

Abstract
Background
The outcome of the surgical repair in congenital heart disease (CHD) correlates with the degree of myocardial damage. In this study we determined whether mitochondrial DNA depletion is a sensitive marker of right ventricular (RV) damage and whether impaired mitochondrial DNA (mtDNA) replication contributes to the transition from compensated hypertrophy to failure.
Methods and Results
RV samples obtained from 31 patients undergoing cardiac surgery were compared to 5 RV samples from non-failing hearts (control). Patients were divided into compensated hypertrophy and failure groups based on preoperative echocardiography, catheterization and/or MRI data. Mitochondrial enzyme activities (citrate synthase and succinate dehydrogenase) were maintained during hypertrophy and decreased by ~40% (p<0.05 vs. control) at the stage of failure. In contrast, mtDNA content was progressively decreased in the hypertrophied RV through failure (by 28±8% and 67±11% respectively, p<0.05 for both), whereas mtDNA encoded gene expression was sustained by increased transcriptional activity during compensated hypertrophy but not in failure. MtDNA depletion was attributed to reduced mtDNA replication in both hypertrophied and failing RV and it was independent of PGC-1 down-regulation but was accompanied by reduced expression of proteins constituting the mtDNA replication fork. Decreased mtDNA content in compensated hypertrophy was also associated with pathological changes of mitochondria ultrastructure.
Conclusions
Impaired mtDNA replication causes early and progressive depletion of mtDNA in the RV of the CHD patients during the transition from hypertrophy to failure. Decreased mtDNA content is likely a sensitive marker of mitochondrial injury in this patient population.
Keywords: mtDNA, mitochondrial biogenesis, PGC-1, congenital heart disease, RV failure
Patients with tetralogy of Fallot, pulmonary atresia, truncus arteriosus, hypoplastic left heart syndrome and other congenital heart defects, frequently suffer from the chronic consequences of a volume and/or pressure overloaded RV. The adaptive mechanisms of the RV to compensate for the hemodynamic overload may lead to hypertrophy and ultimately failure1. The wide variability in clinical status, extent of right ventricular dilatation, and dysfunction at the time of presentation for surgical intervention has resulted in disparate results after surgical repair2, 3. While there is a group of patients that responds favorably to surgical repair, there is also a large subgroup that does not. Clinical studies evaluating recovery of RV function after pulmonary valve insertion have indicated that despite elimination of the hemodynamic overload, the RV function and moreover, the prognosis of these patients does not necessarily normalize4, 5. Therefore, it is critical to intervene on those patients before irreversible RV myocardial damage happens, and the appropriate timing requires further understanding of the cellular and molecular mechanisms underlying the myocardial damage caused by the hemodynamic overload in this population6, 7. Accumulating evidence have suggested that mitochondrial dysfunction plays a critical role in the development of heart failure8. We have previously reported that mitochondrial mass and DNA (mtDNA) content was decreased in the left ventricle in end-stage human failing hearts suggesting impaired mitochondrial biogenesis9. In the present study, we took advantage of the unique opportunity to determine the relationship of RV function and the alterations of mitochondrial biogenesis in children with CHD undergoing surgical repair or transplant. Analysis of the RV tissue from these patients allowed us to assess the mitochondrial mass, enzyme activities, gene expression, mtDNA content and mtDNA replication in a broad spectrum of cardiac pathologies during the transition from cardiac hypertrophy to failure. We found that impaired mtDNA replication and depletion of mtDNA were early events in RV hypertrophy preceding the clinical diagnosis of heart failure. MtDNA depletion also paralleled the pathological changes of mitochondrial ultra-structure suggesting that patients with lower mtDNA content are at a greater risk of developing heart failure and, thus, an early surgical relief may improve the outcomes of these patients.
Methods
Study Population
Thirty-one RV samples were collected from discarded myocardial tissue of patients undergoing cardiac surgery at Children’s Hospital Boston. Cardiac tissue was obtained intraoperatively and stored immediately in liquid nitrogen or fixed for electron microscopy examination. Five non-failing samples were obtained from the RV wall of donor hearts with no history, macroscopic or laboratory signs of cardiac diseases. This study was approved by Boston Children’s Hospital Institutional Review Board.
Clinical information as well as pre and postoperative echocardiography, MRIs and the clinical diagnosis of heart failure were retrospectively reviewed. RV ejection fraction (EF) calculated from the echocardiography and MRI was within the normal range in the hypertrophy group (n=25) while RV failure group showed diminished RV EF (<30%; n=6). In addition, the presence of an increased RV mass, exuberant muscle bundles and a thick RV wall were considered indicators of hypertrophy. The presence of RV dilatation and the clinical diagnosis of RV failure were used to include patients in the heart failure group. Patients with (suspected) mitochondrial disease were excluded in order to study the mitochondrial pathology caused by the cardiac stress due to congenital heart malformation.
Biochemical Assays
Citrate Synthase (CS) and Succinate Dehydrogenase (SDH) enzyme activities were measured in tissue homogenate as previously described10, 11. Total RNA was isolated from frozen RV tissue using the RNeasy Kit (Qiagen). Real Time PCR was performed using SYBR green (Bio-Rad) and the results of each gene were normalized to 18S rRNA levels. The primers are described in Supplemental Table 1. The mtDNA content was measured in a total cell lysate or isolated DNA using the DNeasy kit (Qiagen), by Real Time PCR using primers amplifying the Cytochrome C Oxidase subunit I (COI) region, and it was normalized to nDNA (18S) or mg of tissue protein. To assess the replication of mtDNA, we determined the amount of single stranded DNA generated from the extension of the 7S DNA in the D-loop region, a committed step in mtDNA replication, as previously described12, 13.
Electron Microscopy
Mitochondrial ultrastructure was studied in freshly collected RV specimens by electron microscopy. Samples were dissected in 1–2 mm3 and immediately fixed with 2% glutaraldehyde, post-fixed with 1% osmium tetroxide, and embedded in epon resin. For each sample, ten randomly chosen fields at the magnification of 5000× were used for the quantification of the mitochondrial number. To estimate the mitochondrial cristae density a computerized point grid was digitally layered over the micrographic images at 25000× magnification. The densities of dots on the point grid, as well as the spacing of selected sections were designed to result in 960 total point probes overlying each image. To estimate the mitochondria size distribution the longest mitochondrial dimension was measured in randomly chosen micrographs at 25000× magnification. At least ten images were analyzed for each sample in a blinded fashion.
Statistical analysis
Citrate synthase and succinate dehydrogenase data were expressed as medians in scatter plots whereas the age of the patients is given as median and min-max. The rest of the data as shown as mean of the fold changes ± SEM. Comparisons among groups were analyzed by Mann-Whitney test and regressions analysis by Spearman’s rank correlation. A value of p ≤0.05 was considered significant (2 tailed). All analyses were performed using GraphPad Prism 5.0.
Results
General Clinical Characteristics
The diagnoses of the patients in the study population are described in the Table. Among the 31 patients undergoing surgery 25 were classified in the RV hypertrophy and 6 in the RV failure group. The group with RV hypertrophy (RVH) consists of pressure or volume overload patients as primary diagnosis whereas the RV-failure group consists of patients with hypoplastic left heart syndrome (HLHS) which causes RV pressure overload. Due to the severity of this disease, this is the only group of patients in our study that was presented with heart failure at such an early stage of life.
Table 1.
Patient characteristics and clinical data.
| Non-Failing | Hypertrophy | Failure | |
|---|---|---|---|
| n | 5 | 25 | 6 |
| Male (n) | 3 | 16 | 2 |
| Age in years (median; min-max) | 12 (4–18) | 6.3 (0.1–19) | 0.8 (0.1–1.5) |
| Pressure Overload (n) | N/A | 11 | 6 |
| TOF (n) | N/A | 3 | 0 |
| TA (n) | N/A | 3 | 0 |
| DORV (n) | N/A | 1 | 0 |
| DCRV (n) | N/A | 2 | 0 |
| PS/PA (n) | N/A | 2 | 0 |
| HLHS (n) | N/A | 0 | 6 |
| Volume Overload (n) | N/A | 14 | 0 |
| VSD/AVC (n) | N/A | 2 | 0 |
| DORV (n) | N/A | 3 | 0 |
| TOF (n) | N/A | 7 | 0 |
| PS/PA (n) | N/A | 2 | 0 |
| RV pressure mmHg (mean;SEM) | No data | 67.8 (4.8) | 79.5 (6.2) |
| RV EF % (mean;SEM) | No data | 57.9 (2.1) | 20 (2.6) |
RV= Right Ventricle, EF= Ejection Fraction, TOF= Tetralogy of Fallot, TA= Truncus Arteriosus, DORV=Double Outlet RV, DCRV=Double Chambered RV, PS/PA=Pulmonary Stenosis/Atresia, HLHS=Hypoplastic Left Heart Syndrome, AVC/VSD=Atrioventricular Canal/Ventricular Septal Defect
Progressive depletion of mtDNA during the transition from cardiac hypertrophy to failure
Citrate synthase (CS) is a key enzyme in the Krebs cycle and its activity is a widely used marker of mitochondrial mass14, 15. The CS enzyme activity in whole tissue extracts was preserved in RVH whereas in the RV failure it was reduced by 46% (Figure 1A). Similarly, the activity of another key enzyme of the TCA cycle, Succinate dehydrogenase (SDH), was also decreased only in RV failure by 35% (Figure 1B). The mtDNA copy number has been previously used as an indicator of mitochondrial biogenesis16, 17. Interestingly, in our study the mtDNA content normalized to total tissue protein was progressively decreased in RV hypertrophy and failure (Figure 1C). Similar significant decrease was also observed when mtDNA was normalized to nuclear DNA (nDNA, Figure 1D) and there was no change in the nDNA copy number (Figure 1E). Despite the decrease in mtDNA, the expression of the mtDNA encoded mRNA, ND6, CYTB, COI and 16S rRNA was maintained in RVH but not in RV failure (Figure 1F). Similarly the protein levels of ND6 were unaltered in RVH but significantly reduced in RV failure (Figure 1G). These results suggest that mtDNA depletion precedes the decreases in mitochondrial enzyme activities or protein levels as well as the clinical manifestation of heart failure.
Figure 1.
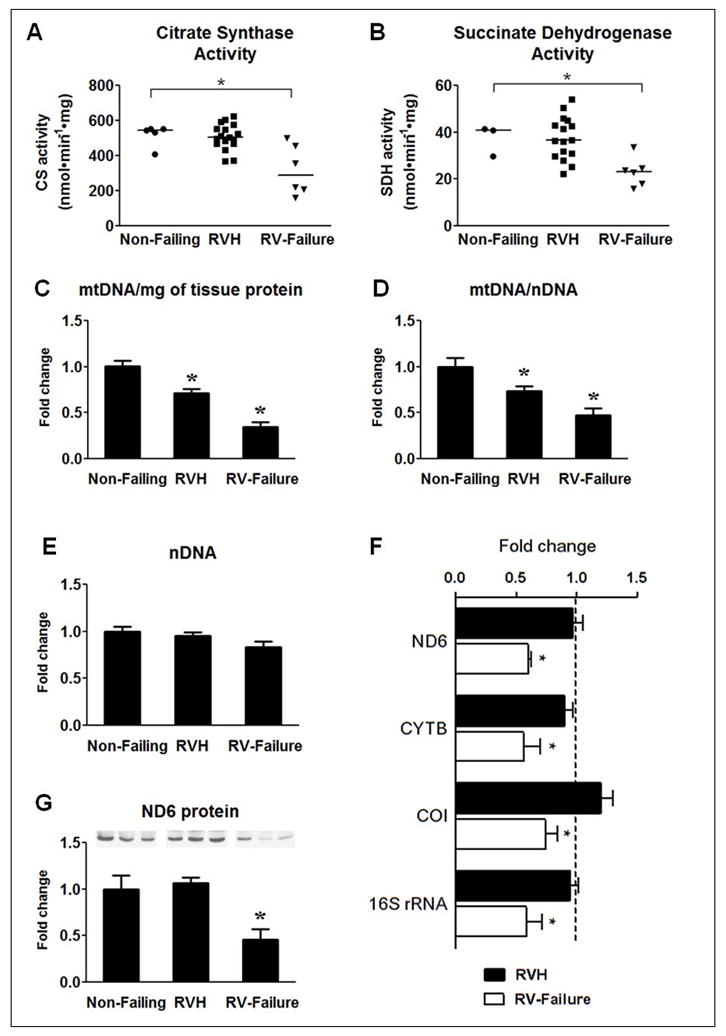
Assessment of mitochondrial enzyme activity and mtDNA content. (A) Citrate Synthase activity and (B) Succinate Dehydrogenase activity. Symbols in the scatter plots represent individual measurements and the continuous line indicates the group median. Mean of the fold changes ± SEM for (C) mtDNA content per mg of total protein, (D) mtDNA per nDNA and (E) nDNA per total DNA in RVH (n=17) and RV-Failure (n=6) group compared to Non-Failing RV controls (n=5). (F) Mean of the fold changes ± SEM of mRNA levels of ND6, CYTB, COI and 16S rRNA in the RVH (n=25) and RV-failure (n=6) relative to Non-Failing controls (indicated by the dashed line; n=5; * p≤0.05 vs. Non-Failing). (G) Mean of the fold changes ± SEM and a representative western blot for the ND6 (n=5 per group; * p≤0.05 vs. Non-Failing).
Decreases in the CS activity and mtDNA content were also observed in the LV of children with advanced stage of heart failure (Supplemental Figure 1). However, we were unable to obtain samples at the stage of LV hypertrophy to assess the progression of mtDNA depletion in the LV.
Defective mtDNA replication leads to mtDNA depletion
We subsequently tested the hypothesis that mtDNA replication was impaired in RV hypertrophy and failure by measuring the mtDNA replication intermediates in the tissue samples. We found that the extension of single strand DNA beyond the D-loop region, normalized to the mtDNA content, was reduced to a similar extent (by 50%) in both RV hypertrophy and failure (Figure 2A). Additionally, the expression of genes involved in the mtDNA replication fork was also down-regulated at the stage of hypertrophy and persisted in RV failure (Figure 2B). These findings suggest that defective mtDNA replication is an early event that contributes to impaired mitochondrial biogenesis and dysfunction in pathological hypertrophy and failure. Similar reduction of mtDNA replication intermediates was also observed in the LV of advanced heart failure in both adult9 and pediatric patients (Supplemental Figure 2).
Figure 2.
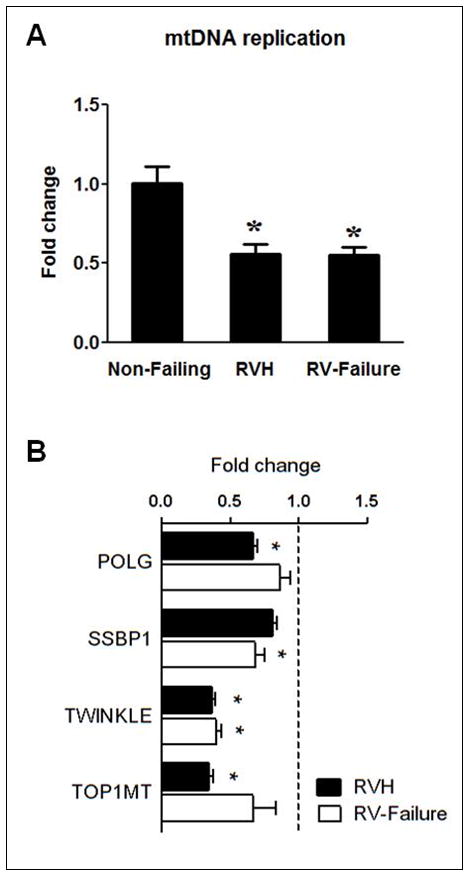
mtDNA replication. mtDNA replication was assessed by measuring the extension of 7S DNA beyond the D-Loop and normalized to the mtDNA content (A) and mRNA levels of POLG, SSBP1, TWINKLE and TOP1MT (B) in Non-Failing RV (indicated by the dashed line; n=5), RVH (n=25) and RV-failure (n=6). Data are shown as the mean of the fold changes ± SEM over the Non-Failing RV (* p≤0.05 vs. Non-Failing).
Transcriptional regulation of mitochondrial biogenesis
The evidence of impaired mtDNA replication in RV hypertrophy prompted us to examine the expression and activity of the PGC-1 pathway, the master regulators of many genes involved mitochondrial biogenesis. Interestingly, both PGC-1α and PGC-1β mRNA was increased at the stage of hypertrophy and returned to normal levels at the stage of RV failure (Figure 3). There were no changes in the expression of the PGC-1 interacting partner NRF1, while there was a switch in the expression of the NRF2 isoforms, NRF2A expression was increased whereas NRF2B2 was decreased. Among the genes regulated by NRF1/2, TFAM, an important regulator of mtDNA transcription as well as mtDNA maintenance, was modestly decreased; TFB1M expression was increased whereas TFB2M was decreased. Despite the changes at the mRNA levels, the protein levels of these genes were not reduced in RV hypertrophy (Figure 4). Similarly, the expression of several other nDNA-encoded mitochondrial proteins not related to mtDNA maintenance was also unchanged in RVH (Supplemental Figure 3). These findings suggest that mtDNA depletion in the RV hypertrophy is not due to the down-regulation of the PGC-1 levels, instead, the higher PGC-1 expression likely contributes to the compensatory increases in the transcription of mtDNA resulting in a sustained level of the mtDNA encoded proteins at the stage of compensated RV hypertrophy despite the decreased mtDNA content (Figure 1F).
Figure 3.
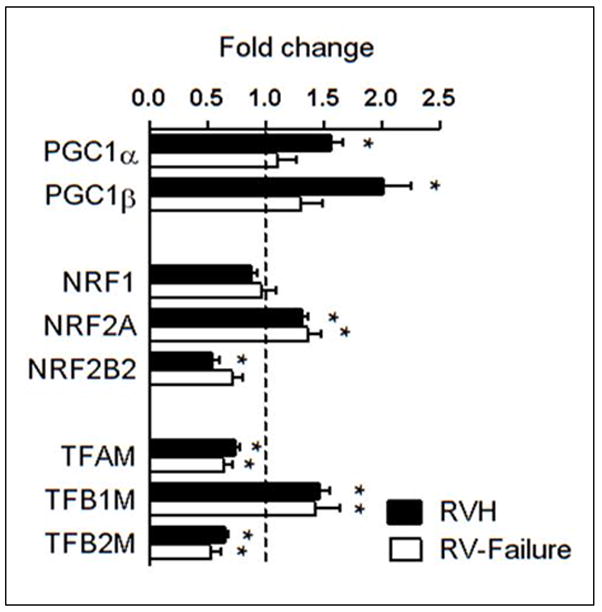
mRNA levels of the PGC-1 pathway in overloaded RV. Mean of the fold changes ± SEM in gene expressions for the PGC-1 pathway in the RVH (n=25) and RV-failure (n=6) relative to Non-Failing controls (indicated by the dashed line; n=5; * p≤0.05 vs. Non-Failing).
Figure 4.
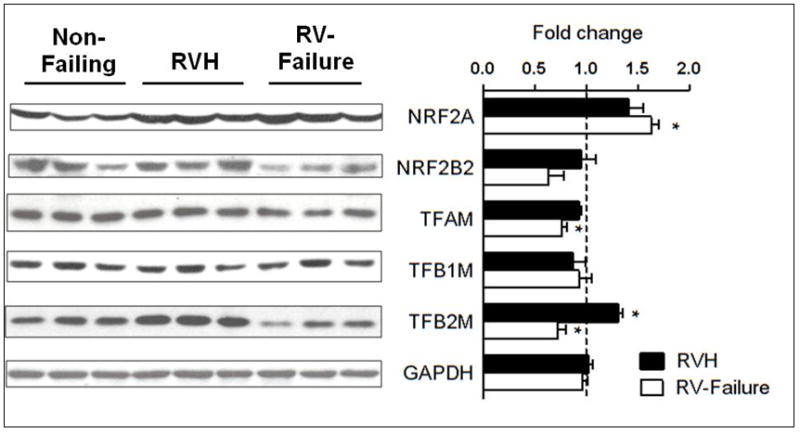
Protein levels of the PGC-1 downstream targets in overloaded RV. Representative western blots and mean of the fold changes ± SEM in gene expressions for the PGC-1 pathway in the RVH (n=5) and RV-failure (n=5) relative to Non-Failing controls (indicated by the dashed line; n=5; *p≤0.05 vs. Non-Failing).
Changes in mitochondrial ultra structure are associated with lower mtDNA content
We compared electron microscopic images of cardiac tissues from patients with compensated hypertrophy with different mtDNA content. There were no differences in the size and density of mitochondria in the hearts of patients with normal or low mtDNA levels (Figure 5). However, there were striking differences in the mitochondrial ultrastructure. In the tissue samples with low mtDNA content, the mitochondria were swollen; the cristae density was reduced, disorganized and oriented in varying oblong and oblique directions in the matrix (Figure 5). The parallel changes of mtDNA content and the mitochondrial ultra structure suggests that mtDNA depletion in RVH is a meaningful indicator of mitochondrial injury.
Figure 5.
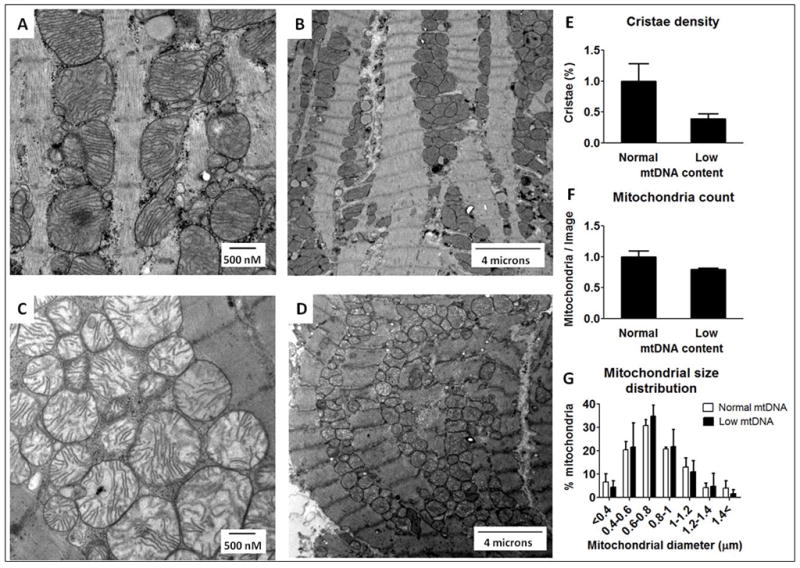
Mitochondrial ultrastructure in heart samples of compensated RV hypertrophy with normal and decreased mtDNA content. Representative electron micrographs of high and low magnifications a patient with normal mtDNA levels (A and B) and a patient with reduced mtDNA levels (C and D). Quantitative morphometric measurements of the cellular volume density for the mitochondrial cristae density (E), total mitochondria number (F) and mitochondrial size distribution (G) based on analysis of ten electron micrograph sections per patient from 2 patients per group. The bars represent mean of the fold changes ± SD.
RV pressure is a determinant of mtDNA gene expression
Within the RV hypertrophy group, the expression of the mtDNA encoded genes, ND6, CYTB, COI and 16S rRNA, was significantly lower in patients with higher than average RV pressure (RVp >70 mmHg) compared to those with RVp <70 mmHg (Figure 6A). Furthermore, the mtDNA copy number was inversely correlated with the RVp in the RVH group (Figure 6B) while the CS activity did not correlate with RVp (Figure 6C). These results collectively suggest that as the RV pressure overload develops there is progressive mtDNA depletion leading to the eventual failure of maintaining the mtDNA-encoded gene expression. Taken together with the parallel changes of ultrastrature and mtDNA content, our observations suggest that pathological stresses associated with the pressure overload have progressively affected the mitochondrial reserve predisposing the heart to failure.
Figure 6.
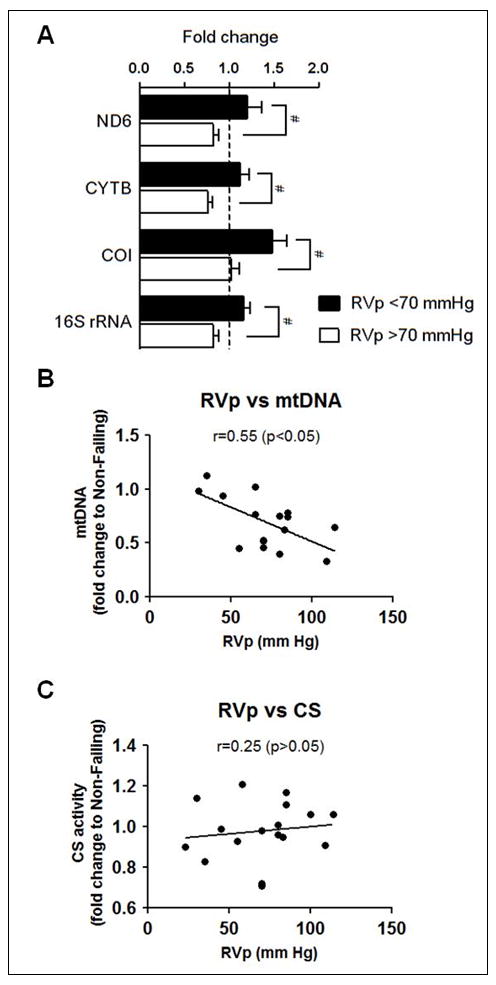
Pressure dependent changes of mtDNA gene expression, mtDNA content and CS activity in the RVH. (A) mRNA levels of ND6, CYTB, COI and 16S rRNA in the RVH group with RV pressure (RVp) <70mmHg (n=7) and RVp≥70mmHg (n=10) compared to Non-Failing RV controls (indicated by the dashed line in (A); n=5). Data are given as the mean of the fold changes ± SEM relative to the Non-Failing RV (* p≤0.05). (B) Correlation coefficient (Spearman’s rank correlation) between RV pressure (RVp) and mtDNA normalized to nDNA, and (C) RV pressure (RVp)-CS activity relationship in the RVH group. Data are given as the mean of the fold changes relative to the Non-Failing RV (n=17).
Discussion
In the present study we have assessed different aspects of mitochondrial biogenesis in the right ventricle of children and young adults with congenital heart disease who were presented with a varying degree of hemodynamic overload with and without RV failure. An important finding is that mtDNA replication was impaired and mitochondrial ultra-structure becomes abnormal in the hypertrophied RV before the patients meet the clinical and functional criteria of heart failure. These observations indicate that mtDNA depletion is a sensitive and meaningful marker of mitochondrial injury in congenital heart diseases presented with RV hemodynamic overload. We have also shown that mtDNA depletion occurs in the absence of down-regulation of the PGC-1 family genes but is coupled to defective mtDNA replication mechanism, and mtDNA encoded gene expression decreases in an RV pressure overload dependent manner. These findings provide novel information for understanding the pathogenesis of mitochondrial dysfunction in human heart failure.
The role of mitochondrial biogenesis in the transition from cardiac hypertrophy to failure
It has been well documented that marked changes of energy metabolism have occurred in cardiac hypertrophy and failure, including glycolysis, fatty acid oxidation, and overall ATP synthesis and turnover8, 18, 19. Although animal studies strongly indicate a role of mitochondrial dysfunction in the development of heart failure, mitochondrial studies in human heart failure have been challenging due to inherent difficulties in obtaining cardiac tissue samples from patients at different stages of the disease. In the present study, we were able to examine the mitochondrial biogenesis in patients with CHD with a broad spectrum of severity at the time of surgery. Our findings suggest that mtDNA replication and maintenance is impaired in the hemodynamically overloaded RV even though the standard functional assessment considers the heart at the stage of compensated hypertrophy. It is noteworthy that not only does the mtDNA level track with the development of heart failure it also parallels the pathological changes in mitochondrial ultra-structure in hypertrophied hearts, thus reliably reflecting the severity of the disease. The importance of mtDNA depletion or mutation in the development of mitochondrial dysfunction and cardiomyopathy has been well documented in mitochondrial diseases or drug toxicity20–22. Preservation of the mtDNA copy number in mouse hearts significantly delays the development of heart failure post myocardial infarction23–25. These pieces of evidence collectively show that the maintenance of mtDNA is critical for the normal heart function although unlikely to be the sole mechanism of heart failure.
Mitochondrial dysfunction and depletion of mtDNA has been associated with down-regulation of the PGC-1α gene and its downstream targets in animal models of heart failure26, 27. In contrast to rodent models, we have previously shown in end-stage human heart failure PGC-1 gene expression is not decreased in the failing LV9. In support of this it has been previously reported that in dogs with tachycardia induced heart failure there was no change in the PGC-1α protein levels28. Here we show that the expression of the PGC-1 genes is increased at the stage of hypertrophy and it returns to normal at the stage of failure. As discussed above, the increased expression of the PGC-1 genes might play a very important role in sustaining the transcriptional activities of the mtDNA in RV hypertrophy although it does not prevent the impairment of mtDNA replication.
Mitochondrial injury as a maker for the timing of surgery in children with CHD
Results of RV surgery in patients with congenital heart disease are controversial with some patients not improving RV size or function despite a technically successful operation4, 7. The long-term outcomes of the surgery highly varies with some centers reporting significant improvement in RV end-diastolic volume and systolic function, as measured by ejection fraction29, and other centers not seeing any improvement in RV function. There is increasing evidence suggesting that the timing of surgery rather than the removal of associated defects is critical for the reversibility of RV dysfunction4. For example, it has been shown that myocardial fibrosis or the development of pulmonary regurgitation post-surgery, both indicative of irreversible pathological remodeling of the RV, correlate closely with the adverse clinical status and the poor recovery of the repaired hearts30, 31, 4. In this study we have shown that impaired replication and depletion of mtDNA are initiated at the stage of clinically compensated hypertrophy and progressively affect mitochondrial protein expression and enzyme activity in a pressure depended manner in RV hypertrophy and during the transition to failure. Furthermore, the depletion of mtDNA is accompanied by significant morphological changes in the mitochondria ultra-structure suggesting that it is a sensitive and early marker of mitochondrial damage. Given the vital roles of mitochondria as the powerhouse as well as the key regulator of cell death in cardiac myocytes these findings may assist to identify a group of patients who should benefit from early surgery.
Limitations
Due to the differences in the cardiac developmental defect and its severity, we are able to examine cardiac tissue from the RV with varying degrees of dysfunction in this pediatric population. This unique opportunity also presents a challenge in finding age-match control RV tissue. As the result, the median age of the control group in this study is older than the patient group and the age range of the controls only partially overlap with the age range of the patients. There is a possibility that the content and the regulation mtDNA transcription and replication of the infant hearts are different from older children. However, the main focus of this study is the mitochondrial biogenesis in RV hypertrophy without clinical failure. The RV hypertrophy patients have considerable age overlap with the control group, and we did not observe age-dependent changes of mtDNA content and replication in control hearts (non-failing) between 4 to 18 years old.
Due to limited number of samples used for electron microscopic examination, we were quite limited in the power of the statistical analysis. Since our study population is small and consists of a mixture of diagnosis and stages of the disease, future studies with a larger number of patients in each subcategory are needed to identify strong clinical surrogates for mitochondrial damage which may serve as a useful guide to establish appropriate timing for surgery.
Conclusions
Our findings suggest that impaired mtDNA replication and maintenance is integral to the development and transition from RV hypertrophy to heart failure. It provides an important basis for the future development of “mitochondrial oriented” clinical and surgical management of patients with overloaded RV due to congenital heart disease. Specifically, the mtDNA depletion pattern could help to establish adequate timing for intervention. To this end, large clinical studies determining the link of mitochondrial impairment at the time of surgery and the outcome in this patient population is warranted.
Supplementary Material
Acknowledgments
We thank Dr. Jeffrey Saffitz for his advice regarding the sample preparations for the electron microscopy and for his critical evaluation of the quality of the electron micrographs. We would also like to express our special gratitude to Dr. Federica del Monte for her valuable input as well as for providing non-failing heart samples. Last but not least we would like to thank Dr. Pantelis Xofis and William Jen Hoe Koh for assisting us with the statistical analysis.
Sources of Funding
This study is supported by grants from the National Heart Lung and Blood Institute to Dr. Tian and from the National Institute of Aging T32 training grant to Dr. Karamanlidis.
Non-standard Abbreviations and Acronyms
- CS
Citrate Synthase
- EF
Ejection Fraction
- GAPDH
Glyceraldehyde-3-Phosphate Dehydrogenase
- LV
Left Ventricle
- mtDNA
mitochondrial DNA
- nDNA
nuclear DNA
- NRF
Nuclear Respiratory Factor
- POLG
Polymerase Gamma Mitochondrial
- RV
Right Ventricle
- RVOTO
Right Ventricular Outflow Track Obstruction
- SDH
Succinate Dehydrogenase
- SSBP1
Single Strand Binding Protein 1, Mitochondrial
- TFAM
Transcription Factor A, Mitochondrial
- TFB1/2M
Transcription Factor B1/2, Mitochondrial
- TOP1MT
Topoisomerase 1 Mitochondrial
Footnotes
Disclosures
None.
References
- 1.Murphy JG, Gersh BJ, Mair DD, Fuster V, McGoon MD, Ilstrup DM, McGoon DC, Kirklin JW, Danielson GK. Long-term outcome in patients undergoing surgical repair of tetralogy of Fallot. N Engl J Med. 1993;329:593–599. doi: 10.1056/NEJM199308263290901. [DOI] [PubMed] [Google Scholar]
- 2.Harrild DM, Berul CI, Cecchin F, Geva T, Gauvreau K, Pigula F, Walsh EP. Pulmonary valve replacement in tetralogy of Fallot: impact on survival and ventricular tachycardia. Circulation. 2009;119:445–451. doi: 10.1161/CIRCULATIONAHA.108.775221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bautista-Hernandez V, Hasan BS, Harrild DM, Prakash A, Porras D, Mayer JE, Jr, Del Nido PJ, Pigula FA. Late pulmonary valve replacement in patients with pulmonary atresia and intact ventricular septum: a case-matched study. Ann Thorac Surg. 2011;91:555–560. doi: 10.1016/j.athoracsur.2010.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Therrien J, Siu SC, McLaughlin PR, Liu PP, Williams WG, Webb GD. Pulmonary valve replacement in adults late after repair of tetralogy of fallot: are we operating too late? J Am Coll Cardiol. 2000;36:1670–1675. doi: 10.1016/s0735-1097(00)00930-x. [DOI] [PubMed] [Google Scholar]
- 5.Bouzas B, Kilner PJ, Gatzoulis MA. Pulmonary regurgitation: not a benign lesion. Eur Heart J. 2005;26:433–439. doi: 10.1093/eurheartj/ehi091. [DOI] [PubMed] [Google Scholar]
- 6.Geva T. Indications and timing of pulmonary valve replacement after tetralogy of Fallot repair. Semin Thorac Cardiovasc Surg Pediatr Card Surg Annu. 2006:11–22. doi: 10.1053/j.pcsu.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 7.Therrien J, Provost Y, Merchant N, Williams W, Colman J, Webb G. Optimal timing for pulmonary valve replacement in adults after tetralogy of Fallot repair. Am J Cardiol. 2005;95:779–782. doi: 10.1016/j.amjcard.2004.11.037. [DOI] [PubMed] [Google Scholar]
- 8.Neubauer S. The failing heart--an engine out of fuel. N Engl J Med. 2007;356:1140–1151. doi: 10.1056/NEJMra063052. [DOI] [PubMed] [Google Scholar]
- 9.Karamanlidis G, Nascimben L, Couper GS, Shekar PS, del Monte F, Tian R. Defective DNA replication impairs mitochondrial biogenesis in human failing hearts. Circ Res. 2010;106:1541–1548. doi: 10.1161/CIRCRESAHA.109.212753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rockl KS, Hirshman MF, Brandauer J, Fujii N, Witters LA, Goodyear LJ. Skeletal muscle adaptation to exercise training: AMP-activated protein kinase mediates muscle fiber type shift. Diabetes. 2007;56:2062–2069. doi: 10.2337/db07-0255. [DOI] [PubMed] [Google Scholar]
- 11.Kerner J, Turkaly PJ, Minkler PE, Hoppel CL. Aging skeletal muscle mitochondria in the rat: decreased uncoupling protein-3 content. Am J Physiol Endocrinol Metab. 2001;28:E1054–1062. doi: 10.1152/ajpendo.2001.281.5.E1054. [DOI] [PubMed] [Google Scholar]
- 12.Brown TA, Clayton DA. Release of replication termination controls mitochondrial DNA copy number after depletion with 2′,3′-dideoxycytidine. Nucleic Acids Res. 2002;30:2004–2010. doi: 10.1093/nar/30.9.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gensler S, Weber K, Schmitt WE, Perez-Martos A, Enriquez JA, Montoya J, Wiesner RJ. Mechanism of mammalian mitochondrial DNA replication: import of mitochondrial transcription factor A into isolated mitochondria stimulates 7S DNA synthesis. Nucleic Acids Res. 2001;29:3657–3663. doi: 10.1093/nar/29.17.3657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Holloszy JO, Oscai LB, Don IJ, Mole PA. Mitochondrial citric acid cycle and related enzymes: adaptive response to exercise. Biochem Biophys Res Commun. 1970;40:1368–1373. doi: 10.1016/0006-291x(70)90017-3. [DOI] [PubMed] [Google Scholar]
- 15.Hood DA, Zak R, Pette D. Chronic stimulation of rat skeletal muscle induces coordinate increases in mitochondrial and nuclear mRNAs of cytochrome-c-oxidase subunits. Eur J Biochem. 1989;179:275–280. doi: 10.1111/j.1432-1033.1989.tb14551.x. [DOI] [PubMed] [Google Scholar]
- 16.Nisoli E, Tonello C, Cardile A, Cozzi V, Bracale R, Tedesco L, Falcone S, Valerio A, Cantoni O, Clementi E, Moncada S, Carruba MO. Calorie restriction promotes mitochondrial biogenesis by inducing the expression of eNOS. Science. 2005;310:314–317. doi: 10.1126/science.1117728. [DOI] [PubMed] [Google Scholar]
- 17.Davis JM, Murphy EA, Carmichael MD, Davis B. Quercetin increases brain and muscle mitochondrial biogenesis and exercise tolerance. Am J Physiol Regul Integr Comp Physiol. 2009;296:R1071–1077. doi: 10.1152/ajpregu.90925.2008. [DOI] [PubMed] [Google Scholar]
- 18.Sambandam N, Lopaschuk GD, Brownsey RW, Allard MF. Energy metabolism in the hypertrophied heart. Heart Fail Rev. 2002;7:161–173. doi: 10.1023/a:1015380609464. [DOI] [PubMed] [Google Scholar]
- 19.Kolwicz SC, Jr, Tian R. Glucose metabolism and cardiac hypertrophy. Cardiovasc Res. 2011;90:194–201. doi: 10.1093/cvr/cvr071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.DiMauro S, Schon EA. Mitochondrial respiratory-chain diseases. N Engl J Med. 2003;348:2656–2668. doi: 10.1056/NEJMra022567. [DOI] [PubMed] [Google Scholar]
- 21.L’Ecuyer T, Sanjeev S, Thomas R, Novak R, Das L, Campbell W, Heide RV. DNA damage is an early event in doxorubicin-induced cardiac myocyte death. Am J Physiol Heart Circ Physiol. 2006;29:H1273–1280. doi: 10.1152/ajpheart.00738.2005. [DOI] [PubMed] [Google Scholar]
- 22.Lewis W. Defective mitochondrial DNA replication and NRTIs: pathophysiological implications in AIDS cardiomyopathy. Am J Physiol Heart Circ Physiol. 2003;284:H1–9. doi: 10.1152/ajpheart.00814.2002. [DOI] [PubMed] [Google Scholar]
- 23.Ikeuchi M, Matsusaka H, Kang D, Matsushima S, Ide T, Kubota T, Fujiwara T, Hamasaki N, Takeshita A, Sunagawa K, Tsutsui H. Overexpression of mitochondrial transcription factor a ameliorates mitochondrial deficiencies and cardiac failure after myocardial infarction. Circulation. 2005;112:683–690. doi: 10.1161/CIRCULATIONAHA.104.524835. [DOI] [PubMed] [Google Scholar]
- 24.Matsushima S, Ide T, Yamato M, Matsusaka H, Hattori F, Ikeuchi M, Kubota T, Sunagawa K, Hasegawa Y, Kurihara T, Oikawa S, Kinugawa S, Tsutsui H. Overexpression of mitochondrial peroxiredoxin-3 prevents left ventricular remodeling and failure after myocardial infarction in mice. Circulation. 2006;113:1779–1786. doi: 10.1161/CIRCULATIONAHA.105.582239. [DOI] [PubMed] [Google Scholar]
- 25.Dai DF, Santana LF, Vermulst M, Tomazela DM, Emond MJ, MacCoss MJ, Gollahon K, Martin GM, Loeb LA, Ladiges WC, Rabinovitch PS. Overexpression of catalase targeted to mitochondria attenuates murine cardiac aging. Circulation. 2009;119:2789–2797. doi: 10.1161/CIRCULATIONAHA.108.822403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Arany Z, Novikov M, Chin S, Ma Y, Rosenzweig A, Spiegelman BM. Transverse aortic constriction leads to accelerated heart failure in mice lacking PPAR-gamma coactivator 1alpha. Proc Natl Acad Sci U S A. 2006;103:10086–10091. doi: 10.1073/pnas.0603615103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ventura-Clapier R, Garnier A, Veksler V. Transcriptional control of mitochondrial biogenesis: the central role of PGC-1alpha. Cardiovasc Res. 2008;79:208–217. doi: 10.1093/cvr/cvn098. [DOI] [PubMed] [Google Scholar]
- 28.Lionetti V, Linke A, Chandler MP, Young ME, Penn MS, Gupte S, d’Agostino C, Hintze TH, Stanley WC, Recchia FA. Carnitine palmitoyl transferase-I inhibition prevents ventricular remodeling and delays decompensation in pacing-induced heart failure. Cardiovasc Res. 2005;66:454–461. doi: 10.1016/j.cardiores.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 29.Vliegen HW, van Straten A, de Roos A, Roest AA, Schoof PH, Zwinderman AH, Ottenkamp J, van der Wall EE, Hazekamp MG. Magnetic resonance imaging to assess the hemodynamic effects of pulmonary valve replacement in adults late after repair of tetralogy of fallot. Circulation. 2002;106:1703–1707. doi: 10.1161/01.cir.0000030995.59403.f8. [DOI] [PubMed] [Google Scholar]
- 30.Wald RM, Haber I, Wald R, Valente AM, Powell AJ, Geva T. Effects of regional dysfunction and late gadolinium enhancement on global right ventricular function and exercise capacity in patients with repaired tetralogy of Fallot. Circulation. 2009;119:1370–1377. doi: 10.1161/CIRCULATIONAHA.108.816546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Babu-Narayan SV, Kilner PJ, Li W, Moon JC, Goktekin O, Davlouros PA, Khan M, Ho SY, Pennell DJ, Gatzoulis MA. Ventricular fibrosis suggested by cardiovascular magnetic resonance in adults with repaired tetralogy of fallot and its relationship to adverse markers of clinical outcome. Circulation. 2006;113:405–413. doi: 10.1161/CIRCULATIONAHA.105.548727. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.