

Abstract
Vascular endothelial cells lining the blood vessels form the interface between the bloodstream and the vessel wall and as such they are continuously subjected to shear and cyclic stress from the flowing blood in the lumen. Additional mechanical stimuli are also imposed on these cells in the form of substrate stiffness transmitted from the extracellular matrix components in the basement membrane, and additional mechanical loads imposed on to lung endothelium as result of respiration or mechanical ventilation in clinical settings. Focal adhesions (FAs) are complex structures assembled at the abluminal endothelial plasma membrane which connect the extracellular filamentous meshwork to the intracellular cytoskeleton and hence constitute the ideal checkpoint capable of controlling or mediating transduction of bidirectional mechanical signals. In this review we focus on focal adhesion kinase (FAK), a component of FAs, which has been studied for a number of years with regards to its involvement in mechanotransduction. We analyzed the recent advances in the understanding of the role of FAK in the signaling cascade(s) initiated by various mechanical stimuli with particular emphasis on potential implications on endothelial cell functions.
Keywords: cyclic stretch, shear stress, migration, permeability, apoptosis, cytoskeleton, focal adhesions, protein phosphorylation
1. Focal adhesions in mechanotransduction
1.1. Focal adhesions: definition and dynamics
Focal adhesions (FAs) are complex structures that assemble at the plasma membrane in discrete regions of integrin-mediated recognition of extracellular matrix (ECM) components. They connect the extracellular filamentous meshwork to the intracellular cytoskeleton (Figure 1) conferring them the status of an ideal checkpoint capable of controlling or mediating bidirectional mechanical transduction through endothelial cell (Geiger et al., 2009). Such signals are now believed to regulate a variety of cellular processes including cell migration, proliferation, apoptosis and detachment from the ECM (Mofrad et al., 2004). However, the precise biochemical mechanism by which FAs control mechanical signal transduction is still unclear. Several hypotheses have been proposed in the recent years to account for this transmission of mechanical information including conformational change of FA components, cellular phosphorylation cascades triggered by signaling kinases induced in mechanically stimulated cells, and transactivation of other membrane-bound proteins such as ion channels. FAs are far from being static or uniform entities. They are dynamic structures as they have been most notably reported during cell migration, where an asymmetry between the front and the back of the cell correlates with the directionality of movement. Newly formed adhesions at the leading edge are much smaller in size than pre-existing mature focal adhesions.
Figure 1. Focal adhesions as ideal checkpoints controlling inside-out and inwards transduction of mechanical signals.
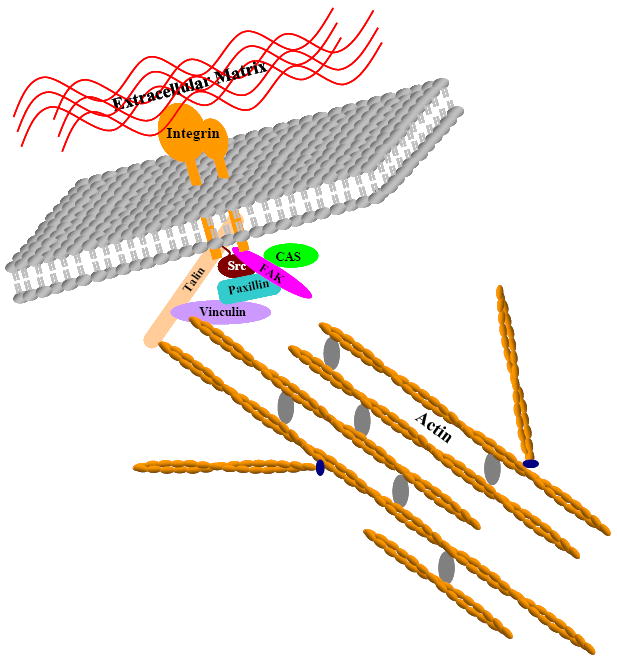
Focal adhesion components including transmembrane integrins and intracellular Src, FAK, Paxillin, Talin, Vinculin and CAS all assemble at the cell surface to create a physical anchorage point linking the extracellular matrix to the intracellular meshwork of actin filaments.
In turn, cell movement on the substrate requires a coordinated cell attachment at the front and detachment at the back of the cell, and FAs at the rear and at the front edges of a migrating cell have to exhibit different levels of adhesivity to the substratum (Shan et al., 2009; Shemesh et al., 2009).
1.2. Involvement of focal adhesions in the two-way mechanotransduction at the cell surface
Although the detailed biochemical events involved in FA mechanosensory processes are yet to be clarified, it is well established that internal mechanical forces generated by the actomyosin machinery as well as external mechanical inputs in the form of stretch or rigidity of the extracellular matrix (ECM) do affect focal adhesion dynamics (Wolfenson et al., 2009). The intracellular modulation of FA by the acto-myosin axis was demonstrated using a variety of approaches which alter myosin-II activity such as inhibitors which downregulate myosin-II light chain phosphorylation or block its ATPase activity in addition to RNAi-mediated knockdown of myosin II subsequently leading to the inhibition of the acto-myosin contractile activity (Alahyan et al., 2006; Alexandrova et al., 2008; Chrzanowska-Wodnicka and Burridge, 1996; Even-Ram et al., 2007; Geiger and Bershadsky, 2001; Riveline et al., 2001; Vicente-Manzanares et al., 2007; Volberg et al., 1994; Zhao et al., 2008). The implication of FAs in mechanosensitivity of extracellular cues was demonstrated by applying mechanical stimuli to these structures in various systems and settings. Cells were indeed subjected to stretch by micromanipulation and edge-pulling (Riveline et al., 2001), by magnetic traction using integrin-linked magnetic beads (Mack et al., 2004), by applying “stretching” forces to elastic substrate (Kaverina et al., 2002; Sniadecki et al., 2007) or to engineered micropillars (Sniadecki and Chen, 2007) underneath a cell. Such external pulling stimuli resulted in a net increase in the size of the focal adhesions (Wolfenson et al., 2009). Using engineered nanopatterned surfaces, a number of studies have shown that the cell is not only sensitive to the chemical composition of such surfaces, but also to their spatial distribution as well as their stiffness (see (Geiger et al., 2009) for review).
Although most of these approaches were reductionist in that they used simplified in vitro systems utilizing single cells or at most two-dimensional monolayer cell cultures, the authors attempted to apply forces in the same order of magnitude as those estimated from in vivo settings. It is well established that different tissues sense and respond to different levels of applied external forces. For instance, while chondrocytes and osteocytes experience stresses (force per unit area) of 20MPa (Ehrlich and Lanyon, 2002; Grodzinsky et al., 2000; Janmey and Weitz, 2004), endothelial cells and neutrophils are capable of responding to stresses lower than 1Pa (Dewey et al., 1981; Fukuda and Schmid-Schonbein, 2003; Garcia-Cardena et al., 2001). Furthermore, according to Chen et al, individual cell contact structures are subjected to stresses of 0.01-0.1 atm (1-10nN) in vivo (Chen et al., 2004), providing a framework for the relative physiological relevance of the forces used in the in vitro systems used in the studies above.
The cell’s “mechanobehaviour” in living tissues is bound to be more complicated as it involves not only one mechanical input but rather a combination of inputs of different types and with various intensities, frequencies and directions. Further studies and more importantly more sophisticated materials and technologies will be needed in the future in order to depict these functions in an integrated 3D system such as a living multicellular tissue or organ.
1.3. Physiological relevance of FA’s mechanotransduction
The role of focal adhesion kinase (FAK) in particular and focal adhesion components in general in the mecanotransduction has been mostly studied in organs and tissues where mechanical inputs are an integral part of the physiological function. Examples of such organs and tissues include bone, heart, lungs, myometrium and vasculature among other tissues.
Bones are constantly exposed to mechanical loading which is in fact important for the maintenance of bone mass and for the structural stability of the skeleton. Such mechanical loading results in a displacement of interstitial fluid within the spaces surrounding bone cells generating fluid shear stress (FSS) that stimulates osteoblasts and osteocytes. Although the mechanisms by which bone cells transduce the external mechanical stimulation into intracellular biochemical signals are poorly understood at present, accumulating evidence is implicating focal adhesions as prime candidates (Wozniak et al., 2000; Young et al., 2010; Young et al., 2009).
Myometrial remodeling in the uterus occurs during pregnancy and is associated with increased mechanical distension leading to increased expression of the dense plaque-associated proteins FAK and paxillin as well as enzymatic activation of FAK, paxillin, Src and extracellular signal-regulated (ERK1/2) kinases in myometrial cells (Wu et al., 2008).
Another major organ where mechanotransduction is of extreme importance is the heart where this process affects not only the regulation of cardiac performance but also the proliferation, differentiation, growth, and survival of the cells within the myocardium (Russell et al., ; Russell et al., 2010; Samarel, 2005). It was established very early on that focal adhesions are directly responsible for the transmission of contractile forces generated within the cardiomyocyte to the surrounding ECM (Danowski et al., 1992). Furthermore, in primary cardiomyocyte cultures, focal adhesion assembly was directly induced by the mechanical forces placed on the cell. It was also noticed that external stretch and intracellular contractility increased the number and size of FAs (Sharp et al., 1997; Simpson et al., 1993).
When blood is pumped through the vasculature in a pulsatile fashion due the heart contractile activity, blood vessels are submitted to mechanical forces in the form of stretch and shear stress (Lehoux et al., 2006; Lehoux et al., 2005). Smooth muscle cells and endothelial cells of the arterial wall “sense” and “react” to these stimuli by remodeling their cytoskeleton and initiating a series of adaptive signal modifications to accommodate for the level of force exerted on the vessel. This is further accentuated in the lungs where blood vessels as well as other tissues are submitted to further compressions, stretches and distortions due to the normal rhythmic respiratory activity as a result of the alternate repetitions of inflations and deflations of the alveolar cavity. As a gas exchange organ, the lung is designed in such a way as to accommodate air entry and exit into the airways which are fueled by the inflation-deflation cycles. As a result the pulmonary tissues are constantly exposed to external mechanical stimulations due to the specialized respiratory function of this organ. Unlike systemic vasculature, pulmonary vasculature experiences mechanical forces resulting from blood circulation as well as from the rhythmic respiratory cycles. Furthermore and due to the highly branched structure of the pulmonary circulation which ensures efficient gas exchange, large and small caliber pulmonary vessels experience reduced resistance and intraluminal pressure and comparable levels of shear stress (5-10 dynes/cm2) compared to 15-40 dynes/cm2 in systemic circulation (Davies, 1995; Resnick et al., 2003). Mechanical stretch experienced by vascular cells is a combination of pulsatile (heart beat-driven) and tonic (balance between hydrostatic pressure and tonic contraction of vascular smooth muscle) components. Pulsatile distension of the arterial wall in systemic circulation normally does not exceed 10-12%, but various vasomotor reactions may change diameter of smaller caliber “resistance” arteries to 60% of initial diameter or more and last minutes or hours (Koller and Bagi, 2002). Pulmonary circulation is characterized by lower hydrostatic pressure, but is subjected to additional mechanical stretch due to respiratory cycles or mechanical ventilation. Thus, cyclic stretch is more prominent factor in the lung alveolar capillaries, while shear forces may have differential effects on various potions of the pulmonary vascular bed. The examples of pathologic conditions associated with increased mechanical stress in the lung are briefly discussed below.
Focal adhesions play a major role in such a biophysical “communication” between the cells and their environment (Lehoux et al., 2006; Lehoux et al., 2005). Vascular endothelial cells lining the blood vessels form the interface between the bloodstream and the vessel walls and as such they are continuously subjected to shear and cyclic stresses generated by flowing blood (Katanosaka and Naruse, 2010). As result, the endothelial cells undergo significant morphological changes, which appear to be regulated by focal adhesion-associated proteins paxillin, FAK and zyxin (Ngu et al., 2010). Exposure of endothelial cells to uniaxial cyclic stretch leads to cell alignment perpendicular to the axis of stretch (Shirinsky et al., 1989). Cyclic uni-axial stretching of endothelial cells also leads to activation of Src tyrosine kinase leading to increased protein tyrosine phosphorylation of FA proteins including FAK, p130Cas, and paxillin (Katanosaka et al., 2008; Naruse et al., 1998a; Naruse et al., 1998b; Naruse et al., 1998c; Sai et al., 1999; Suzuki et al., 1997; Wang et al., 2001). FAK therefore seems to play a central role in controlling integrin-mediated cell adhesion and possibly force-dependent FA dynamics and will be discussed in more detail in the following sections.
2. Structure and regulation of FAK
2.1. The Gene
The FAK (Focal Adhesion Kinase) gene, known as PTK2 (Protein Tyrosine Kinase 2) gene is located on chromosome 8 (8q24.3; Entrez Gene ID: 5747) where it encodes for a 125 kDa non membrane-bound protein tyrosine kinase which, over the years was given different names including FAK1, FAK; FADK; FAK1; FRNK; pp125FAK; PTK2 (Fiedorek and Kay, 1995; Zhao and Guan, 2009). Focal adhesion kinase (pp125FAK or FAK) was first identified as one of a number of proteins which became phosphorylated on tyrosine residues upon integrin-mediated adhesion to a substratum or following transformation by the oncogenic tyrosine kinase v-src (Guan and Shalloway, 1992; Guan et al., 1991; Kanner et al., 1990; Kornberg et al., 1991; Schaller et al., 1992). Focal Adhesion Kinase exhibits a high degree of evolutionary conservation between species at the amino acid level (>94%) suggesting that it might play a crucial function in the cell.(Corsi et al., 2006; Whitney et al., 1993).
2.2. Domain structure and organization
Focal adhesion kinase can simplistically be subdivided into 3 major parts or domains. The central catalytic domain, is flanked by two structurally and functionally distinct domains, namely the N-terminal FERM (band 4.1, Ezrin, Fadixin, Moesin homology) domain and the C-terminal FAT (Focal Adhesion Targeting) domain. The catalytic domain exhibits a classical tyrosine kinase activity and its sequence is homologous to a number of other receptor and non-receptor protein tyrosine kinases (Parsons, 2003). Phosphorylation of two highly conserved tyrosine residues (Y576 and Y577) within the ‘catalytic loop’ is crucial for maximal activation of FAK (Parsons, 2003). FAT domain is involved, as its designation states, in the targeting of the kinase to focal adhesion sites through interaction with other components of the focal adhesions such as paxillin, talin and growth factor receptor-bound protein 2 (Grb2) (Chen et al., 1995; Ezratty et al., 2005; Hildebrand et al., 1993; Hildebrand et al., 1995; Lulo et al., 2009; Prutzman et al., 2004; Scheswohl et al., 2008). Unlike the FAT domain, FERM domain “provides” an auto-regulatory mechanism of FAK (reviewed in (Frame et al., 2010). Fluorescence resonance energy transfer (FRET)-based approaches (Papusheva et al., 2009) as well as structural studies (Lietha et al., 2007) have revealed that the N-terminal FERM domain directly binds the catalytic domain via a polybasic KAKTLR sequence in the FERM domain (Papusheva et al., 2009). Such an intramolecular interaction blocks access to the catalytic cleft and protects the FAK activation loop from Src phosphorylation. This conformation also sequesters the Y397 autophosphorylation and Src recruitment site, which lies in the linker connecting the FERM and kinase domains (Lietha et al., 2007). FERM domain was also shown to be involved in the nuclear translocation of FAK and its subsequent kinase-independent regulation of cell proliferation and survival through its interaction and regulation of the tumor suppressor p53 (Lim et al., 2008). In addition to the 3 major domains, FAK has 3 prolin-rich (PR), Src homology 3 (SH3)-domain interacting sequences. One between FERM and KD and 2 between KD and FAT providing additional protein-protein interaction interfaces. (Corsi et al., 2006).
2.3. FAK activation and phosphorylation sites
Upon cell attachment to ECM proteins, integrins become activated and form clusters at the cell surface thus initiating the formation of adhesion complexes which will stabilize and become mature focal adhesions. These events enhance FAK’s catalytic activity and increase FAK tyrosine phosphorylation (Mitra et al., 2005; Parsons, 2003). FAK autophosphorylation at tyrosine 397 (Y397) leads to the recruitment of Src and Src-family kinases as well as to an increased phosphorylation of other proteins present in the adhesion complex such as paxillin and p130Cas (Mitra et al., 2005). Subsequent phosphorylation of specific tyrosine residues leads to the recruitment of additional SH2-domain-containing signaling proteins such as phosphatidylinositol 3 (PI 3)-kinase and Grb2 (Mitra et al., 2005; Parsons, 2003; Parsons et al., 2000). Activated FAK becomes phosphorylated not only on tyrosine but also on serine and threonine residues at multiple sites, the distribution of which was analyzed by mass spectrometry (Ciccimaro et al., 2006; Grigera et al., 2005). In addition to a few single phosphorylation sites, three phosphorylation “hot spots” were identified near the major site of autophosphorylation (Y397) and within the C-terminal linker region between the kinase domain and the FAT domain (Grigera et al., 2005). Site-specific FAK phosphorylation is also triggered by mechanical forces and determines physiological cellular responses to shear stress and cyclic stretch. These mechanisms will be discussed below.
2.4. FAK binding partners
As discussed above, in addition to its kinase activity, FAK consists of a combination of protein-protein interaction motifs such as FERM, FAT, PR as well as multitude of phosphorylation sites, making it an ideal anchor for participating in mechanotransduction. FAK associates with multiple cell surface receptors and signaling proteins through which it can modulate the mechanical response. FAK is now considered as a point of convergence of integrin and growth factor mechanical signaling pathway. Direct or indirect interactions of FAK with these membrane-bound entities has been well documented (Cabodi et al., 2010; Chen and Chen, 2006; Long et al., 2010; Morgan et al., 2009; Zheng et al., 2009).
2.4.1. Growth Factor Receptors
A recent study identified a splicing isoform of the SRC-3 oncogene, SRC-3Delta4, as a signaling adaptor that links epidermal growth factor receptor (EGFR) and FAK and promotes EGF-induced phosphorylations of FAK and c-Src in the signaling cascade leading to epidermal growth factor (EGF)-induced cell migration, an event that requires mechanical signal co-ordination by FA (Long et al., 2010). In other instances, FAK can interact directly with growth factor receptors. One example is the hepatocyte growth factor receptor c-Met which upon its activation and phosphorylation at Y1349 and Y1356 interacts with the FERM domain of FAK (Chen and Chen, 2006). Met-FAK interaction leads to FAK activation and the initiation of the signaling cascade contributing to hepatocyte growth factor-induced cell motility and cell invasion (Chen and Chen, 2006). In the same line of evidence, FAK also interacts directly through a region contained within aa127-243 with the N-terminal portion of the insulin-like growth factor-1 receptor (IGF-1R)(Zheng et al., 2009).
2.4.2. Other membrane-bound proteins
Direct interaction of FAK with the C terminus of the hSlo alpha-subunit of BK cation channels via its Pro-1-rich domain has been implicated in an alternative mechanism of mechanotransduction in osteoblasts (Rezzonico et al., 2003). A less well characterized interaction at the cell surface involves epithelial membrane protein 2 (EMP2), which was proposed to control collagen gel contraction (Morales et al., 2009).
2.4.3. Cytoplasmic partners
In addition to Src kinase, a number of important intracellular partners have been reported that include the Src family member Fyn which binds FAK’s proline-rich domain (Baillat et al., 2008; Teutschbein et al., 2009). p190 Rho guanine nucleotide exchange factor (p190RhoGEF), a RhoA-specific guanosine diphosphate/guanosine triphosphate (GDP/GTP) exchange factor, was shown to interact directly with the FAT domain of FAK providing a link between FAK activity and actin dynamics (Zhai et al., 2003). In endothelial cells, FAK associates with and phosphorylates a Rho-specific GTPase activating protein, p190RhoGTPase activating protein (p190RhoGAP), leading to dowregulation of RhoA and subsequent restoration of the endothelial barrier function after thrombin challenge (Holinstat et al., 2006). Arp2/3 complex which is an important modulator of actin dynamics was shown to associate and colocalize with FAK at those transient structures formed early after adhesion. This interaction involves FERM domain of FAK and the Arp3 subunit, but does not occur when FAK is phosphorylated on Y397 (Serrels et al., 2007). These findings suggest important regulation of cortical cytoskeletal remodeling by FAK phosphorylation.
2.4.4. Nuclear interactors
As mentioned earlier, FAK directly interacts with the N-terminal transactivation domain of p53 and modulates p53-mediated survival/apoptotic signaling (Golubovskaya et al., 2005; Golubovskaya et al., 2008). Additional nuclear functions of FAK have been recently evaluated. It was shown that FAK plays a role in the regulation of specific genes through a new mechanism involving FAK interaction with MBD2 (Methyl CpG binding domain protein 2), which then regulates heterochromatin remodeling in the promoter region of Myogenin (Mei and Xiong, 2010). The recently reported interactions of FAK with the C-terminal domain of the tumor suppressor neurofibromin (NF1) (Kweh et al., 2009) and with the nuclear transcription factor Elk-1 (Mamali et al., 2008) strongly suggest that FAK exhibits additional functional activities yet to be uncovered.
Thus, although it is not completely clear which of these modulatory activities of FAK are involved in mechanotransduction per se, some indications related to FAK phosphorylation and redistribution and their involvement in mechanosensing have been reported and will be discussed later in this article.
3. Regulation of FAK by mechanical forces
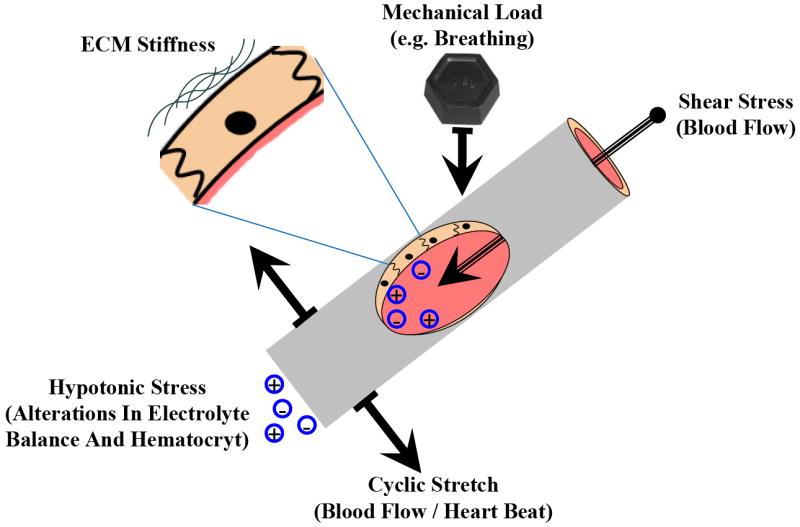
It is now well established that a number of load-sensitive cells including fibroblasts, chondrocytes, smooth muscle cells, and endothelial cells are able to “sense” a variety of mechanical forces generated by gravity, tension, compression, hydrostatic pressure, fluid shear stress, etc. (Figure 2). They can also themselves generate mechanical forces known as cell traction forces (CTFs) which influence many biological processes such as wound healing, angiogenesis, and metastasis (Califano and Reinhart-King, 2009; Gov, 2009; Kniazeva and Putnam, 2009; Li and Wang, 2009; Rosel et al., 2008; Sieminski et al., 2004; Stevenson et al., 2010). It is also now well accepted that mechanical loads applied to stem cells can regulate their self-renewal and induce controlled cell lineage differentiation (Wang and Thampatty, 2008). The sections below will discuss regulation of FAK activity and physiologic functions by different types of mechanical forces acting on the cell in vivo.
Figure 2. Mechanical perturbations experienced by blood vessels and in particular by the inner endothelial monolayer.
3.1. Cyclic Stretch
Exposure of bovine aortic endothelial cells subjected to 10% average strain at 60 cycles/min induces rapid tyrosine phosphorylation and reorganization of FAK and paxillin suggesting that focal adhesion proteins play a specific role in cyclic strain-induced endothelial cell morphological change and migration (Yano et al., 1996). A number of groups including our lab have reported similar cellular effects. Of note, exposure of human pulmonary endothelial cells to cyclic stretch induced FAK phosphorylation at Y397 and Y576 (Shikata et al., 2005). Pressure overload, which induced a time-dependent and amplitude-dependent increase in FAK tyrosine phosphorylation and FAK association with F-actin in the myocardium (Franchini et al., 2000), was shown to stimulate FAK phosphorylation at multiple tyrosine residues (Y397, Y576, Y861 and Y925). Such FAK hyperphosphorylation led to increases in FAK/Src association and Src activity, as well as enabled FAK association with myosin heavy chain in cardiac myocytes (Fonseca et al., 2005).
Another study by Albinsson and Hellstrand (Albinsson and Hellstrand, 2007) used the organ tissue model of increased mechanical load in the mouse portal vein. The results show that stretch causes a biphasic activation of FAK, which may reflect first a direct phosphorylation of preexisting focal adhesions followed by a rearrangement of focal adhesions. Cyclic mechanical stretch also increases beta (1D)-integrin protein levels, which mediate cyclic stretch-dependent FAK and RhoA activities. Silencing of beta(1D)-integrin in immature developing myoblasts abolished stretch-induced increases in FAK phosphorylation and further downregulated RhoA activity (Zhang et al., 2007). Finally, stretch-induced FAK phosphorylation at Y397 led to activation of FAK kinase activity resulting in proliferation and differentiation of neonatal cardiac fibroblasts into myofibroblasts. The exact mechanism of action is not clear but involves mammalian target of rapamycin (mTOR) and potentially Akt activation mediated by PI3-kinase. Reduction in FAK expression suppressed fibroblast activation invoked by cyclic stretch through a mechanism involving mTOR (Dalla Costa et al., 2010).
Surprisingly, mechanical forces may also induce FAK translocation to the nucleus (Senyo et al., 2007), suggesting direct role of FAK in the nuclear processes and possibly in gene regulation.
3.2. Shear stress
Shear stress increases the activity of a number of kinases and modulates the phosphorylation of signaling proteins in endothelial cells (Chien et al., 1998; Jalali et al., 1998; Shikata et al., 2005). Exposure of human umbilical cord vein endothelial cells (HUVEC) to laminar shear stress induces the recruitment of FAK to FAs and to increased FAK phosphorylation at Y397 (Li et al., 2002). This is in contrast with cell migration observed in response to serum stimulation, which is accompanied by increased number of FAs and lack of directional preference of FA formation and migration. The total level of FAK(Y397) phosphorylation after shear stress in that study was lower than that after serum treatment, suggesting that the polarized change at cell periphery rather than the total level of FAK(Y397) phosphorylation is important for directional migration (Li et al., 2002). In human pulmonary EC shear stress in the range of 5-10 dynes/cm2 induced preferential FAK phosphorylation at Y576, as opposed to phosphorylation at Y397 and Y576 induced by pathologically relevant magnitude of cyclic stretch (18% equibiaxial stretch) (Shikata et al., 2005).
Similar studies in bovine aortic endothelial cells indicate that shear stress of 12 dyn/cm2 increased the FAK tyrosine phosphorylation and enzymatic activity and facilitated a rapid and transient FAK association with Grb2. These data suggest that shear stress-activated FAK may regulate the mitogen-activated protein (MAP) kinase signaling pathways through a Grb2 - Son of sevenless (Sos) complex. The data also demonstrated that shear stress-induced FAK(Y397) phosphorylation is critical for subsequent dual activation of ERK2 and c-Jun N-terminal kinase 1 (JNK1) MAP kinases (Li et al., 1997). The differences between site specific shear stress-induced FAK phosphorylation reported in these studies may indicate different sensitivity to shear stress magnitudes by endothelial cells from different vascular beds. Indeed, exposure of microvascular EC to low laminar shear stress induced significantly higher activation of ELR(+) Cys-X-Cys (CXC) chemokines interleukin (IL)-8 and growth regulated oncogene (GRO)-α in comparison to macrovascular endothelium (Shaik et al., 2009). These chemokines play an important role in leukocyte trafficking into the tissues, and differential activation of IL-8 and GRO-α in micro- and microvascular endothelium is highly consistent with in vivo observations. They indicate that for reasons that are not well elucidated, circulating leukocytes are recruited into the tissues mainly in small vessels such as capillaries and venules. Importantly, shear stress-induced IL-8 and GRO-α upregulation involved activation of focal adhesion kinase, which required cell surface heparan sulfates, beta(3)-integrins and induced downstream activation of stress-induced MAP kinases p38beta and stress-activated protein kinase (SAPK)-1. Because microvascular and macrovascular endothelium exhibit different spectra of lectins and other heparin sulfates expressed at the cell surface (King et al., 2004), specific lectins or heparin sulfate sub-types may account for differential responses of micro- and macrovascular EC to shear stress mediated by FAK signaling. These interesting possibilities require further investigations.
More recent studies using multi-wavelength four-dimensional fluorescence microscopy have shown that unidirectional shear stress (SS) causes formation of new FAs and leads to a rapid remodeling and reorganization of pre-existing mature FAs in a cell density-dependent manner (Mott and Helmke, 2007). In general, analysis of shear stress- and cyclic stretch-induced signaling suggests dynamic interactions between integrin molecules present in focal adhesion complexes and membrane events involved in mechanotransduction, which are integrated by calcium-dependent and calcium-independent mechanisms. Calcium-dependent pathway includes activation of phospholipase C, hydrolysis of phosphatidylinositol 4,5-bisphosphate (PIP2) leading to increases in intracellular calcium and stimulation of kinases such as calcium-calmodulin and C kinases (PKC), while calcium-independent signaling involves activation of a small GTP-binding proteins Rac and Rho, and stimulation of calcium-independent PKC and MAP kinases (Berk et al., 1995; Davies et al., 1997; Schwartz and DeSimone, 2008). Thus, focal adhesion complexes would link the two pathways by regulating activity of phosphatidylinositol 4-phosphate (PIP) 5-kinase and FAK, and both molecules trigger remodeling of cell contacts and cytoskeleton and induce other intracellular signals leading to integrated endothelial cell response to mechanical stimuli.
3.3. Cell traction forces
A recent study used FAK-null cells or cells expressing wild-type or mutant (Y397F and Y576F/Y577F) FAK under the control of an inducible promoter to establish the role of FAK in the generation of steady-state adhesive forces on micropatterned substrates. It was established that FAK reduces steady-state adhesion strength through a mechanism involving vinculin recruitment to focal adhesions (Dumbauld et al., 2010a; Dumbauld et al., 2010b). Studies using FAK-null or Y397F-expressing fibroblasts plated on flexible polyacrylamide substrates showed similar effects (Wang et al., 2001a).
3.4. Substrate stiffness
A role of cell matrix stiffness in FA dynamics in endothelial cells remains to be investigated. However, analysis of matrix stiffness effects on FAs signaling has been reported in epithelial cancer cell lines. Provenzano et al. assessed the effect of stiffer matrices on epithelial cell growth in order to explain the mechanism responsible for increased epithelial proliferation and cancer risk in human patients with high breast tissue density (Provenzano et al., 2009). They observed that increased collagen-matrix density which enhances matrix stiffness resulted in an increased formation of activated three-dimensional (3D)-matrix adhesions in non transformed mammary epithelial cells and a chronically elevated outside-in/inside-out FAK-Rho signaling loop which is necessary for the generation and the maintenance of an invasive phenotype. This activation also led to cell proliferation through a hyper-activation of the Ras-mitogen-activated protein kinase (MAPK) pathway providing a compelling evidence for the importance of the mechanically activated FAK and Rho in upregulation of MAPK cascade and following cell proliferation (Provenzano et al., 2009). Consistent with this finding, low rigidity collagen gel downregulated beta(1)-integrin activation and focal adhesion kinase (FAK) Y397 phosphorylation in epithelial cells (Wei et al., 2008). Furthermore, cell exposure to increased extracellular pressure enhances FAK phosphorylation at Y397 and Y576 in a number of human cell lines in an alpha-actinin-1 phosphorylation-dependent manner (Craig et al., 2007) suggesting once again that FAK is a key player in the mechanotransduction initiated by substrate stiffness.
3.5. Hypotonic Stress
Hypotonic stress (HTS) and cell swelling imposes additional mechanical strain on cell plasma membrane and underlying cytoskeletal meshwork. HTS has been shown to activate the RhoA/Rho-kinase pathway followed by tyrosine phosphorylation of FAK and paxillin, which led to ATP release and actin reorganization in HUVECs (Hirakawa et al., 2004). Because HTS-induced signaling events resembled EC responses to lysophosphatydic acid, the authors concluded that HTS-induced mechanosensitive responses are not directly activated by mechanical stress, but may involve intermediate signaling steps.
However, other reports suggest involvement of specific mechanosensing molecules in the HTS-induced signaling activation. HTS-induced transient reorganization of endothelial actin cytoskeleton through sequential activation of RhoA and FAK was selectively reversed by anti-integrin alpha5beta1 blocking antibody. In those experiments, HTS-induced RhoA translocation, tyrosine phosphorylation of FAK and paxillin as well as actin reorganization were inhibited by blocking alpha5beta1 integrin (Hirakawa et al., 2006). The other study showed that HTS-induced tyrosine phosphorylation of FAK in endothelial cells was abolished by heparinase III (Oike et al., 2008). These results clearly suggest that heparan sulfate proteoglycan expressed at the cell surface is one of the sensory molecules of hypotonic cell swelling in endothelial cells.
Taken together, these data demonstrate that hypotonic stress represents a clinically relevant example of mechanically induced signaling in circulation, which may trigger various cellular responses including changes in permeability, inflammatory responses, increased leukocyte adhesion, or apoptotic signaling. These possibilities remain to be elucidated.
4. Role of FAK in regulating mechanical stress-induced EC functions
All tissues and cells in human body experience certain level of mechanical stimulation. Mechanical forces modulate many aspects of cell/tissue physiology such as cell/tissue remodeling, proliferation, apoptosis, etc. These events apparently require FAK in the integration of external mechanical stimuli and their translation into physiological responses. FAs are very dynamic structures. For example, in migrating cells the FAs at the rear edge undergo disassembly with coordinated and rapid FA assembly in the cell leading edge (Broussard et al., 2008; Easley et al., 2008; Goetz et al., 2008; Owen et al., 2007; Xu et al., 2009). FAK inactivation can ‘freeze” this dynamics (Chan et al., 2009; Hamadi et al., 2005; Orr et al., 2004; Schober et al., 2007) and depending on the experimental setting and the nature of specific cell type or tissue it can result in either activation or downregulation of a particular physiological function. FAK as regulator of FA dynamics plays a role in vascular endothelial mechanotransduction triggered by various patterns of cyclic stretch and shear stress, or cell swelling associated with hypotonic stress. Vascular cell proliferation, apoptosis, remodeling and migration described in the following sections are the best characterized functions mediated by mechanical forces and regulated by FAK. FAK activation by disturbed flow also causes phosphorylation of NFkB p65 subunit leading to induction of ICAM-1 (Petzold et al., 2009) associated with endothelial activation, increased leukocyte adhesion, and vascular inflammation.
It was demonstrated that in human pulmonary EC, pathologic amplitudes of cyclic stretch and laminar shear stress induce different patterns of site-specific FAK phosphorylation and intracellular FA localization. FAK preferentially localized at the cell periphery in EC exposed to flow while FAK containing focal adhesions became more central and enlarged in EC exposed to 18% cyclic stretch (Chien et al., 1998; Jalali et al., 1998; Shikata et al., 2005). The differential FAK arrangement and site-specific FAK phosphorylation correlated with differential EC permeability responses evoked by these mechanical stimulations (Chien et al., 1998; Jalali et al., 1998; Shikata et al., 2005). Although the role of FAK in control of permeability by mechanical factors is highly suggestive, existing methodological limitations do not allow precise characterization of the FAK involvement in vascular endothelial permeability changes induced by mechanical forces. More detailed studies await further developments in experimental methodologies and techniques.
4.1. Cell/tissue remodeling
At later stages of vascular development and in adult tissues, both the endothelial cytoskeleton and extracellular matrix underlying endothelial lining of blood vessels become oriented with regard to a vector of external mechanical forces implied by circulating blood, heart propulsions, lung inflation, etc. The focal adhesions play important role in this process by sensing the mechanical forces and re-applying them to cytoskeleton. When cultured EC subjected to uniaxial cyclic stretch, F-actin stress fibers tend to align perpendicularly to the stretch direction and this alignment depends on amplitude, frequency and time of cyclic stretch (Hsu et al., 2010; Shirinsky et al., 1989; Sokabe et al., 1997). This cytoskeleton remodeling further drives re-alignment of the whole cell in the same direction. FAK involvement in stress fiber re-orientation is controversial. Although the early observation implicated FAK in the cyclic stretch-induced stress fiber alignment (Sokabe et al., 1997), the recent data state otherwise. Ngu and colleagues (Ngu et al., 2010) found that neither FAK nor paxillin knock-down prevented stress fiber orientation in response to uniaxial cyclic stretch in human aortic endothelial cells. Similarly, uniaxial cyclic stretch induced stress fiber alignment and the phosphorylation of JNK, ERK and p38 were comparable in both, FAK-null and FAK-expressing mouse embryonic fibroblasts (Hsu et al., 2010).
4.2. Cell proliferation and apoptosis
Mechanical stimulation affects cell proliferation in almost all types of tissues including the endothelium (Ando et al., 1987; Birukov et al., 1995; Shyu, 2009; Sumpio et al., 1987). Proliferation response upon mechanical stimulation involves FAK activation. Uniaxial cyclic stretch of intestinal and pulmonary epithelial cells (Chaturvedi et al., 2008; Chaturvedi et al., 2007a; Chaturvedi et al., 2007b) or skeletal myocytes (Kumar et al., 2004) as well as oscillatory shear stress of osteoblasts (Lee et al., 2010) all cause FAK activation followed by cell proliferation. Cyclic stretch causes rapid Src-independent phosphorylation of FAK at Y397 and Y925, leading to the activation of the Ras/Raf/MEK/ERK-1,2 pathway (Boutahar et al., 2004). Stretch-induced FAK phosphorylation at Y576 appears to be important for stimulation of ERK-1,2 and stretch-induced activation of proliferation (Chaturvedi et al., 2008; Chaturvedi et al., 2007a; Chaturvedi et al., 2007b). These studies show that stretch effects on proliferation are blocked by siRNA-mediated FAK depletion or by overexpression of phosphorylation-deficient or dominant negative mutants.
FAK may directly participate in mitosis. In mitotic endothelial cells, FAK phosphorylated at Ser732 has been co-localized with centrosomes (Park et al., 2009). FAK deletion caused increased numbers of centrosomes, multipolar and disorganized spindles, and unaligned chromosomes during mitosis which were rescued by re-expression of wild-type FAK but not the S732A mutant (Park et al., 2009).
Pharmacological inhibition of FAK and p38-MAPK signaling causes caspase-3 activation and apoptosis in endothelial cells (Boosani et al., 2009). In turn, caspase activation causes degradation of components of FA complexes leading to disruption of FAs and subsequent apoptosis. Overexpression of FAK but not the catalytically dead, Y397F or D395A FAK mutants protected cells from adenosine/homocysteine-induced apoptosis (Bellas et al., 2002). Information about direct role of FAK in mechanical force-mediated control of apoptosis is limited. Application of pulsatile shear stress (12+/-4 dyn/cm2) parallel to the axis of orientation in HUVECs grown on fibronectin micropatterned strips caused cell elongation, enhanced stress fiber formation and FAK phosphorylation FAK, and led to a reduction in apoptosis. In contrast, apoptosis was not inhibited by flow perpendicular to EC orientation. These data indicate that mechanical forces may modulate EC apoptosis in a direction-dependent manner (Wu et al., 2007). Ahn and Park found that in EC X-chromosome linked inhibitor of apoptosis (XIAP) physically interacts with FAK, this interaction is activated by shear stress, and further promotes shear stress-induced FAK phosphorylation at Y576 leading to activation of ERK and endothelial proliferation (Ahn and Park, 2010). XIAP knockdown reduces shear stress-enhanced phosphorylation of FAK at Y576 and induces shear stress-triggered translocation of FAK into nucleus. Such spatial segregation of FAK from Src upon inhibition of XIAP decreases Y576 phosphorylation and shear-stimulated ERK activation and may lead to enhanced apoptotic signaling (Ahn and Park, 2010).
4.3. Angiogenesis and cell migration
FAK plays an important role in the response of migrating cells to mechanical input (Wang et al., 2001a). Exposure of endothelial cells to laminar shear stress induces lamellipodial protrusion at the cell periphery in the flow direction within 10 min with the recruitment of FAK at FAs. It also stimulates migration in the same direction which is accompanied by polarized formation of new FAs. Such fluid shear stress-enhanced EC migration in the flow direction as hence defined as “mechanotaxis.” (Li et al., 2002).
Experiments using cells cultured on flexible polyacrylamide substrates show that FAK is involved in the guidance of cell migration on mechanically stimulated substrates. Compared with control cells expressing wild-type FAK cells, FAK knockout decreases migration speed and directional persistence. In addition, FAK knockout impairs cell ability to respond to exerted forces by reorienting their movements and forming prominent focal adhesions (Li et al., 2002; Wang et al., 2001a). FAK tyrosine phosphorylation at Y397 appears to be a critical for control of force-induced cell migration and reorientation, because FAK knockouts and phosphorylation deficient FAK F397F mutants show similar defects in migration pattern and force-induced reorientation. Importantly, FAK-null cells showed impaired responses to decreases in substrate flexibility, which causes control cells to generate weaker traction forces and migrate away from soft substrates.
Regulation of cell spreading and migration in cells exposed to fluid flow occurs via dynamic dephosphorylation of FAK by focal adhesion localized dual-specific phosphatase PTEN (Haier and Nicolson, 2002). If expression of PTEN was reduced by siRNA approach, cell adhesion under dynamic conditions of laminar flow, but not under static conditions was significantly increased, whereas cell spreading was increased.
In turn, mechanical ventilation combined with hyperoxia promotes changes in focal adhesion proteins in lung alveolar type 2 cells that may be deleterious for cell adhesion and migration (Desai et al., 2007). Although not yet demonstrated, similar changes in response to VILI-relevant mechanical stimulation may occur in pulmonary capillary endothelium leading to barrier compromise and diminished migration capacity, which may further lead to delayed capillary barrier resealing and contribute to worsening vascular leak
Repetitive deformation of underlying substrate stimulated intestinal epithelial motility across fibronectin in a manner that requires both Src activation and FAK(Y925)-dependent pathway that activates ERK (Chaturvedi et al., 2007b). Other study shows that cyclic stretch impairs signaling of cell migration after injury induced by increased cyclic mechanical stretch via a pathway that involves FAK-JIP3-JNK (Desai et al., 2009).
4.4. Barrier function
A body of compelling evidence from a number of groups including ours has now established that mechanical stimulation of endothelial and epithelial cells at certain levels does influence their ability to form tight monolayers and to maintain junctional integrity both in vivo and in vitro (Birukov et al., 2003; Birukova et al., 2006; Birukova et al., 2010; Birukova et al., 2008a; Birukova et al., 2008b; Breslin and Kurtz, 2009; Cavanaugh et al., 2006; Cohen et al., 2008; Colgan et al., 2007; Muller-Marschhausen et al., 2008; Nonas et al., 2008; Sidhaye et al., 2008). Although FAK was known to be involved in the regulation of barrier function for a number of years (reviewed in (Wu, 2005)), the most direct evidence of its involvement in the maintenance of the endothelial barrier integrity was only reported recently by Zhao et al. using an EC-specific FAK kinase-defective (KD) mutant knockin (conditional FAK knockin [CFKI]) mouse model (Zhao et al., 2010). Endothelial cells lacking FAK or expressing dominant negative FAK exhibited a significant loss in barrier integrity accompanied by an abnormal distribution of vascular endothelial cadherin (VE-cadherin) and decreased VE-cadherin Y658 phosphorylation suggesting that FAK plays a key role in the in the maintenance of the barrier integrity in a kinase-dependent manner (Zhao et al., 2010). In another study, siRNA-mediated reduction in FAK expression led to a significant inhibition of sphingosine-1-phosphate-induced EC barrier enhancement providing an indirect evidence for the role of FAK in the maintenance of the barrier integrity (Zhao et al., 2009). Similarly, overexpression of the FAK inhibitor, FAK-related non-kinase (FRNK) in human bronchial epithelial cells (HBEpCs) inhibited lysophosphatidic acid-induced increases in TER and E-cadherin accumulation at cell-cell junctions (He et al., 2009). When monolayers of rat lung microvascular ECs are exposed to an oxidative stress (H2O2), TER undergoes a biphasic process: a transient and short decrease lasting 10 min followed by a recovery phase despite the continuous presence of H2O2. The two phases being indicative of EC junctional opening and resealing respectively (Quadri and Bhattacharya, 2007). More importantly, inhibition of FAK activity through expression of the FAK mutant del-FAK blocked the junctional recovery phase (Quadri and Bhattacharya, 2007) suggesting that FAK is a key player in the reassembly of cell-cell junction and the reestablishment of tight and impermeable monolayer. Other indirect studies implicating FAK in the vascular endothelial growth factor (VEGF)-mediated modulation of endothelial barrier function using similar approaches have been reported (Avraham et al., 2003; Wu et al., 2003)
On the other hand, and as discussed earlier in this review, FAK is regulated by diverse mechanical stimulations. Taken together, these observations suggest that FAK may play a key role in the regulation of barrier function as a result of mechanostimulation of endothelial cells. Further studies are required to directly evaluate mechanisms of EC permeability regulation by mechanically-activated FAK.
Summary and Future Perspective
The current data suggest that, despite the considerable overlap between signaling pathways activated by different types of mechanical stimulation (shear stress, cyclic stretch, substrate stiffness, hypotonic stress) FAK activation and its involvement in specific cell responses (Figure 3) is still dictated by the nature and magnitude of mechanical signal. Such specificity may be explained in part by cell type differences, but may be also achieved via unique combinations of FAK activation with other mechanically induced signaling cascades specific to particular type or the magnitude of mechanical stimulus. Integral analysis of mechano-sensitive signaling networks activated in specific settings of mechanical stimulation will help clarify the roles of FAK in endothelial functions under physiologic and pathologic mechanical environment.
Figure 3. Role of Focal Adhesion Kinase (FAK) in the integration of endothelial cell response to mechanical forces.

Different types of mechanical stimulations (cyclic stretch, shear forces, pressure, extra cellular matrix stiffness, hypotonic stress, etc) modulate FAK activity and interactions through phosphorylation and re-localization in the cell. By affecting focal adhesion dynamics and localization, FAK controls directional endothelial cell migration and maintains cell polarity. Signaling through ERK/p38 MAP kinase cascades to NF-κB activates or inhibits cell proliferation depending on physiological state of the cell. In parallel, FAK activation suppresses caspase-dependent apoptosis. By reconciling directional migration, proliferation/differentiation and apoptosis, FAK mediates physical force directed angiogenesis. FAK activation by disturbed flow is essential for phosphorylation of NFkB p65 subunit leading to induction of ICAM-1, endothelial activation, increased leukocyte adhesion and vascular inflammation. Depending on the type and magnitude, mechanical forces induce differential patterns of site-specific FAK phosphorylation and intracellular localization and may promote both, enhancement or weakening of vascular endothelial barrier.
The understanding of the involvement of FAK in mechanoperception per se is still sketchy. Increased phosphorylation at selective sites and increased expression of the kinase have been reported, although the exact sequence of signaling events induced by mechanically activated FAK remains to be elucidated. Genetic approaches using a battery of FAK mutants may help pinpoint the exact amino acids or protein domains involved in FAK-mediated mechanotransduction. At the same time, it is unclear at this point how FAK may contribute to sometimes opposite cell responses to different types of mechanical stimulation (i.e. low vs high magnitude cyclic stretch, or laminar vs turbulent flow), and whether the same pathways are involved in transmitting different types of mechanical stimuli. Further analyses are needed in order to clarify these important issues.
Highlights.
We focus on focal adhesion kinase (FAK), a component of FAs.
FAK plays a role in mechanotransduction and influences endothelial cell functions.
Structure/function relationship of FAK domains is discussed.
FAK plays a role in“sensing” physiologic and pathologic mechanical perturbations.
Acknowledgments
Supported by NIH grants NIH/NHLBI PO1-058064, HL076259, HL087823
Abbreviations
- CTF
cell traction force
- EC
Endothelial Cell
- ECM
Extracellular matrix
- FA
Focal adhesion
- FAK
Focal adhesion kinase
- FERM
band 4.1, Ezrin, Fadixin, Moesin homology
- FAT
Focal Adhesion Targeting
- FSS
Fluid shear stress
- Grb2
Growth factor receptor-bound protein 2
- HUVEC
Human umbilical cord vein endothelial cells
- HTS
Hypotonic stress
- mTOR
Mammalian target of rapamycin
- PR
prolin-rich
- PTK2
Protein tyrosine kinase 2
- SH2/3
Src homology 2/3
- Sos
Son of sevenless
- SS
Shear stress
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alahyan M, et al. The mechanism of smooth muscle caldesmon-tropomyosin inhibition of the elementary steps of the actomyosin ATPase. J Biol Chem. 2006;281:19433–48. doi: 10.1074/jbc.M507602200. [DOI] [PubMed] [Google Scholar]
- Alexandrova AY, et al. Comparative dynamics of retrograde actin flow and focal adhesions: formation of nascent adhesions triggers transition from fast to slow flow. PLoS One. 2008;3:e3234. doi: 10.1371/journal.pone.0003234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avraham HK, et al. Vascular endothelial growth factor regulates focal adhesion assembly in human brain microvascular endothelial cells through activation of the focal adhesion kinase and related adhesion focal tyrosine kinase. J Biol Chem. 2003;278:36661–8. doi: 10.1074/jbc.M301253200. [DOI] [PubMed] [Google Scholar]
- Berk BC, et al. Protein kinases as mediators of fluid shear stress stimulated signal transduction in endothelial cells: a hypothesis for calcium-dependent and calcium-independent events activated by flow. J Biomech. 1995;28:1439–50. doi: 10.1016/0021-9290(95)00092-5. [DOI] [PubMed] [Google Scholar]
- Birukov KG, et al. Magnitude-dependent regulation of pulmonary endothelial cell barrier function by cyclic stretch. Am J Physiol Lung Cell Mol Physiol. 2003;285:L785–97. doi: 10.1152/ajplung.00336.2002. [DOI] [PubMed] [Google Scholar]
- Birukova AA, et al. Differential regulation of pulmonary endothelial monolayer integrity by varying degrees of cyclic stretch. Am J Pathol. 2006;168:1749–61. doi: 10.2353/ajpath.2006.050431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birukova AA, et al. Mechanotransduction by GEF-H1 as a novel mechanism of ventilator-induced vascular endothelial permeability. Am J Physiol Lung Cell Mol Physiol. 2010;298:L837–48. doi: 10.1152/ajplung.00263.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birukova AA, et al. Magnitude-dependent effects of cyclic stretch on HGF- and VEGF-induced pulmonary endothelial remodeling and barrier regulation. Am J Physiol Lung Cell Mol Physiol. 2008a;295:L612–23. doi: 10.1152/ajplung.90236.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birukova AA, et al. Long-term cyclic stretch controls pulmonary endothelial permeability at translational and post-translational levels. Exp Cell Res. 2008b;314:3466–77. doi: 10.1016/j.yexcr.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breslin JW, Kurtz KM. Lymphatic endothelial cells adapt their barrier function in response to changes in shear stress. Lymphat Res Biol. 2009;7:229–37. doi: 10.1089/lrb.2009.0015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabodi S, et al. Integrins and signal transduction. Adv Exp Med Biol. 2010;674:43–54. doi: 10.1007/978-1-4419-6066-5_5. [DOI] [PubMed] [Google Scholar]
- Cavanaugh KJ, et al. Stretch increases alveolar epithelial permeability to uncharged micromolecules. Am J Physiol Cell Physiol. 2006;290:C1179–88. doi: 10.1152/ajpcell.00355.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CS, et al. Mechanotransduction at cell-matrix and cell-cell contacts. Annu Rev Biomed Eng. 2004;6:275–302. doi: 10.1146/annurev.bioeng.6.040803.140040. [DOI] [PubMed] [Google Scholar]
- Chen HC, et al. Interaction of focal adhesion kinase with cytoskeletal protein talin. J Biol Chem. 1995;270:16995–9. doi: 10.1074/jbc.270.28.16995. [DOI] [PubMed] [Google Scholar]
- Chen SY, Chen HC. Direct interaction of focal adhesion kinase (FAK) with Met is required for FAK to promote hepatocyte growth factor-induced cell invasion. Mol Cell Biol. 2006;26:5155–67. doi: 10.1128/MCB.02186-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chrzanowska-Wodnicka M, Burridge K. Rho-stimulated contractility drives the formation of stress fibers and focal adhesions. J Cell Biol. 1996;133:1403–15. doi: 10.1083/jcb.133.6.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen TS, et al. Frequency and peak stretch magnitude affect alveolar epithelial permeability. Eur Respir J. 2008;32:854–61. doi: 10.1183/09031936.00141007. [DOI] [PubMed] [Google Scholar]
- Colgan OC, et al. Regulation of bovine brain microvascular endothelial tight junction assembly and barrier function by laminar shear stress. Am J Physiol Heart Circ Physiol. 2007;292:H3190–7. doi: 10.1152/ajpheart.01177.2006. [DOI] [PubMed] [Google Scholar]
- Craig DH, et al. Alpha-actinin-1 phosphorylation modulates pressure-induced colon cancer cell adhesion through regulation of focal adhesion kinase-Src interaction. Am J Physiol Cell Physiol. 2007;293:C1862–74. doi: 10.1152/ajpcell.00118.2007. [DOI] [PubMed] [Google Scholar]
- Danowski BA, et al. Costameres are sites of force transmission to the substratum in adult rat cardiomyocytes. J Cell Biol. 1992;118:1411–20. doi: 10.1083/jcb.118.6.1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies PF. Flow-mediated endothelial mechanotransduction. Physiol Rev. 1995;75:519–60. doi: 10.1152/physrev.1995.75.3.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies PF, et al. Spatial relationships in early signaling events of flow-mediated endothelial mechanotransduction. Annu Rev Physiol. 1997;59:527–49. doi: 10.1146/annurev.physiol.59.1.527. [DOI] [PubMed] [Google Scholar]
- Dewey CF, Jr, et al. The dynamic response of vascular endothelial cells to fluid shear stress. J Biomech Eng. 1981;103:177–85. doi: 10.1115/1.3138276. [DOI] [PubMed] [Google Scholar]
- Ehrlich PJ, Lanyon LE. Mechanical strain and bone cell function: a review. Osteoporos Int. 2002;13:688–700. doi: 10.1007/s001980200095. [DOI] [PubMed] [Google Scholar]
- Even-Ram S, et al. Myosin IIA regulates cell motility and actomyosin-microtubule crosstalk. Nat Cell Biol. 2007;9:299–309. doi: 10.1038/ncb1540. [DOI] [PubMed] [Google Scholar]
- Ezratty EJ, et al. Microtubule-induced focal adhesion disassembly is mediated by dynamin and focal adhesion kinase. Nat Cell Biol. 2005;7:581–90. doi: 10.1038/ncb1262. [DOI] [PubMed] [Google Scholar]
- Fiedorek FT, Jr, Kay ES. Mapping of the focal adhesion kinase (Fadk) gene to mouse chromosome 15 and human chromosome 8. Mamm Genome. 1995;6:123–6. doi: 10.1007/BF00303256. [DOI] [PubMed] [Google Scholar]
- Frame MC, et al. The FERM domain: organizing the structure and function of FAK. Nat Rev Mol Cell Biol. 2010;11:802–14. doi: 10.1038/nrm2996. [DOI] [PubMed] [Google Scholar]
- Fukuda S, Schmid-Schonbein GW. Regulation of CD18 expression on neutrophils in response to fluid shear stress. Proc Natl Acad Sci U S A. 2003;100:13152–7. doi: 10.1073/pnas.2336130100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Cardena G, et al. Biomechanical activation of vascular endothelium as a determinant of its functional phenotype. Proc Natl Acad Sci U S A. 2001;98:4478–85. doi: 10.1073/pnas.071052598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger B, Bershadsky A. Assembly and mechanosensory function of focal contacts. Curr Opin Cell Biol. 2001;13:584–92. doi: 10.1016/s0955-0674(00)00255-6. [DOI] [PubMed] [Google Scholar]
- Geiger B, et al. Environmental sensing through focal adhesions. Nat Rev Mol Cell Biol. 2009;10:21–33. doi: 10.1038/nrm2593. [DOI] [PubMed] [Google Scholar]
- Grigera PR, et al. FAK phosphorylation sites mapped by mass spectrometry. J Cell Sci. 2005;118:4931–5. doi: 10.1242/jcs.02696. [DOI] [PubMed] [Google Scholar]
- Grodzinsky AJ, et al. Cartilage tissue remodeling in response to mechanical forces. Annu Rev Biomed Eng. 2000;2:691–713. doi: 10.1146/annurev.bioeng.2.1.691. [DOI] [PubMed] [Google Scholar]
- He D, et al. Lysophosphatidic acid enhances pulmonary epithelial barrier integrity and protects endotoxin-induced epithelial barrier disruption and lung injury. J Biol Chem. 2009;284:24123–32. doi: 10.1074/jbc.M109.007393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildebrand JD, et al. Identification of sequences required for the efficient localization of the focal adhesion kinase, pp125FAK, to cellular focal adhesions. J Cell Biol. 1993;123:993–1005. doi: 10.1083/jcb.123.4.993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildebrand JD, et al. Paxillin, a tyrosine phosphorylated focal adhesion-associated protein binds to the carboxyl terminal domain of focal adhesion kinase. Mol Biol Cell. 1995;6:637–47. doi: 10.1091/mbc.6.6.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirakawa M, et al. Pivotal role of integrin alpha5beta1 in hypotonic stress-induced responses of human endothelium. Faseb J. 2006;20:1992–9. doi: 10.1096/fj.05-5580com. [DOI] [PubMed] [Google Scholar]
- Hsu HJ, et al. Stretch-induced stress fiber remodeling and the activations of JNK and ERK depend on mechanical strain rate, but not FAK. PLoS One. 2010:5. doi: 10.1371/journal.pone.0012470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janmey PA, Weitz DA. Dealing with mechanics: mechanisms of force transduction in cells. Trends Biochem Sci. 2004;29:364–70. doi: 10.1016/j.tibs.2004.05.003. [DOI] [PubMed] [Google Scholar]
- Katanosaka Y, et al. Analysis of cyclic-stretching responses using cell-adhesion-patterned cells. J Biotechnol. 2008;133:82–9. doi: 10.1016/j.jbiotec.2007.09.017. [DOI] [PubMed] [Google Scholar]
- Katanosaka Y, Naruse K. Musculoskeletal rehabilitation and bone. A novel approach to mechanotransduction using cell-adhesion-patterned cells. Clin Calcium. 2010;20:514–9. [PubMed] [Google Scholar]
- Kaverina I, et al. Tensile stress stimulates microtubule outgrowth in living cells. J Cell Sci. 2002;115:2283–91. doi: 10.1242/jcs.115.11.2283. [DOI] [PubMed] [Google Scholar]
- King J, et al. Structural and functional characteristics of lung macro- and microvascular endothelial cell phenotypes. Microvasc Res. 2004;67:139–51. doi: 10.1016/j.mvr.2003.11.006. [DOI] [PubMed] [Google Scholar]
- Koller A, Bagi Z. On the role of mechanosensitive mechanisms eliciting reactive hyperemia. Am J Physiol Heart Circ Physiol. 2002;283:H2250–9. doi: 10.1152/ajpheart.00545.2002. [DOI] [PubMed] [Google Scholar]
- Kweh F, et al. Neurofibromin physically interacts with the N-terminal domain of focal adhesion kinase. Mol Carcinog. 2009;48:1005–17. doi: 10.1002/mc.20552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehoux S, et al. Molecular mechanisms of the vascular responses to haemodynamic forces. J Intern Med. 2006;259:381–92. doi: 10.1111/j.1365-2796.2006.01624.x. [DOI] [PubMed] [Google Scholar]
- Lehoux S, et al. Differential regulation of vascular focal adhesion kinase by steady stretch and pulsatility. Circulation. 2005;111:643–9. doi: 10.1161/01.CIR.0000154548.16191.2F. [DOI] [PubMed] [Google Scholar]
- Lietha D, et al. Structural basis for the autoinhibition of focal adhesion kinase. Cell. 2007;129:1177–87. doi: 10.1016/j.cell.2007.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long W, et al. SRC-3Delta4 mediates the interaction of EGFR with FAK to promote cell migration. Mol Cell. 2010;37:321–32. doi: 10.1016/j.molcel.2010.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lulo J, et al. Crystal structures of free and ligand-bound focal adhesion targeting domain of Pyk2. Biochem Biophys Res Commun. 2009;383:347–52. doi: 10.1016/j.bbrc.2009.04.011. [DOI] [PubMed] [Google Scholar]
- Mack PJ, et al. Force-induced focal adhesion translocation: effects of force amplitude and frequency. Am J Physiol Cell Physiol. 2004;287:C954–62. doi: 10.1152/ajpcell.00567.2003. [DOI] [PubMed] [Google Scholar]
- Mei L, Xiong WC. FAK interaction with MBD2: A link from cell adhesion to nuclear chromatin remodeling? Cell Adh Migr. 2010;4:77–80. doi: 10.4161/cam.4.1.10343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mofrad MR, et al. Force-induced unfolding of the focal adhesion targeting domain and the influence of paxillin binding. Mech Chem Biosyst. 2004;1:253–65. [PubMed] [Google Scholar]
- Morgan MR, et al. Giving off mixed signals--distinct functions of alpha5beta1 and alphavbeta3 integrins in regulating cell behaviour. IUBMB Life. 2009;61:731–8. doi: 10.1002/iub.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller-Marschhausen K, et al. Physiological hydrostatic pressure protects endothelial monolayer integrity. Am J Physiol Cell Physiol. 2008;294:C324–32. doi: 10.1152/ajpcell.00319.2007. [DOI] [PubMed] [Google Scholar]
- Naruse K, et al. Uni-axial cyclic stretch induces c-src activation and translocation in human endothelial cells via SA channel activation. FEBS Lett. 1998a;441:111–5. doi: 10.1016/s0014-5793(98)01528-2. [DOI] [PubMed] [Google Scholar]
- Naruse K, et al. Pp125FAK is required for stretch dependent morphological response of endothelial cells. Oncogene. 1998b;17:455–63. doi: 10.1038/sj.onc.1201950. [DOI] [PubMed] [Google Scholar]
- Naruse K, et al. Involvement of SA channels in orienting response of cultured endothelial cells to cyclic stretch. Am J Physiol. 1998c;274:H1532–8. doi: 10.1152/ajpheart.1998.274.5.H1532. [DOI] [PubMed] [Google Scholar]
- Ngu H, et al. Effect of focal adhesion proteins on endothelial cell adhesion, motility and orientation response to cyclic strain. Ann Biomed Eng. 2010;38:208–22. doi: 10.1007/s10439-009-9826-7. [DOI] [PubMed] [Google Scholar]
- Nonas S, et al. Oxidized phospholipids reduce ventilator-induced vascular leak and inflammation in vivo. Crit Care. 2008;12:R27. doi: 10.1186/cc6805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons JT. Focal adhesion kinase: the first ten years. J Cell Sci. 2003;116:1409–16. doi: 10.1242/jcs.00373. [DOI] [PubMed] [Google Scholar]
- Provenzano PP, et al. Matrix density-induced mechanoregulation of breast cell phenotype, signaling and gene expression through a FAK-ERK linkage. Oncogene. 2009;28:4326–43. doi: 10.1038/onc.2009.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prutzman KC, et al. The focal adhesion targeting domain of focal adhesion kinase contains a hinge region that modulates tyrosine 926 phosphorylation. Structure. 2004;12:881–91. doi: 10.1016/j.str.2004.02.028. [DOI] [PubMed] [Google Scholar]
- Quadri SK, Bhattacharya J. Resealing of endothelial junctions by focal adhesion kinase. Am J Physiol Lung Cell Mol Physiol. 2007;292:L334–42. doi: 10.1152/ajplung.00228.2006. [DOI] [PubMed] [Google Scholar]
- Resnick N, et al. Hemodynamic forces as a stimulus for arteriogenesis. Endothelium. 2003;10:197–206. doi: 10.1080/10623320390246289. [DOI] [PubMed] [Google Scholar]
- Riveline D, et al. Focal contacts as mechanosensors: externally applied local mechanical force induces growth of focal contacts by an mDia1-dependent and ROCK-independent mechanism. J Cell Biol. 2001;153:1175–86. doi: 10.1083/jcb.153.6.1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell B, et al. Mechanical stress-induced sarcomere assembly for cardiac muscle growth in length and width. J Mol Cell Cardiol. 48:817–23. doi: 10.1016/j.yjmcc.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell B, et al. Mechanical stress-induced sarcomere assembly for cardiac muscle growth in length and width. J Mol Cell Cardiol. 2010;48:817–23. doi: 10.1016/j.yjmcc.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sai X, et al. Activation of pp60(src) is critical for stretch-induced orienting response in fibroblasts. J Cell Sci. 1999;112(Pt 9):1365–73. doi: 10.1242/jcs.112.9.1365. [DOI] [PubMed] [Google Scholar]
- Samarel AM. Costameres, focal adhesions, and cardiomyocyte mechanotransduction. Am J Physiol Heart Circ Physiol. 2005;289:H2291–301. doi: 10.1152/ajpheart.00749.2005. [DOI] [PubMed] [Google Scholar]
- Scheswohl DM, et al. Multiple paxillin binding sites regulate FAK function. J Mol Signal. 2008;3:1. doi: 10.1186/1750-2187-3-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz MA, DeSimone DW. Cell adhesion receptors in mechanotransduction. Curr Opin Cell Biol. 2008;20:551–6. doi: 10.1016/j.ceb.2008.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaik SS, et al. Low intensity shear stress increases endothelial ELR+ CXC chemokine production via a focal adhesion kinase-p38{beta} MAPK-NF-{kappa}B pathway. J Biol Chem. 2009;284:5945–55. doi: 10.1074/jbc.M807205200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan Y, et al. Nudel and FAK as antagonizing strength modulators of nascent adhesions through paxillin. PLoS Biol. 2009;7:e1000116. doi: 10.1371/journal.pbio.1000116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp WW, et al. Mechanical forces regulate focal adhesion and costamere assembly in cardiac myocytes. Am J Physiol. 1997;273:H546–56. doi: 10.1152/ajpheart.1997.273.2.H546. [DOI] [PubMed] [Google Scholar]
- Shemesh T, et al. Role of focal adhesions and mechanical stresses in the formation and progression of the lamellipodium-lamellum interface [corrected] Biophys J. 2009;97:1254–64. doi: 10.1016/j.bpj.2009.05.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirinsky VP, et al. Mechano-chemical control of human endothelium orientation and size. J Cell Biol. 1989;109:331–9. doi: 10.1083/jcb.109.1.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidhaye VK, et al. Shear stress regulates aquaporin-5 and airway epithelial barrier function. Proc Natl Acad Sci U S A. 2008;105:3345–50. doi: 10.1073/pnas.0712287105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson DG, et al. Contractile activity and cell-cell contact regulate myofibrillar organization in cultured cardiac myocytes. J Cell Biol. 1993;123:323–36. doi: 10.1083/jcb.123.2.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sniadecki NJ, et al. Magnetic microposts as an approach to apply forces to living cells. Proc Natl Acad Sci U S A. 2007;104:14553–8. doi: 10.1073/pnas.0611613104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sniadecki NJ, Chen CS. Microfabricated silicone elastomeric post arrays for measuring traction forces of adherent cells. Methods Cell Biol. 2007;83:313–28. doi: 10.1016/S0091-679X(07)83013-5. [DOI] [PubMed] [Google Scholar]
- Sokabe M, et al. Mechanotransduction and intracellular signaling mechanisms of stretch-induced remodeling in endothelial cells. Heart Vessels. 1997;Suppl 12:191–3. [PubMed] [Google Scholar]
- Suzuki M, et al. Up-regulation of integrin beta 3 expression by cyclic stretch in human umbilical endothelial cells. Biochem Biophys Res Commun. 1997;239:372–6. doi: 10.1006/bbrc.1997.7364. [DOI] [PubMed] [Google Scholar]
- Vicente-Manzanares M, et al. Regulation of protrusion, adhesion dynamics, and polarity by myosins IIA and IIB in migrating cells. J Cell Biol. 2007;176:573–80. doi: 10.1083/jcb.200612043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volberg T, et al. Effect of protein kinase inhibitor H-7 on the contractility, integrity, and membrane anchorage of the microfilament system. Cell Motil Cytoskeleton. 1994;29:321–38. doi: 10.1002/cm.970290405. [DOI] [PubMed] [Google Scholar]
- Wang JG, et al. Uniaxial cyclic stretch induces focal adhesion kinase (FAK) tyrosine phosphorylation followed by mitogen-activated protein kinase (MAPK) activation. Biochem Biophys Res Commun. 2001;288:356–61. doi: 10.1006/bbrc.2001.5775. [DOI] [PubMed] [Google Scholar]
- Wang JH, Thampatty BP. Mechanobiology of adult and stem cells. Int Rev Cell Mol Biol. 2008;271:301–46. doi: 10.1016/S1937-6448(08)01207-0. [DOI] [PubMed] [Google Scholar]
- Wolfenson H, et al. The heel and toe of the cell’s foot: a multifaceted approach for understanding the structure and dynamics of focal adhesions. Cell Motil Cytoskeleton. 2009;66:1017–29. doi: 10.1002/cm.20410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wozniak M, et al. Mechanically strained cells of the osteoblast lineage organize their extracellular matrix through unique sites of alphavbeta3-integrin expression. J Bone Miner Res. 2000;15:1731–45. doi: 10.1359/jbmr.2000.15.9.1731. [DOI] [PubMed] [Google Scholar]
- Wu MH. Endothelial focal adhesions and barrier function. J Physiol. 2005;569:359–66. doi: 10.1113/jphysiol.2005.096537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu MH, et al. Focal adhesion kinase mediates porcine venular hyperpermeability elicited by vascular endothelial growth factor. J Physiol. 2003;552:691–9. doi: 10.1113/jphysiol.2003.048405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, et al. Myometrial mechanoadaptation during pregnancy: implications for smooth muscle plasticity and remodelling. J Cell Mol Med. 2008;12:1360–73. doi: 10.1111/j.1582-4934.2008.00306.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young SR, et al. Activation of NF-kappaB by fluid shear stress, but not TNF-alpha, requires focal adhesion kinase in osteoblasts. Bone. 2010;47:74–82. doi: 10.1016/j.bone.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young SR, et al. Focal adhesion kinase is important for fluid shear stress-induced mechanotransduction in osteoblasts. J Bone Miner Res. 2009;24:411–24. doi: 10.1359/JBMR.081102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang SJ, et al. Effect of cyclic stretch on beta1D-integrin expression and activation of FAK and RhoA. Am J Physiol Cell Physiol. 2007;292:C2057–69. doi: 10.1152/ajpcell.00493.2006. [DOI] [PubMed] [Google Scholar]
- Zhao FQ, et al. Blebbistatin stabilizes the helical order of myosin filaments by promoting the switch 2 closed state. Biophys J. 2008;95:3322–9. doi: 10.1529/biophysj.108.137067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Guan JL. Signal transduction by focal adhesion kinase in cancer. Cancer Metastasis Rev. 2009;28:35–49. doi: 10.1007/s10555-008-9165-4. [DOI] [PubMed] [Google Scholar]
- Zhao J, et al. Phosphotyrosine protein dynamics in cell membrane rafts of sphingosine-1-phosphate-stimulated human endothelium: role in barrier enhancement. Cell Signal. 2009;21:1945–60. doi: 10.1016/j.cellsig.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, et al. Role of kinase-independent and -dependent functions of FAK in endothelial cell survival and barrier function during embryonic development. J Cell Biol. 2010;189:955–65. doi: 10.1083/jcb.200912094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng D, et al. Targeting of the protein interaction site between FAK and IGF-1R. Biochem Biophys Res Commun. 2009;388:301–5. doi: 10.1016/j.bbrc.2009.07.156. [DOI] [PMC free article] [PubMed] [Google Scholar]