoroacetimidate 12 58 (25 mg, 44 μmol) in CH2Cl2 (1 mL) containing activated 4 Å molecular sieves (120 mg) was stirred under an atmosphere of argon at room temperature for 30 min. After cooling to −20 °C, a solution of BF3·OEt2 in CH2Cl2 (0.1 M, 87 μL, 8.7 μmol) was added and the resulting mixture was stirred at room temperature overnight. Triethylamine (10 μL) was then added, and the mixture was filtered through a Celite pad. The filtrate was sequentially washed with saturated NaHCO3 and brine, dried over MgSO4, filtered, and concentrated. The residue was subjected to flash silica gel column chromatography (hexanes/EtOAc, 5:2) to provide 13 (25 mg, 76%) as a white amorphous powder. ESI-MS: calcd. for C82H90N2O22Si, M = 1482.57; Found (m/z), 1505.81 [M+Na]+; 1H NMR (400 MHz, CDCl3, TMS) δ 7.86–6.59 (m, 32H), 5.76 (dd, 1H, J = 10.8, 9.0 Hz), 5.56 (d, 1H, J = 8.7 Hz), 5.11 (t, 1H, J = 9.6 Hz), 5.03 (m, 1H), 4.79–4.69 (m, 4H), 4.63–4.58 (m, 2H), 4.47–4.36 (m, 5H), 4.26 (dd, 1H, J = 10.8, 8.5 Hz), 4.15–4.06 (m, 4H), 3.95–3.89 (m, 2H), 3.82 (s, 3H), 3.69–3.55 (m, 5H), 3.46 (m, 1H), 3.27–3.21 (m, 2H), 2.97 (m, 1H), 1.99, 1.93, 1.84 (3s, 9H), 0.77 (s, 9H), −0.04, −0.14 (2s, 6H); 13C NMR (100 MHz, CDCl3) δ 170.69, 170.04, 169.48, 167.52, 158.80, 138.96, 138.90, 137.84, 137.15, 134.35, 133.26, 131.60, 131.20, 130.97, 128.40, 128.15, 128.02, 127.92, 127.82, 127.72, 127.51, 127.47, 127.43, 127.04, 126.47, 123.62, 122.97, 113.66, 100.95, 97.23, 97.07, 79.86, 78.73, 76.86, 76.23, 75.68, 74.65, 74.26, 73.99, 73.49, 73.41, 71.32, 71.13, 70.69, 70.57, 68.92, 68.60, 61.84, 61.78, 55.58, 55.31, 55.22, 25.68, 20.60, 20.56, 20.39, 17.98, −5.41, −5.56.
Synthesis of benzyl 3,4,6-tri-O-acetyl-2-deoxy-2-phthalimido-β-D-glucopyranosyl-(1→4)-2-O-benzyl-β-D-mannopyranosyl-(1→4)-3,6-di-O-benzyl-2-deoxy-2-phthalimido-β-D-glucopyranoside (14)
To a stirred solution of compound 13 (302 mg, 0.20 mmol) in CH2Cl2 (20 mL) was added TFA (3.75 mL) at 0 °C. The solution was stirred at 0 °C for 1 h, and MeOH (2 mL) was added. The mixture was sequentially washed with saturated NaHCO3 and brine. The organic layer was then dried over MgSO4 and filtered. The filtrate was concentrated by evaporation and the residue was purified by column chromatography on silica gel (hexanes/EtOAc, 1:1) to yield 14 (224 mg, 88%) as a white solid. ESI-MS: calcd. for C68H68N2O21, M = 1248.43; Found (m/z), 1271.58 [M+Na]+; 1H NMR (400 MHz, CDCl3, TMS) δ 7.88–6.73 (m, 28H), 5.78 (dd, 1H, J = 10.6, 9.2 Hz), 5.42 (d, 1H, J = 8.7 Hz), 5.12 (t, 1H, J = 9.6 Hz), 5.05 (m, 1H), 4.86–4.65 (m, 5H), 4.47–4.41 (m, 3H), 4.36–4.30 (m, 2H), 4.27 (dd, 1H, J = 12.4, 2.3 Hz), 4.27 (dd, 1H, J = 12.4, 6.5 Hz), 4.16–4.12 (m, 2H), 3.98 (m, 1H), 3.89 (m, 1H), 3.85 (d, 1H, J = 2.8 Hz), 3.73 (t, 1H, J = 9.2 Hz), 3.67–3.63 (m, 2H), 3.56 (dd, 1H, J = 11.0, 3.3 Hz), 3.44–3.38 (m, 2H), 3.14 (m, 1H), 2.08, 2.04, 1.85 (3s, 9H); 13C NMR (100 MHz, CDCl3) δ 170.58, 169.91, 169.44, 167.68, 138.70, 138.43, 137.56, 137.09, 134.47, 133.49, 131.50, 128.48, 128.05, 127.92, 127.71, 127.50, 127.46, 127.34, 127.28, 126.80, 123.64, 123.08, 100.66, 98.53, 97.23, 79.13, 78.62, 77.43, 76.69, 74.85, 74.54, 74.26, 74.06, 73.51, 72.96, 71.84, 70.59, 70.34, 68.81, 68.02, 61.94, 61.15, 55.54, 54.53, 20.55, 20.52, 20.32.
Synthesis of benzyl 2,3,4,6-tetra-O-acetyl-α-D-mannopyranosyl-(1→3)-[3,4,6-tri-O-acetyl-2-deoxy-2-phthalimido-β-D-glucopyranosyl-(1→4)]-[2,3,4,6-tetra-O-acetyl-α-D-mannopyranosyl-(1→6)]-2-O-benzyl-β-D-mannopyranosyl-(1→4)-3,6-di-O-benzyl-2-deoxy-2-phthalimido-β-D-glucopyranoside (16)
A solution of 14 (50 mg, 40 μmol) and 2,3,4,6-tetra-O-acetyl-α-D-mannopyranosyl tricholoroacetimidate 15 77 (197 mg, 0.40 mmol) in CH2Cl2 (2.5 mL) containing activated 4 Å molecular sieves (296 mg) was stirred under an atmosphere of argon at room temperature for 30 min. After cooling to −20 °C, a solution of TMSOTf in CH2Cl2 (1 M, 80 μL, 80 μmol) was added and the resulting mixture was stirred at room temperature overnight. Triethylamine (10 μL) was then added, and the mixture was filtered through a Celite pad. The filtrate was sequentially washed with saturated NaHCO3 and brine, dried over MgSO4, and filtered. The filtrate was concentrated and the residue was purified by silica gel column chromatography (hexanes/EtOAc, 2:3) to provide 16 (41 mg, 54%) as a white amorphous powder. ESI-MS: calcd for C96H104N2O39, M = 1909.84; Found (m/z): 1910.68 [M+H]+; 1H NMR (400 MHz, CDCl3, TMS) δ 7.92–6.61 (m, 28H), 5.77 (dd, 1H, J = 10.5, 8.7 Hz), 5.64 (m, 1H), 5.45–5.39 (m, 2H), 5.34–5.20 (m, 4H), 5.11–5.04 (m, 3H), 4.93–4.81 (m, 3H), 4.77–4.73 (m, 2H), 4.64 (d, 1H, J = 12.8 Hz), 4.50–4.39 (m, 3H), 4.35–4.25 (m, 3H), 4.23–4.10 (m, 6H), 4.09–3.92 (m, 5H), 3.73 (m, 1H), 3.68–3.62 (m, 2H), 3.60–3.55 (m, 3H), 3.47 (m, 1H), 3.22 (dd, 1H, J = 9.6, 2.7 Hz), 2.32, 2.13, 2.10, 2.08, 2.06, 2.02, 2.01, 1.96, 1.92, 1.90, 1.86 (11s, 33H); 13C NMR (100 MHz, CDCl3) δ 170.59, 170.56, 170.39, 170.30, 170.08, 169.60, 169.56, 169.39, 168.09, 167.50, 167.11, 138.11, 138.04, 137.67, 136.96, 134.96, 134.49, 133.51, 133.15, 131.53, 131.39, 131.08, 130.94, 128.37, 128.35, 128.23, 127.93, 127.73, 127.65, 127.52, 127.38, 127.31, 126.92, 123.61, 123.47, 123.18, 122.84, 99.58, 99.41, 97.20, 97.11, 96.88, 78.08, 77.12, 77.07, 75.85, 74.85, 74.59, 74.20, 74.05, 73.29, 72.31, 70.66, 70.46, 69.53, 69.48, 69.22, 68.98, 68.65, 68.40, 68.16, 67.86, 66.67, 65.62, 65.32, 62.67, 62.51, 62.44, 55.52, 54.16, 20.98, 20.93, 20.77, 20.66, 20.63, 20.56, 20.53, 20.50, 20.39, 20.32.
Synthesis of 2,3,4,6-tetra-O-acetyl-α-D-mannopyranosyl-(1→3)-[2-acetamido-3,4,6-tri-O-acetyl-2-deoxy-β-D-glucopyranosyl-(1→4)]-[2,3,4,6-tetra-O-acetyl-α-D-mannopyranosyl-(1→6)]-2-O-acetyl-β-D-mannopyranosyl-(1→4)-2-acetamido-1,3,6-tri-O-acetyl-2-deoxy-D-glucopyranose (18)
Compound 16 (173 mg, 91 μmol) was dissolved in EtOH/H2O/NH2NH2 monohydrate (10:1:1, 12 mL), and the mixture was stirred at 80 °C overnight. The mixture was concentrated in vacuo, and the residue was treated with Ac2O-pyridine (1:1, 10 mL) at room temperature overnight. The mixture was concentrated, diluted with CH2Cl2, and washed sequentially with 1 M HCl, saturated NaHCO3, and brine. The organic layer was dried over MgSO4 and filtered. The filtrate was concentrated. Silica gel column chromatography (CH2Cl2/MeOH, 30:1) of the residue afforded compound 17 (122 mg, 77%) as a white powder. Without further purification, compound 17 thus obtained was dissolved in CH2Cl2–MeOH–AcOH (20:80:1, v/v, 12 mL), and 20% palladium(II) hydroxide on activated carbon (120 mg) was added. The mixture was vigorously stirred at room temperature under hydrogen atmosphere overnight and then filtered through a Celite pad. The filtrate was concentrated in vacuo. Pyridine (10 mL) and Ac2O (10 mL) were added, and the mixture was stirred at room temperature overnight. The mixture was concentrated, diluted with CH2Cl2, washed sequentially with 1 M HCl, saturated NaHCO3, and brine. The organic layer was dried over MgSO4 and filtered. The filtrate was concentrated. Silica gel column chromatography (CH2Cl2/MeOH, 45:1) of the residue afforded 18 (97 mg, 89% from 17) as a white amorphous powder. ESI-MS: calcd for C64H88N2O41, M = 1541.37; Found (m/z): 1563.75 [M+Na]+; 1H NMR (400 MHz, CDCl3, TMS, selected signals) δ 6.07 (d, 1H, J = 3.7 Hz), 5.96–5.88 (m, 2H), 4.69 (d, 1H, J = 8.7 Hz), 2.21–1.94 (m, 51H); 13C NMR (100 MHz, CDCl3) δ 171.02, 170.88, 170.55, 170.40, 170.35, 170.27, 170.23, 170.00, 169.83, 169.68, 169.60, 169.49, 169.06, 168.98, 100.07, 97.39, 96.80, 90.46, 75.13, 73.60, 73.14, 72.39, 72.05, 71.86, 71.20, 69.78, 68.87, 68.74, 68.48, 68.40, 66.37, 65.72, 65.23, 62.56, 62.06, 61.75, 61.67, 54.06, 50.59, 22.85, 22.61, 20.73, 20.57, 20.45, 20.36, 20.29, 20.20.
Synthesis of 2-methyl-[2,3,4,6-tetra-O-acetyl-α-D-mannopyranosyl-(1→3)-[2-acetamido-3,4,6-tri-O-acetyl-2-deoxy-β-D-glucopyranosyl-(1→4)]-[2,3,4,6-tetra-O-acetyl-α-D-mannopyranosyl- (1→6)]-2-O-acetyl-β-D-mannopyranosyl-(1→4)-3,6-di-O-acetyl-1,2-dideoxy-α-D-glucopyrano]-[2,1-d]-2-oxazoline (19)
To a solution of 18 (55 mg, 36 μmol) in dichloroethane (3.5 mL) containing activated 4 Å molecular sieves (420 mg) were added 2,4,6-collidine (71 μL, 0.53 mmol), TMSBr (69 μL, 0.53 mmol), and BF3·OEt2 (67 μL, 0.53 mmol). The reaction mixture was stirred at room temperature overnight. The mixture was diluted with CH2Cl2, filtered through a Celite pad, washed sequentially with saturated NaHCO3 and brine. The organic layer was dried over MgSO4 and filtered. The filtrate was concentrated and the residue was subjected to silica gel chromatography (CH2Cl2/MeOH, 50:1) on silica gel to give 19 (25 mg, 48%) as a white amorphous powder. ESI-MS: calcd for C62H84N2O39, M = 1480.47; Found (m/z): 1481.69 [M+H]+; 1H NMR (400 MHz, CDCl3, TMS) δ 6.00 (d, 1H, J = 9.1 Hz), 5.91 (d, 1H, J = 7.4 Hz), 5.54 (m, 1H), 5.36–5.04 (m, 10H), 5.01 (d, 1H, J = 1.4 Hz), 4.75 (s, 1H), 4.66 (d, 1H, J = 8.7 Hz), 4.39–3.76 (m, 17H), 3.59 (m, 1H), 3.50 (m, 1H), 3.43 (m, 1H), 2.21–1.94 (m, 48H); 13C NMR (100 MHz, CDCl3) δ 171.14, 170.84, 170.64, 170.60, 170.55, 169.98, 169.86, 169.73, 169.60, 169.57, 169.55, 169.49, 169.25, 165.93, 100.36, 99.08, 99.03, 98.41, 97.71, 76.07, 74.48, 74.38, 72.65, 72.40, 72.11, 69.77, 69.60, 69.39, 68.97, 68.93, 68.84, 68.81, 68.59, 68.48, 67.78, 66.76, 65.98, 65.79, 64.60, 63.46, 63.18, 62.35, 62.22, 54.36, 23.03, 20.92, 20.82, 20.78, 20.71, 20.68, 20.64, 20.61, 20.56, 20.54, 20.47, 13.60.
Synthesis of 2-methyl-[α-D-mannopyranosyl-(1→3)]-[2-acetamido-2-deoxy-β-D-glucopyranosyl- (1→4)]-[α-D-mannopyranosyl-(1→6)]-β-D-mannopyranosyl-(1→4)-1,2-dideoxy-α-D-glucopyrano-[2,1-d]-2-oxazoline (1)
To a solution of 19 (23 mg, 16 μmol) in MeOH (2 mL) was added MeONa in MeOH (0.5 M, 3.2 μL, 1.6 μmol). After stirring at room temperature overnight, the reaction mixture was concentrated to dryness. The residue was dissolved in water and lyophilized to give sugar oxazoline 1 (14 mg, quantitative). ESI-MS: calcd for C34H56N2O25, M = 892.32; Found (m/z), 893.78 [M+H]+; MALDI-TOF HRMS: calcd for [M + Na]+, 915.3070; found (m/z), 915.3048; 1H NMR (400 MHz, D2O) δ 5.96 (d, 1H, J = 7.3 Hz), 5.10 (s, 1H), 4.89 (s, 1H), 4.59 (m, 1H), 4.40 (d, 1H, J = 8.2 Hz), 4.24 (m, 1H), 4.09–3.23 (m, 29H), 1.94–1.92 (m, 6H); 13C NMR (100 MHz, D2O) δ 174.61, 168.73, 101.83, 101.18, 101.06, 100.31, 99.93, 77.82, 77.24, 76.43, 74.26, 73.44, 73.15, 70.85, 70.48, 70.40, 70.23, 69.88, 69.84, 69.14, 66.91, 66.87, 65.70, 65.10, 61.70, 61.47, 61.13, 56.16, 22.19, 13.00.
Preparation of free bisecting GlcNAc-containing oligosaccharide 20
A solution of peracetylated pentasaccharide 18 (50 mg, 32.4 μmol) in MeOH (2 mL) containing catalytic amount of MeONa was stirred at room temperature overnight. The reaction mixture was neutralized with Dowex 50W-X8 (H+ form) and filtered. The filtrate was concentrated to dryness. The residue was dissolved in water and lyophilized to give free pentasaccharide 20 (30 mg, quantitative). ESI-MS: calcd for C34H58N2O26, M = 910.33, Found (m/z): 933.37 [M+Na]+; 1H NMR (400 MHz, D2O) δ 5.10 (s, 1H), 5.07 (m, 0.7H, αH-1 of GlcNAc-1), 4.82 (s, 1H), 4.63 (m, 1H), 4.40 (m, 1H), 4.16–3.23 (m, 30H), 1.93 (m, 6H); 13C NMR (100 MHz, D2O) δ 174.61, 101.93, 101.18, 100.86, 100.11, 100.03, 94.92, 79.92, 77.34, 76.43, 74.56, 73.44, 73.15, 70.65, 70.48, 70.24, 70.03, 69.98, 69.84, 69.14, 66.91, 66.67, 65.80, 65.10, 61.40, 61.17, 60.03, 56.16, 22.19, 21.90.
Synthesis of sugar oxazoline 1 by the DMC-based one-pot method
A solution of pentasaccharide 20 (15 mg, 16.5 μmol), 2-chloro-1,3-dimethylimidazolinium chloride (DMC) (50 mg, 295 μmol) and Et3N (90 μL, 648 μmol) in water (400 μL) was stirred at 4 °C for 1h. The reaction mixture was subjected to gel filtration chromatography on a Sephadex G-10 column eluted by 0.05% aqueous Et3N. The fractions containing the product were combined and lyophilized to give oxazoline 1 as a white powder (15 mg, quantitative yield). The sugar oxazoline obtained by this method was identical to the above synthesized compound as confirmed by 1H NMR, 13C NMR, and ESI-MS.
Synthesis of bisecting LacNAc-containing hexasaccharide 21
A solution of pentasaccharide 20 (15 mg, 16.5 μmol) and UDP-Gal (15 mg, 24.9 μmol) in a HEPES buffer (50 mM, pH 7.5, 1.5 mL) containing α-lactalbumin (0.2 mg/mL) and Mn2+ (20 mM) was incubated with β-1,4-galactosyltransferase (1 U) at 37 °C for 6 h. The reaction mixture was subject to gel filtration chromatography on a Sephadex G-10 column eluted by 0.05% aqueous Et3N. The fractions containing the product were combined and lyophilized to give hexasaccharide 21 as a white solid (18 mg, 1quantitative yield). ESI-MS: calcd for C40H68N2O31, M = 1072.38, Found (m/z): 1095.03 [M+Na]+. 1H NMR (400 MHz, D2O) δ 5.10 (s, 1H), 5.08 (m, 0.65H, αH-1 of GlcNAc-1), 4.81 (s, 1H), 4.61 (m, 1H), 4.42 (d, 1H, J = 6.8 Hz), 4.32 (d, 1H, J = 7.6 Hz), 4.24–3.36 (m, 35H), 3.08 (m, 1H), 1.93 (m, 6H). 13C NMR (100 MHz, D2O) δ 174.62, 101.95, 101.02, 100.89, 100.28, 100.14, 100.07, 94.92, 79.96, 77.38, 76.40, 74.59, 74.19, 73.86, 73.56, 73.42, 73.33, 73.10, 72.24, 70.50, 70.41, 70.27, 70.21, 70.02, 69.92, 69.88, 69.68, 69.12, 67.65, 66.90, 66.83, 65.78, 65.14, 61.47, 61.18, 60.06, 56.12, 53.56, 22.16, 21.95.
Synthesis of bisecting LacNAc-containing sugar oxazoline 2
A solution of hexasaccharide 21 (15 mg, 14.0 μmol), 2-chloro-1,3-dimethylimidazolinium chloride (DMC) (50 mg, 295 μmol) and Et3N (90 μL, 648 μmol) in water (400 μL) was stirred at 4 °C for 1h. The reaction mixture was subjected to gel filtration chromatography on a Sephadex G-10 column eluted by 0.05% aqueous Et3N. The fractions containing the product were combined and lyophilized to give oxazoline 2 (15 mg, quantitative yield) as a white powder. ESI-MS: calcd for C40H66N2O30, M = 1054.37, Found (m/z): 1055.42 [M+H]+. MALDI-TOF HRMS: calcd for [M + Na]+, 1077.3598; found (m/z), 1077.3638; 1H NMR (400 MHz, D2O) δ 5.97 (d, 1H, J = 6.8 Hz), 5.10 (s, 1H), 4.88 (d, 1H, J = 1.6 Hz), 4.59 (s, 1H), 4.43 (d, 1H, J = 7.6 Hz), 4.32 (d, 1H, J = 8.0 Hz), 4.24 (m, 1H), 1.94-1.92 (m, 6H). 13C NMR (100 MHz, D2O, selected from 1H-13C HMQC) δ 102.35, 101.85, 101.33, 100.46, 99.67, 99.56, 82.58, 79.23, 77.87, 76.02, 74.33, 74.18, 72.68, 72.17, 71.25, 70.97, 70.57, 70.12, 69.27, 68.87, 68.53, 67.32, 66.56, 65.65, 61.32, 56.02, 53.88, 22.56, 13.27.
Synthesis of benzyl 2-O-benzoyl-4,6-O-benzylidene-3-O-p-methoxybenzyl-β-D-glucopyranosyl-(1→4)-2-azido-3,6-di-O-benzyl-2-deoxy-β-D-glucopyranoside (24)
Ethyl 2-O-benzyl-4,6-O-benzylidene-3-O-p-methoxybenzyl-1-thio-β-D-glucopyranoside 22 62 (677 mg, 1.26 mmol), benzyl 2-azido-3,6-di-O-benzyl-2-deoxy-β-D-glucopyranoside 23 (500 mg, 1.05 mmol) and 4Å molecular sieves (1.44 g) were stirred in dichloroethane (12 mL) at room temperature for 30 min and the mixture was then cooled to 0 °C. A solution of NIS (290 mg, 1.29 mmol) and TfOH (12.6 μL, 142 μmol) in CH2Cl2–Et2O (1:1, 12.6 mL) was added to the mixture and the resulting mixture was stirred for 30 min at 0 °C. Et3N (60 μL) was added, the reaction mixture was filtered through a Celite pad. The filtrate was diluted with CH2Cl2, and washed sequentially with 10% Na2S2O3, saturated NaHCO3, and brine. The organic layer was dried over MgSO4, filtered, and concentrated. The residue was subjected to silica gel column chromatography (hexanes/EtOAc, 6:1) to give 24 (632 mg, 63%) as a white amorphous powder. ESI-MS: calcd for C55H55N3O12, M = 949.3786; Found (m/z): 950.51 [M+H]+. 1H NMR (400 MHz, CDCl3, TMS) δ 7.86–6.58 (m, 29H), 5.52 (s, 1H), 5.19 (t, 1H, J = 8.5 Hz), 4.93 (d, 1H, J = 10.6 Hz), 4.84 (d, 1H, J = 11.9 Hz), 4.73 (d, 1H, J = 10.5 Hz), 4.72 (d, 1H, J = 11.5 Hz), 4.68 (d, 1H, J = 7.8 Hz), 4.66 (d, 1H, J = 12.4 Hz), 4.58 (d, 1H, J = 11.9 Hz), 4.57 (d, 1H, J = 11.9 Hz), 4.30 (d, 1H, J = 12.4 Hz), 4.20–4.16 (m, 2H), 3.99 (t, 1H, J = 9.4 Hz), 3.75–3.63 (m, 5H), 3.59 (dd, 1H, J = 11.3, 3.0 Hz), 3.49–3.41 (m, 3H), 3.33–3.22 (m, 2H), 3.08 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 164.70, 159.06, 138.34, 137.91, 137.23, 136.72, 133.24, 129.89, 129.79, 129.63, 129.43, 129.03, 128.52, 128.37, 128.26, 128.21, 128.03, 127.97, 127.85, 127.81, 127.60, 126.01, 113.51, 101.21, 100.66, 100.31, 81.85, 81.11, 77.21, 76.13, 75.30, 74.50, 73.78, 73.54, 73.46, 70.80, 68.50, 67.29, 66.04, 65.71, 55.07.
Synthesis of benzyl 2-O-benzoyl-4,6-O-benzylidene-β-D-glucopyranosyl-(1→4)-2-azido-3,6-di-O-benzyl-2-deoxy-β-D-glucopyranoside (25)
To a solution of 24 (47 mg, 50 μmol) in CH2Cl2-H2O (17.5/1, 2.1 mL) was added DDQ (26 mg, 115 μmol) at 0 °C. After 30 min, the reaction mixture was warmed to room temperature and further stirred for 2 h. The reaction mixture was diluted with CH2Cl2, washed with saturated NaHCO3 and brine, and dried over MgSO4. The mixture was filtered and the filtrate was concentrated. The residue was subjected to silica gel column chromatography (hexanes/EtOAc, 4:1) to provide 25 (36 mg, 87%) as a white amorphous powder. ESI-MS: calcd for C47H47N3O11, M = 829.3211; Found (m/z): 830.58 [M+H]+. 1H NMR (400 MHz, CDCl3, TMS) δ 7.99–7.26 (m, 25H), 5.48 (s, 1H), 5.16 (t, 1H, J = 8.7 Hz), 4.93 (d, 1H, J = 10.5 Hz), 4.86 (d, 1H, J = 12.4 Hz), 4.79 (d, 1H, J = 8.3 Hz), 4.75 (d, 1H, J = 11.5 Hz), 4.71 (d, 1H, J = 12.4 Hz), 4.59 (d, 1H, J = 11.9 Hz), 4.21–4.17 (m, 2H), 4.03 (t, 1H, J = 9.2 Hz), 3.89–3.83 (m, 1H), 3.69 (dd, 1H, J = 11.0, 3.2 Hz), 3.58–3.43 (m, 4H), 3.35–3.25 (m, 2H), 3.14 (m, 1H), 2.51 (d, 1H, J = 3.2 Hz); 13C NMR (100 MHz, CDCl3) δ 165.35, 138.33, 137.93, 136.81, 136.70, 133.44, 129.80, 129.34, 129.24, 128.53, 128.50, 128.37, 128.34, 128.21, 128.03, 127.91, 127.87, 127.81, 127.78, 127.62, 126.25, 101.86, 100.45, 100.32, 81.12, 80.87, 76.24, 75.26, 74.78, 74.52, 73.53, 72.29, 70.81, 68.40, 67.41, 66.01, 65.73.
Synthesis of benzyl 2-O-benzoyl-4-O-benzyl-β-D-glucopyranosyl-(1→4)-2-azido-3,6-di-O-benzyl-2-deoxy-β-D-glucopyranoside (26)
To a suspension of 25 (50 mg, 60 μmol) and 4 Å molecular sieves (192 mg) in CH2Cl2 (1.6 mL) were added successively Et3SiH (29 μL, 181 μmol) and PhBCl2 (27 μL, 205 μmol) at −78 °C. After stirring at −78 °C for 1 h, Et3N (116 μL) and MeOH (116 μL) were added. The reaction mixture was diluted with CH2Cl2, filtered through a Celite pad, and washed sequentially with saturated NaHCO3 and brine. The organic layer was dried over MgSO4, filtered, and concentrated. The resulting residue was subjected to silica gel column chromatography (hexanes/EtOAc, 3:1) to afford 26 (47 mg, 94%) as a white amorphous powder. ESI-MS: calcd for C47H49N3O11, M = 831.3367; Found (m/z), 832.50 [M+H]+. 1H NMR (400 MHz, CDCl3, TMS) δ 7.95–7.26 (m, 25H), 5.02 (dd, 1H, J = 9.2, 8.3 Hz), 4.95 (d, 1H, J = 11.0 Hz), 4.86 (d, 1H, J = 12.4 Hz), 4.79–4.67 (m, 5H), 4.60 (d, 1H, J = 11.9 Hz), 4.41 (d, 1H, J = 11.9 Hz), 4.20 (d, 1H, J = 7.8 Hz), 3.97 (t, 1H, J = 9.3 Hz), 3.75 (dt, 1H, J = 9.2, 3.7 Hz), 3.70–3.65 (m, 2H), 3.53 (dd, 1H, J = 11.0, 1.4 Hz), 3.49–3.43 (m, 2H), 3.41–3.29 (m, 2H), 3.21 (ddd, 1H, J = 9.5, 4.5, 2.6 Hz), 3.15 (m, 1H), 2.31 (d, 1H, J = 4.1 Hz), 1.40 (t, 1H, J = 7.1 Hz); 13C NMR (100 MHz, CDCl3) δ 165.65, 138.36, 137.88, 137.79, 136.69, 133.38, 129.71, 129.25, 128.53, 128.51, 128.45, 128.39, 128.34, 127.99, 127.96, 127.85, 127.78, 127.69, 127.10, 100.26, 99.88, 81.06, 77.97, 76.13, 75.51, 75.13, 75.02, 74.70, 74.65, 74.48, 73.54, 70.75, 67.33, 65.79, 61.54.
Synthesis of benzyl 6-O-acetyl-2,3,4-tri-O-benzoyl-α-D-mannopyranosyl-(1→3)-[6-O-acetyl-2,3,4-tri-O-benzoyl-α-D-mannopyranosyl-(1→6)]-2-O-benzoyl-4-O-benzyl-β-D-glucopyranosyl-(1→4)-2-azido-3,6-di-O-benzyl-2-deoxy-β-D-glucopyranoside (28)
A solution of compound 26 (75 mg, 90 μmol) and 2,3,4-tri-O-acetyl-6-O-benzoyl-α-D-mannopyranosyl tricholoroacetimidate 27 78 (305 mg, 449 μmol) in CH2Cl2 (3.4 mL) containing activated 4 Å molecular sieves (456 mg) was stirred under an atmosphere of argon at room temperature for 30 min. After cooling to −40 °C, a solution of TMSOTf in CH2Cl2 (0.1 M, 450 μL, 45 μmol) was added and the resulting mixture was stirred at room temperature overnight. The mixture was filtered through a Celite pad. The filtrate was poured into saturated NaHCO3, and extracted with CH2Cl2. The organic layer was washed with brine, dried over MgSO4, and filtered. The filtrate was concentrated and the residue was subjected to silica gel column chromatography (hexanes/EtOAc, 5:2) to provide 28 (159 mg, 95%) as a white amorphous. ESI-MS: calcd for C105H97N3O29; M = 1864.90; Found (m/z): 1865.83 [M+H]+. 1H NMR (400 MHz, CDCl3, TMS) δ 8.10–7.16 (m, 55H), 5.95–5.83 (m, 3H), 5.77–5.74 (m, 2H), 5.61 (t, 1H, J = 2.0 Hz), 5.51 (s, 1H), 5.37 (t, 1H, J = 8.7 Hz), 5.13 (d, 1H, J = 0.9 Hz), 5.07 (d, 1H, J = 11.0 Hz), 4.90 (d, 1H, J = 11.5 Hz), 4.84–4.80 (m, 2H), 4.76–4.69 (m, 3H), 4.54 (d, 1H, J = 11.9 Hz), 4.36 (d, 1H, J = 11.9 Hz), 4.28–4.02 (m, 7H), 3.89 (dd, 1H, J = 12.1, 2.6 Hz), 3.81 (d, 1H, J = 11.0 Hz), 3.73–3.61 (m, 3H), 3.54–3.43 (m, 4H), 3.28 (t, 1H, J = 9.4 Hz), 3.11 (d, 1H, J = 9.6 Hz), 2.08 (s, 3H), 1.99 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 170.61, 170.32, 165.44, 165.32, 165.26, 165.12, 165.04, 165.01, 164.65, 138.33, 137.90, 137.06, 136.82, 133.56, 133.49, 133.44, 133.43, 133.35, 133.27, 133.23, 133.07, 132.94, 130.05, 129.80, 129.76, 129.67, 129.59, 129.33, 129.23, 129.07, 128.98, 128.94, 128.80, 128.61, 128.56, 128.42, 128.34, 128.28, 128.23, 128.20, 128.15, 128.11, 128.07, 127.93, 127.88, 127.76, 127.71, 127.51, 100.60, 99.71, 98.60, 98.03, 80.77, 80.17, 79.28, 75.64, 75.26, 75.01, 74.70, 74.39, 73.63, 73.02, 70.76, 70.26, 69.95, 69.85, 69.70, 69.12, 68.91, 67.63, 67.49, 66.40, 65.87, 65.40, 62.30, 61.70, 20.66, 20.53.
Synthesis of benzyl 6-O-acetyl-2,3,4-tri-O-benzoyl-α-D-mannopyranosyl-(1→3)-[6-O-acetyl-2,3,4-tri-O-benzoyl-α-D-mannopyranosyl-(1→6)]-2-O-benzoyl-4-O-benzyl-β-D-glucopyranosyl- (1→4)-2-acetamido-3,6-di-O-benzyl-2-deoxy-β-D-glucopyranoside (29)
To a solution of 28 (604 mg, 0.324 mmol) in CHCl3 (6 mL) was added pyridine (6 mL) and thioacetic acid (AcSH) (6 mL) at room temperature. After stirring for 48 h, the mixture was concentrated and the residue was subjected to silica gel column chromatography (hexanes/EtOAc, 1:1) to afford 29 (474 mg, 78%). ESI-MS: calcd for C107H101NO30; M = 1880.94; Found (m/z), 1881.68 [M+H]+. 1H NMR (400 MHz, CDCl3, TMS) δ 8.12–7.12 (m, 55H), 6.01–5.86 (m, 3H), 5.79–5.74 (m, 3H), 5.63 (d, 1H, J = 8.7 Hz), 5.54 (s, 1H), 5.43 (t, 1H, J = 8.9 Hz), 5.26 (s, 1H), 5.15 (d, 1H, J = 11.5 Hz), 4.83 (d, 1H, J = 11.5 Hz), 4.70 (d, 1H, J = 8.2 Hz), 4.65–4.53 (m, 5H), 4.35 (d, 1H, J = 12.4 Hz), 4.29 (dd, 1H, J = 12.2, 4.4 Hz), 4.24–4.07 (m, 6H), 3.96–3.83 (m, 4H), 3.77–3.69 (m, 3H), 3.64–3.62 (m, 2H), 3.51–3.46 (m, 2H), 2.09 (s, 3H), 1.99 (s, 3H), 1.81 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 170.65, 170.33, 170.04, 165.49, 165.46, 165.31, 165.24, 165.14, 165.05, 164.89, 138.44, 138.10, 137.66, 137.32, 133.54, 133.44, 133.33, 133.25, 133.10, 133.06, 129.82, 129.77, 129.65, 129.43, 129.30, 129.25, 129.00, 128.91, 128.76, 128.68, 128.61, 128.42, 128.25, 128.22, 128.06, 128.03, 127.95, 127.89, 127.73, 127.37, 127.31, 99.84, 99.05, 98.52, 98.10, 80.44, 78.77, 77.17, 75.35, 74.75, 74.36, 73.45, 72.80, 70.55, 70.25, 70.04, 69.87, 69.73, 69.01, 68.91, 66.80, 66.63, 65.86, 62.50, 61.77, 53.33, 23.25, 20.66, 20.53.
Synthesis of 2,3,4,6-tetra-O-acetyl-α-D-mannopyranosyl-(1→3)-[2,3,4,6-tetra-O-acetyl-α-D-mannopyranosyl-(1→6)]-2,4-di-O-acetyl-β-D-glucopyranosyl-(1→4)-2-acetamido-1,3,6-tri-O-acetyl-2-deoxy-D-glucopyranose (30)
To a solution of 29 (95 mg, 54 μmol) in MeOH/CH2Cl2 (5:1, 10 mL) was added a solution of MeONa in MeOH (0.5 M, 400 μL). The resulting mixture was stirred at room temperature overnight and then neutralized with Dowex 50W-X8 (H+ form). The mixture was filtered and the filtrate was concentrated. The residue was dissolved in MeOH (5 mL) and palladium(II) hydroxide (20%) on activated carbon (40 mg) was added. The resulting mixture was vigorously stirred at room temperature under hydrogen atmosphere for 24 h. The reaction mixture was filtered through a Celite pad and the filtrate was concentrated in vacuo to dryness. The residue was dissolved in pyridine (3 mL) and Ac2O (3 mL) for acetylation. The mixture was stirred at room temperature for 18 h and then concentrated in vacuo to dryness. The residue was subjected to silica gel column chromatography (CH2Cl2/MeOH, 60:1 to 40:1) to give 30 (62 mg, 92% in three steps) as a white solid. ESI-MS: calcd for C52H71NO34; M = 1253.3858; found (m/z): 1254.68 [M + H+]. 1H NMR (400 MHz, CDCl3, TMS) δ 5.92 (d, 1H, J = 7.3 Hz), 5.78 (d, 1H, J = 2.7 Hz), 5.36–5.21 (m, 4H), 5.14–5.08 (m, 2H), 5.03–4.93 (m, 2H), 4.89 (d, 1H, J = 1.8 Hz), 4.79 (s, 1H), 4.70 (d, 1H, J = 7.8 Hz), 4.39 (dd, 1H, J = 12.3, 4.5 Hz), 4.24–4.18 (m, 2H), 4.16–3.99 (m, 5H), 3.95 (dt, 1H, J = 10.1, 2.8 Hz), 3.89–3.74 (m, 3H), 3.57 (m, 1H), 3.49–3.42 (m, 2H), 2.16–2.13 (m, 9H), 2.11–2.07 (m, 18H), 2.01 (s, 3H), 1.95–1.94 (m, 9H); 13C NMR (100 MHz, CDCl3) δ 170.68, 170.52, 170.49, 170.00, 169.96, 169.87, 169.54, 169.48, 169.33, 169.23, 166.21, 101.56, 99.67, 98.83, 97.46, 81.70, 78.24, 72.53, 71.98, 70.02, 69.52, 69.31, 69.19, 68.92, 68.82, 68.73, 68.40, 67.29, 65.68, 64.99, 64.36, 63.65, 61.97, 61.49, 20.89, 20.85, 20.78, 20.77, 20.68, 20.62, 20.56, 20.47, 20.44, 13.78.
Synthesis of 2-methyl-[α-D-mannopyranosyl-(1→3)]-[α-D-mannopyranosyl-(1→6)]-[β-D-glucopyranosyl-(1→4)]-1,2-dideoxy-α-D-glucopyrano]-[2,1-d]-2-oxazoline (4)
To a solution of 30 (58 mg, 46 μmol) in MeOH (2 mL) was added a catalytic amount of MeONa and the solution was stirred at r.t. overnight. The reaction mixture was neutralized with Dowex 50W-X8 (H+ form) and filtered. The filtrate was concentrated to dryness and the residue was dissolved in water and lyophilized to give the free tetrasaccharide (33 mg, quantitative) (ESI-MS, found (m/z), 730.31 [M + Na]+). The free oligosaccharide was then dissolved in water (1 mL), and 2-chloro-1,3-dimethylimidazolinium chloride (DMC) (78 mg, 460 μmol) and Et3N (128 μL, 920 μmol) were added. The solution was stirred at 4 °C for 1h. Then the reaction mixture was subjected to gel filtration on a Sephadex G-10 column eluted with 0.05% aqueous Et3N. The fractions containing the product were combined and lyophilized to give oxazoline 4 (30 mg, 95%) as a white powder. ESI-MS: calcd for C26H43NO20, M = 689.24; Found (m/z): 690.35 [M + H]+. MALDI-TOF HRMS: calcd for [M + Na]+, 712.2276; found (m/z), 712.2258; 1H NMR (400 MHz, D2O) δ 5.97 (d, 1H, J = 7.3 Hz), 5.08 (s, 1H), 4.79 (s, 1H), 4.40 (d, 1H, J = 7.8 Hz), 4.27 (m, 1H), 4.08 (m, 1H), 3.92 (m, 1H), 3.94–3.46 (m, 19H), 3.31 (m, 1H), 3.21 (m, 1H), 1.94 (s, 3H); 13C NMR (100 MHz, D2O) δ 168.12, 104.60, 101.10, 99.93, 99.70, 82.25, 79.62, 74.07, 72.90, 72.76, 71.91, 71.02, 70.79, 70.66, 70.40, 69.96, 68.92, 66.91, 66.63, 65.60, 65.43, 61.93, 61.03, 60.67, 12.97.
Overproduction of human IgG1-Fc in Chinese Hamster Ovary (CHO) cell lines
The expression of human IgG1-Fc in Pichia pastoris was performed following the procedure that we have previously described.36 For producing recombinant IgG1-Fc in Chinese Hamster Ovary (CHO) cell lines, the CHO cells were transiently transfected with a mammalian expression plasmid pcDNA-Fc encoding the human Fc gene, using Fugene6 transfection reagent (Roche). After transfection, kifunensine was added into the medium to a final concentration of 2 μg/mL. The expression of fusion protein was periodically monitored using an anti-human IgG1 Fc ELISA assay kit (Human IgG ELISA Quantitation Kit, BETHYL Laboratories Inc, Montgomery TX). The culture supernatant was collected 4 days after transfection. The Fc protein was purified by affinity chromatography using a protein A-agarose resin (Pierce). The cell-free culture supernatant was loaded on the protein A column that was pre-equilibrated with IgG binding buffer (pH 8.0, Thermo Scientific, Rockford, IL) After extensive wash with the same buffer, Fc protein was eluted with IgG Elution Buffer (pH 3.0, Thermo Scientific, Rockford, IL). Each elution fraction was immediately neutralized with 1M Tris, pH 8.8. Subsequently, the elution fractions were combined and concentrated by centrifugal filtration (Amicon® Ultra centrifugal filter, Millipore, Billerica, MA). The integrity and purity of the recombinant IgG-Fc that carries high-mannose type N-glycans (HM-Fc) was checked by SDS-PAGE. MALDI-TOF MS of HM-Fc, calcd M = 65444 Da; found (m/z), 65447. MALDI-TOF MS of the PNGase F released N-glycan, calcd for Man9GlcNAc2, M = 1883.67 Da; found (m/z) 1906.40 [M + Na+]. For the production of the natural complex type glycoform of IgG-Fc, the transfected CHO cells were grown in the absence of the α-mannosidase inhibitor kifunensine, and the recombinant IgG-Fc was purified following the same procedure.
Preparation of GlcNAc-Fc homodimer
The recombinant IgG-Fc (HM-Fc) (15 mg) was treated with Endo-H (0.8 unit) in a phosphate buffer (3 mL, 50 mM, pH 6.5) at 30 °C. After 10 h, SDS-PAGE indicated the completion of the de-glycosylation. The GlcNAc-Fc homodimer was then purified by protein A affinity chromatography. MALDI-TOF MS of GlcNAc-Fc: calcd M = 62119 Da; found (m/z), 62112.
Synthesis of the bisecting-GlcNAc Fc glycoform (Fc-1) by the Endo-A catalyzed transglycosylation of GlcNAc-Fc
A solution of GlcNAc-Fc homodimer (1.20 mg, 19.3 nmol) and bi-GlcNAc-Man3GlcNAc oxazoline 1 (0.345 mg, 386 nmol, about 10 equivalent for each of the GlcNAc moiety in the GlcNAc-Fc homodimer) in a phosphate buffer (50 mM, pH 7.0, 200 μL) was incubated with Endo-A (8 μg) at 23°C. The enzymatic reactions were monitored by SDS-PAGE analysis and MALDI-TOF MS analysis of an aliquot of the reaction mixture. When all the GlcNAc-Fc was converted (after 3h), the reaction mixture was subject to an affinity chromatography on a protein A-agarose column, following the procedure described above for purification of the recombinant IgG1-Fc. The fractions containing the IgG-Fc glycoform were combined, desalted by ultracentrifugation, and lyophilized to give Fc-1 (1.1 mg) (quantified using the anti-human IgG1 Fc ELISA quantification kit). The purified IgG-Fc glycoform (Fc-1) was analyzed by MALDI-TOF MS. The homogeneity of the product was confirmed by SDS-PAGE analysis under reducing conditions. The attached N-glycan was released by PNGase F treatment and analyzed by MALDI-TOF MS following the previously reported procedure.36 MALDI-TOF MS of Fc-1, calcd M = 63905 Da; found (m/z), 63909. PNGase F released N-glycan (bisecting-GlcNAc-Man3GlcNAc2): calcd for C42H71N3O31, M = 1113.41 Da; found (m/z), 1136.80 [M + Na]+.
Synthesis of other Fc glycoforms (Fc-2 to Fc-6) by the Endo-A catalyzed transglycosylation of GlcNAc-Fc
The synthesis of glycoforms Fc-2, Fc-3, Fc-4, Fc-5, and Fc-6 was performed in the same way as described for the preparation of Fc-1, using excess of the corresponding sugar oxazoline (10–15 molecular equivalent of each of the GlcNAc moiety in the GlcNAc-Fc homodimer). After SDS-PAGE indicated the completion of the GlcNAc-Fc transformation, the product was purified on a protein A affinity column. The purity of the product was confirmed by SDS-PADE analysis and the identity was characterized by MALDI-TOF MS analysis of the intact Fc glycoforms and the released N-glycans.
Fc-2: calcd M = 64229 Da; found (m/z), 64212; the attached N-glycan (bisecting-LacNAc-Man3GlcNAc2): calcd for C48H81N3O36, M = 1275.46 Da; found (m/z), 1298.40 [M + Na]+.
Fc-3: calcd. M = 63822 Da; found (m/z), 63840; the attached N-glycan (bisecting-Man-Man3GlcNAc2): calcd for C40H68N2O31, M = 1072.38 Da; found (m/z), 1095.10 [M + Na]+.
Fc-4: calcd. M = M = 63498 Da; found (m/z), 63506; the attached N-glycan (Man2GlcGlcNAc2): calcd for C34H58N2O26, M = 910.33 Da; found (m/z), 933.42 [M + Na]+.
Fc-5: calcd. M = M = 63498 Da; found (m/z), 63534; the attached N-glycan (Man3GlcNAc2): calcd for C34H58N2O26, M = 910.33 Da; found (m/z), 933.80 [M + Na]+.
Fc-6: calcd. M = 62849 Da; found (m/z), 62870; the attached N-glycan (ManGlcNAc2): calcd for C22H38N2O16, M = 586.22 Da; found (m/z), 609.70 [M + Na]+.
SPR studies
The binding between different glycoforms of IgG1-Fc and FcγIIIa receptor was measured by surface plasmon resonance (SPR) using a Biacore T100 instrument (GE Healthcare, USA). Protein A was immobilized on a CM5 biosensor chip (GE Healthcare, USA) at 2700 RU using a standard primary amine coupling chemistry at pH 4.5. A reference flow cell was prepared similarly without injecting protein A. To capture the different glycoforms of IgG1-Fc, each of the individual IgG1-Fc glycoforms in HBS-P buffer (10 mM HEPES, 150 mM NaCl, 0.05% surfactant P20, pH 7.4) was injected at 10 μL/min onto the protein A surface until it reached the capture level of 100 RU. A series of dilution of FCγIIIa receptor was injected at 10 μL/min. After each cycle, the surface was regenerated by injecting 20 mM HCl at 10 μL/min for 30 seconds. Data were fitted into a 1:1 steady state binding model using BIAcore T100 evaluation software and the equilibrium constant KD was derived.
Supplementary Material
Acknowledgments
We thank Prof. Kaoru Takegawa (Kyushu University) for providing the pGEX-2T/Endo-A plasmid that was used for expressing the recombinant Endo-A, and the Gliknik Inc. for providing the mammalian expression vector pcDNA-Fc that was used for expressing the high-mannose type glycoform. This work was supported by the National Institutes of Health (NIH grants R01 GM096973 and R01 GM080374)
Footnotes
SUPPORTING INFORMATION AVAILABLE: Complete author list of ref 32; amino acid sequence of the recombinant IgG1-Fc; reaction scheme and SDS-PAGE analysis for the transglycosylation of GlcNAc-Fc with complex-type N-glycan oxazoline. This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.Adams GP, Weiner LM. Nat Biotechnol. 2005;23:1147–1157. doi: 10.1038/nbt1137. [DOI] [PubMed] [Google Scholar]
- 2.Jefferis R. Nat Rev Drug Discov. 2009;8:226–234. doi: 10.1038/nrd2804. [DOI] [PubMed] [Google Scholar]
- 3.Aggarwal S. Nat Biotechnol. 2010;28:1165–1171. doi: 10.1038/nbt1110-1165. [DOI] [PubMed] [Google Scholar]
- 4.Nimmerjahn F, Ravetch JV. Nat Rev Immunol. 2008;8:34–47. doi: 10.1038/nri2206. [DOI] [PubMed] [Google Scholar]
- 5.Takahashi N, Nakagawa H, Fujikawa K, Kawamura Y, Tomiya N. Anal Biochem. 1995;226:139–146. doi: 10.1006/abio.1995.1201. [DOI] [PubMed] [Google Scholar]
- 6.Wormald MR, Rudd PM, Harvey DJ, Chang SC, Scragg IG, Dwek RA. Biochemistry. 1997;36:1370–1380. doi: 10.1021/bi9621472. [DOI] [PubMed] [Google Scholar]
- 7.Jefferis R. Biotechnol Prog. 2005;21:11–16. doi: 10.1021/bp040016j. [DOI] [PubMed] [Google Scholar]
- 8.Arnold JN, Wormald MR, Sim RB, Rudd PM, Dwek RA. Annu Rev Immunol. 2007;25:21–50. doi: 10.1146/annurev.immunol.25.022106.141702. [DOI] [PubMed] [Google Scholar]
- 9.Krapp S, Mimura Y, Jefferis R, Huber R, Sondermann P. J Mol Biol. 2003;325:979–989. doi: 10.1016/s0022-2836(02)01250-0. [DOI] [PubMed] [Google Scholar]
- 10.Sondermann P, Huber R, Oosthuizen V, Jacob U. Nature. 2000;406:267–273. doi: 10.1038/35018508. [DOI] [PubMed] [Google Scholar]
- 11.Crispin M, Bowden TA, Coles CH, Harlos K, Aricescu AR, Harvey DJ, Stuart DI, Jones EY. J Mol Biol. 2009;387:1061–1066. doi: 10.1016/j.jmb.2009.02.033. [DOI] [PubMed] [Google Scholar]
- 12.Ferrara C, Grau S, Jager C, Sondermann P, Brunker P, Waldhauer I, Hennig M, Ruf A, Rufer AC, Stihle M, Umana P, Benz J. Proc Natl Acad Sci USA. 2011;108:12669–12674. doi: 10.1073/pnas.1108455108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yamaguchi Y, Nishimura M, Nagano M, Yagi H, Sasakawa H, Uchida K, Shitara K, Kato K. Biochim Biophys Acta. 2006;1760:693–700. doi: 10.1016/j.bbagen.2005.10.002. [DOI] [PubMed] [Google Scholar]
- 14.Matsumiya S, Yamaguchi Y, Saito J, Nagano M, Sasakawa H, Otaki S, Satoh M, Shitara K, Kato K. J Mol Biol. 2007;368:767–779. doi: 10.1016/j.jmb.2007.02.034. [DOI] [PubMed] [Google Scholar]
- 15.Barb AW, Prestegard JH. Nat Chem Biol. 2011;7:147–153. doi: 10.1038/nchembio.511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Homans SW, Dwek RA, Fernandes DL, Rademacher TW. Biochim Biophys Acta. 1984;798:78–83. doi: 10.1016/0304-4165(84)90012-6. [DOI] [PubMed] [Google Scholar]
- 17.Nimmerjahn F, Ravetch JV. Annu Rev Immunol. 2008;26:513–533. doi: 10.1146/annurev.immunol.26.021607.090232. [DOI] [PubMed] [Google Scholar]
- 18.Shields RL, Lai J, Keck R, O’Connell LY, Hong K, Meng YG, Weikert SH, Presta LG. J Biol Chem. 2002;277:26733–26740. doi: 10.1074/jbc.M202069200. [DOI] [PubMed] [Google Scholar]
- 19.Shinkawa T, Nakamura K, Yamane N, Shoji-Hosaka E, Kanda Y, Sakurada M, Uchida K, Anazawa H, Satoh M, Yamasaki M, Hanai N, Shitara K. J Biol Chem. 2003;278:3466–3473. doi: 10.1074/jbc.M210665200. [DOI] [PubMed] [Google Scholar]
- 20.Niwa R, Shoji-Hosaka E, Sakurada M, Shinkawa T, Uchida K, Nakamura K, Matsushima K, Ueda R, Hanai N, Shitara K. Cancer Res. 2004;64:2127–2133. doi: 10.1158/0008-5472.can-03-2068. [DOI] [PubMed] [Google Scholar]
- 21.Kanda Y, Yamada T, Mori K, Okazaki A, Inoue M, Kitajima-Miyama K, Kuni-Kamochi R, Nakano R, Yano K, Kakita S, Shitara K, Satoh M. Glycobiology. 2007;17:104–118. doi: 10.1093/glycob/cwl057. [DOI] [PubMed] [Google Scholar]
- 22.Ferrara C, Stuart F, Sondermann P, Brunker P, Umana P. J Biol Chem. 2006;281:5032–5036. doi: 10.1074/jbc.M510171200. [DOI] [PubMed] [Google Scholar]
- 23.Kaneko Y, Nimmerjahn F, Ravetch JV. Science. 2006;313:670–673. doi: 10.1126/science.1129594. [DOI] [PubMed] [Google Scholar]
- 24.Anthony RM, Nimmerjahn F, Ashline DJ, Reinhold VN, Paulson JC, Ravetch JV. Science. 2008;320:373–376. doi: 10.1126/science.1154315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Anthony RM, Wermeling F, Karlsson MC, Ravetch JV. Proc Natl Acad Sci USA. 2008;105:19571–19578. doi: 10.1073/pnas.0810163105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Umana P, Jean-Mairet J, Moudry R, Amstutz H, Bailey JE. Nat Biotechnol. 1999;17:176–180. doi: 10.1038/6179. [DOI] [PubMed] [Google Scholar]
- 27.Davies J, Jiang L, Pan LZ, LaBarre MJ, Anderson D, Reff M. Biotechnol Bioeng. 2001;74:288–294. [PubMed] [Google Scholar]
- 28.Hodoniczky J, Zheng YZ, James DC. Biotechnol Prog. 2005;21:1644–1652. doi: 10.1021/bp050228w. [DOI] [PubMed] [Google Scholar]
- 29.Yamane-Ohnuki N, Kinoshita S, Inoue-Urakubo M, Kusunoki M, Iida S, Nakano R, Wakitani M, Niwa R, Sakurada M, Uchida K, Shitara K, Satoh M. Biotechnol Bioeng. 2004;87:614–622. doi: 10.1002/bit.20151. [DOI] [PubMed] [Google Scholar]
- 30.Stanley P, Sundaram S, Tang J, Shi S. Glycobiology. 2005;15:43–53. doi: 10.1093/glycob/cwh136. [DOI] [PubMed] [Google Scholar]
- 31.Cox KM, Sterling JD, Regan JT, Gasdaska JR, Frantz KK, Peele CG, Black A, Passmore D, Moldovan-Loomis C, Srinivasan M, Cuison S, Cardarelli PM, Dickey LF. Nat Biotechnol. 2006;24:1591–1597. doi: 10.1038/nbt1260. [DOI] [PubMed] [Google Scholar]
- 32.Li H, et al. Nat Biotechnol. 2006;24:210–215. doi: 10.1038/nbt1178. [DOI] [PubMed] [Google Scholar]
- 33.Strasser R, Castilho A, Stadlmann J, Kunert R, Quendler H, Gattinger P, Jez J, Rademacher T, Altmann F, Mach L, Steinkellner H. J Biol Chem. 2009;284:20479–20485. doi: 10.1074/jbc.M109.014126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mimura Y, Sondermann P, Ghirlando R, Lund J, Young SP, Goodall M, Jefferis R. J Biol Chem. 2001;276:45539–45547. doi: 10.1074/jbc.M107478200. [DOI] [PubMed] [Google Scholar]
- 35.Watt GM, Lund J, Levens M, Kolli VS, Jefferis R, Boons GJ. Chem Biol. 2003;10:807–814. doi: 10.1016/j.chembiol.2003.08.006. [DOI] [PubMed] [Google Scholar]
- 36.Wei Y, Li C, Huang W, Li B, Strome S, Wang LX. Biochemistry. 2008;47:10294–10304. doi: 10.1021/bi800874y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huang W, Li J, Wang LX. ChemBioChem. 2011;12:932–941. doi: 10.1002/cbic.201000763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Umekawa M, Li C, Higashiyama T, Huang W, Ashida H, Yamamoto K, Wang LX. J Biol Chem. 2010;285:511–521. doi: 10.1074/jbc.M109.059832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schwarz F, Huang W, Li C, Schulz BL, Lizak C, Palumbo A, Numao S, Neri D, Aebi M, Wang LX. Nat Chem Biol. 2010;6:264–266. doi: 10.1038/nchembio.314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huang W, Zhang X, Ju T, Cummings RD, Wang LX. Org Biomol Chem. 2010;8:5224–5233. doi: 10.1039/c0ob00341g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huang W, Yang Q, Umekawa M, Yamamoto K, Wang LX. ChemBioChem. 2010;11:1350–1355. doi: 10.1002/cbic.201000242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huang W, Wang D, Yamada M, Wang LX. J Am Chem Soc. 2009;131:17963–17971. doi: 10.1021/ja9078539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huang W, Li C, Li B, Umekawa M, Yamamoto K, Zhang X, Wang LX. J Am Chem Soc. 2009;131:2214–2223. doi: 10.1021/ja8074677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Umekawa M, Huang W, Li B, Fujita K, Ashida H, Wang LX, Yamamoto K. J Biol Chem. 2008;283:4469–4479. doi: 10.1074/jbc.M707137200. [DOI] [PubMed] [Google Scholar]
- 45.Ochiai H, Huang W, Wang LX. J Am Chem Soc. 2008;130:13790–13803. doi: 10.1021/ja805044x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rising TW, Heidecke CD, Moir JW, Ling Z, Fairbanks AJ. Chem Eur J. 2008;14:6444–6464. doi: 10.1002/chem.200800365. [DOI] [PubMed] [Google Scholar]
- 47.Heidecke CD, Ling Z, Bruce NC, Moir JW, Parsons TB, Fairbanks AJ. ChemBioChem. 2008;9:2045–2051. doi: 10.1002/cbic.200800214. [DOI] [PubMed] [Google Scholar]
- 48.Amin MN, Huang W, Mizanur RM, Wang LX. J Am Chem Soc. 2011;133:14404–14417. doi: 10.1021/ja204831z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Takegawa Y, Deguchi K, Nakagawa H, Nishimura S. Anal Chem. 2005;77:6062–6068. doi: 10.1021/ac050843e. [DOI] [PubMed] [Google Scholar]
- 50.Paulsen H. Angew Chem Int Ed Engl. 1990;29:823–839. [Google Scholar]
- 51.Paulsen H, Heume M, Nurnberger H. Carbohydr Res. 1990;200:127–166. doi: 10.1016/0008-6215(90)84187-y. [DOI] [PubMed] [Google Scholar]
- 52.Weiss H, Unverzagt C. Angew Chem Int Ed. 2003;42:4261–4263. doi: 10.1002/anie.200351625. [DOI] [PubMed] [Google Scholar]
- 53.Eller S, Schuberth R, Gundel G, Seifert J, Unverzagt C. Angew Chem Int Ed. 2007;46:4173–4175. doi: 10.1002/anie.200604788. [DOI] [PubMed] [Google Scholar]
- 54.Eller S, Raps C, Niemietz M, Unverzagt C. Tetrahedron Lett. 2010;51:2648–2651. [Google Scholar]
- 55.Wang P, Zhu J, Yuan Y, Danishefsky SJ. J Am Chem Soc. 2009;131:16669–16671. doi: 10.1021/ja907136d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Crich D, Dudkin V. J Am Chem Soc. 2001;123:6819–6825. doi: 10.1021/ja010086b. [DOI] [PubMed] [Google Scholar]
- 57.Crich D, Li W, Li H. J Am Chem Soc. 2004;126:15081–15086. doi: 10.1021/ja0471931. [DOI] [PubMed] [Google Scholar]
- 58.Grundler G, Schmidt RR. Carbohydr Res. 1985;135:203–218. [Google Scholar]
- 59.Colon M, Staveski MM, Davis JT. Tetrahedron Lett. 1991;32:4447–4450. [Google Scholar]
- 60.Li B, Zeng Y, Hauser S, Song H, Wang LX. J Am Chem Soc. 2005;127:9692–9693. doi: 10.1021/ja051715a. [DOI] [PubMed] [Google Scholar]
- 61.Noguchi M, Tanaka T, Gyakushi H, Kobayashi A, Shoda SI. J Org Chem. 2009;74:2210–2212. doi: 10.1021/jo8024708. [DOI] [PubMed] [Google Scholar]
- 62.Verduyn R, Douwes M, van der Klein PAM, Mösinger EM, van der Marel GA, van Boom JH. Tetrahedron. 1993;49:7301–7316. [Google Scholar]
- 63.Vallee F, Karaveg K, Herscovics A, Moremen KW, Howell PL. J Biol Chem. 2000;275:41287–41298. doi: 10.1074/jbc.M006927200. [DOI] [PubMed] [Google Scholar]
- 64.Chang VT, Crispin M, Aricescu AR, Harvey DJ, Nettleship JE, Fennelly JA, Yu C, Boles KS, Evans EJ, Stuart DI, Dwek RA, Jones EY, Owens RJ, Davis SJ. Structure. 2007;15:267–273. doi: 10.1016/j.str.2007.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Scanlan CN, Ritchie GE, Baruah K, Crispin M, Harvey DJ, Singer BB, Lucka L, Wormald MR, Wentworth P, Jr, Zitzmann N, Rudd PM, Burton DR, Dwek RA. J Mol Biol. 2007;372:16–22. doi: 10.1016/j.jmb.2007.06.027. [DOI] [PubMed] [Google Scholar]
- 66.Zhou Q, Shankara S, Roy A, Qiu H, Estes S, McVie-Wylie A, Culm-Merdek K, Park A, Pan C, Edmunds T. Biotechnol Bioeng. 2008;99:652–665. doi: 10.1002/bit.21598. [DOI] [PubMed] [Google Scholar]
- 67.Yang Q, Li C, Wei Y, Huang W, Wang LX. Bioconjugate Chem. 2010;21:875–883. doi: 10.1021/bc9004238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ochiai H, Huang W, Wang LX. Carbohydr Res. 2009;344:592–598. doi: 10.1016/j.carres.2009.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Umekawa M, Higashiyama T, Koga Y, Tanaka T, Noguchi M, Kobayashi A, Shoda S, Huang W, Wang LX, Ashida H, Yamamoto K. Biochim Biophys Acta. 2010;1800:1203–1209. doi: 10.1016/j.bbagen.2010.07.003. [DOI] [PubMed] [Google Scholar]
- 70.Chu SY, Vostiar I, Karki S, Moore GL, Lazar GA, Pong E, Joyce PF, Szymkowski DE, Desjarlais JR. Mol Immunol. 2008;45:3926–3933. doi: 10.1016/j.molimm.2008.06.027. [DOI] [PubMed] [Google Scholar]
- 71.Shields RL, Namenuk AK, Hong K, Meng YG, Rae J, Briggs J, Xie D, Lai J, Stadlen A, Li B, Fox JA, Presta LG. J Biol Chem. 2001;276:6591–6604. doi: 10.1074/jbc.M009483200. [DOI] [PubMed] [Google Scholar]
- 72.Cartron G, Dacheux L, Salles G, Solal-Celigny P, Bardos P, Colombat P, Watier H. Blood. 2002;99:754–758. doi: 10.1182/blood.v99.3.754. [DOI] [PubMed] [Google Scholar]
- 73.Johnson P, Glennie M. Semin Oncol. 2003;30:3–8. doi: 10.1053/sonc.2003.50025. [DOI] [PubMed] [Google Scholar]
- 74.Koene HR, Kleijer M, Algra J, Roos D, von dem Borne AE, de Haas M. Blood. 1997;90:1109–1114. [PubMed] [Google Scholar]
- 75.Hessell AJ, Hangartner L, Hunter M, Havenith CE, Beurskens FJ, Bakker JM, Lanigan CM, Landucci G, Forthal DN, Parren PW, Marx PA, Burton DR. Nature. 2007;449:101–104. doi: 10.1038/nature06106. [DOI] [PubMed] [Google Scholar]
- 76.Fujita K, Tanaka N, Sano M, Kato I, Asada Y, Takegawa K. Biochem Biophys Res Commun. 2000;267:134–138. doi: 10.1006/bbrc.1999.1963. [DOI] [PubMed] [Google Scholar]
- 77.Ren T, Liu D. Tetrahedron Lett. 1999;40:7621–7625. [Google Scholar]
- 78.Heng L, Ning J, Kong F. Carbohydr Res. 2001;331:431–437. doi: 10.1016/s0008-6215(01)00059-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.