

Abstract
The cyclin-dependent kinase inhibitor p27 plays an important role in cell cycle regulation. Reduced expression of p27 is commonly associated with poor prognosis in many malignancies, including gastric cancer. Cytoplasmic p27 mislocalization may be an additional indicator of high-grade tumors and poor prognosis in cancer. Since chronic infection by Helicobacter pylori is the most important risk factor for gastric cancer development, we evaluated the effects of H. pylori on p27 expression and localization in gastric cancer cells. Co-culture of gastric cells with H. pylori induced cytoplasmic p27 expression and reduced nuclear p27 expression in vitro. Cytoplasmic p27 expression was associated with and dependent upon phosphorylation of p27 at T157 and T198: wild type p27 accumulated in the cytoplasm, but non-phosphorylatable mutants affecting T157 or T198 were nuclear in H. pylori infected cells. These post-translational p27 changes were secondary to activation of cellular PI3K and AKT signaling pathways and dependent upon a functional H. pylori cag pathogenicity island. We investigated the clinical significance of cytoplasmic p27 mislocalization in 164 cases of resected gastric cancer in tissue microarrays. In 97 cases (59%) cytoplasmic p27 mislocalization was observed, and this was associated with increased mortality in multivariate analysis. These results show that H. pylori infection induces AKT/PI3K-mediated phosphorylation of p27 at T157 and T198 to cause cytoplasmic p27 mislocalization in gastric cancer, and that p27 mislocalization is an adverse prognostic feature in gastric cancer. This is the first demonstration of the translocation of a specific bacterial virulence factor that post-translationally regulates a host cell cyclin-dependent kinase inhibitor. This is of particular significance because p27 has both tumor-suppressive and oncogenic activities, depending upon its subcellular localization. Cytoplasmic mislocalization of p27 induced by H. pylori may be an important mechanistic link between H. pylori infection and gastric carcinogenesis.
Keywords: gastric cancer, Helicobacter pylori, cell cycle regulation, cyclin-dependent kinase inhibitor
INTRODUCTION
Gastric cancer is the second most common cause of cancer death worldwide (Hartgrink et al 2009). The most important known risk factor for gastric cancer development is prior infection by Helicobacter pylori, a micro-aerophilic flagellated gram-negative bacterium that promotes a persistent inflammatory state in the gastric mucosa that lasts for decades. Approximately 70% of the global gastric cancer burden has been attributed to H. pylori infection (Parkin 2006). The consequences of H. pylori-associated gastritis include chronic oxidative stress in the gastric mucosa, cell cycle dysregulation and alterations in the normal epithelial cell populations and gastric secretory physiology (Wroblewski et al 2010). Prospective cohort studies and randomized control trials of H. pylori eradication indicate that H. pylori-positive individuals have a 2 to 10-fold increased risk of non-cardia gastric cancer compared with uninfected subjects and that eradication of H. pylori can reduce this risk significantly (Forman and Burley 2006, Fuccio et al 2009). H. pylori infection is necessary but not sufficient for gastric cancer development, additional risk factors for gastric carcinogenesis include dietary components and the host’s genetic background (Fock et al 2008, Liu et al 2009).
The cyclin-dependent kinase inhibitor p27 is an important regular of the G1 to S phase transition in normal cell cycle progression (Chu et al 2008). Mice lacking one or both copies of p27 display increased susceptibility to tumorigenesis (Fero et al 1998), including gastric carcinogenesis following experimental H. pylori infection (Kuzushita et al 2005). A tumor suppressor function for p27 is also supported by the frequent association of low p27 levels with high-grade tumors and poor prognosis in several types of human cancer (Chu et al 2008). Most, though not all, studies also describe an association with p27 loss and poor prognosis in gastric cancer (Chu et al 2008, Feakins et al 2000, Mori et al 1997, Yasui et al 1997).
The regulation of the expression, subcellular localization and activity of p27 is complex and occurs at multiple levels, including at the levels of transcription, translation and post-translationally (Chu et al 2008). Loss of nuclear p27 expression and/or its cytoplasmic mislocalization in tumor cells have both been reported to be associated with poor outcome in cancer of the breast (Liang et al 2002, Shin et al 2002, Viglietto et al 2002), prostate (Li et al 2006), ovary (Duncan et al), and in astrocytomas (Hidaka et al 2009). An oncogenic role for cytoplasmic p27 is supported by evidence from a “knock-in” mouse model in which the cyclin – CDK regulatory domain of p27 appears responsible for tumor suppressor activity and a cyclin-CDK-independent function of cytoplasmic p27 promotes tumor formation (Besson et al 2007). A cytoplasmic, oncogenic role of p27 may be related to its ability to promote cell motility and migration via its binding to and inhibition of RhoA, as shown in cell culture by the use of a mutant p27 that cannot bind to cyclins and CDKs (Besson et al 2004).
Several phosphorylation sites on p27 have been mapped as targets of a variety of cellular kinases. Some of these have been implicated in regulating the subcellular localization of p27 and modifying its function (Vervoorts and Luscher 2008, Wander et al). Among these, phosphorylation of threonine residues at positions 157 and 198 has been linked to cytoplasmic p27 expression in several model systems (Hong et al 2008, Larrea et al 2009, Liang et al 2002, Shin et al 2002, Viglietto et al 2002).
We previously reported that H. pylori infection of gastric cancer cells in vitro and in vivo is associated with p27 loss and resistance to apoptosis (Eguchi et al 2004, Kim et al 2006, Shirin et al 2000). We now provide evidence that H. pylori induces cytoplasmic p27 expression, associated with phosphorylation of p27 at T157 and T198. These post-translational p27 changes are dependent upon cellular PI3K and AKT signaling pathways and upon a functional H. pylori cag pathogenicity island. These data, together with our observation of frequent cytoplasmic p27 mislocalization in primary human gastric cancer, in association with worse prognosis, supports the notion that the cytoplasmic mislocalization of p27 induced by H. pylori may represent an important mechanistic link between H. pylori infection and gastric carcinogenesis.
RESULTS
Effects of H. pylori on the nuclear and cytoplasmic localization of p27
To investigate the effects of H. pylori infection on the subcellular localization of p27, AGS cells were cocultured with H. pylori and protein was extracted from cytoplasmic and nuclear fractions. Western blotting of these subcellular lysates demonstrated that p27 was mainly located in the nucleus before H. pylori infection, whereas p27 was predominantly cytoplasmic after H. pylori infection (Fig. 1A). The cytoplasmic to nuclear ratio of p27 expression was increased by a mean ± standard deviation of 7.7 ± 3.4 fold by H. pylori infection (p=0.01). The altered subcellular localization of p27 after the addition of H. pylori was confirmed by indirect immunofluorescence (Fig. 1B). Similar results were obtained with MKN-28 cells transfected with a wild type YFP p27 fusion construct (data not shown).
Fig. 1. H. pylori increases cytoplasmic localization of p27.

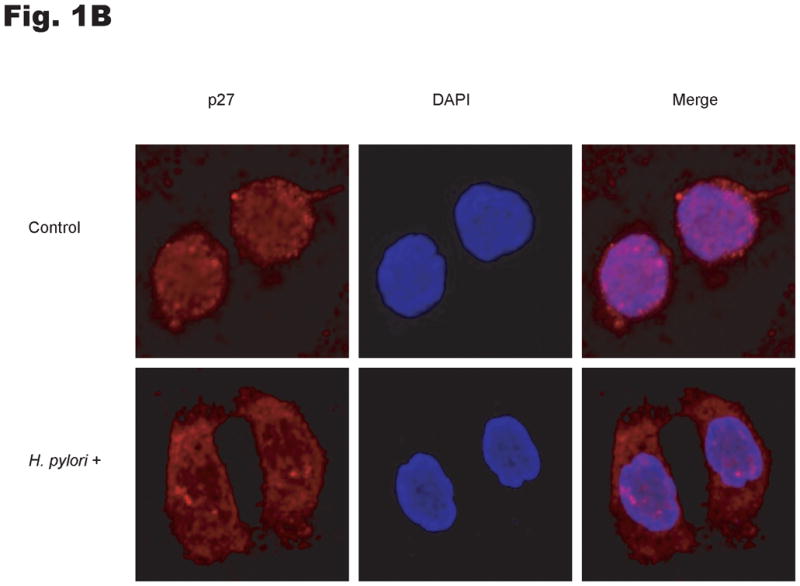
(A) AGS cells were treated with H. pylori strain ATCC 49503 at a multiplicity of infection of 100:1 for 6 hrs and nuclear and cytoplasmic extracts immunoblotted for p27. Lamin A and α-tubulin antibodies were used to confirm enrichment for nuclear and cytoplasmic fractions respectively. (B) Immunofluorescent confocal micrography of p27 distribution after H. pylori infection. AGS cells were incubated without or with H. pylori strain ATCC 49503 at a multiplicity of infection of 100:1 for 10 hours and then processed for indirect immunofluorescence under confocal microscopy. Endogenous p27 stains red and the nuclei were identified by DAPI (blue) staining. Original magnification x 600.
Effects of H. pylori on the phosphorylation of p27 at T157 and T198
Because cytoplasmic localization of p27 has been linked to phosphorylation of p27 at T157 and T198 in breast cancer (Larrea et al 2009, Liang et al 2002, Shin et al 2002, Viglietto et al 2002), we next investigated whether H. pylori could induce phosphorylation of p27 at T157 and T198 in gastric cancer cells. AGS cells were serum-starved for 24 hours and then infected with H. pylori for 6 hours. The phosphorylation of p27 at T157 and T198 was then evaluated by immunoblotting using phospho-specific antibodies to phospho-T157 or phospho-T198. H. pylori increased cytoplasmic phosphorylation of p27 at T198 by a mean of 2.7 ± 1.1 fold (P<0.05) (Fig. 2A). The phosphorylation of T198 was accompanied by phosphorylation of serine 473 of AKT, indicative of AKT activation (2.2 ± 0.4 fold increase, P<0.05) and the cytoplasmic to nuclear ratio of total p27 increased by 6.1 ± 3.6 fold (P<0.05). Available reagents did not permit detection of endogenous cellular p27pT157. However, when AGS cells were transiently transfected with wild type Myc-tagged p27, H. pylori infection was observed to increase exogenous p27pT157 and p27pT198 (3.3 ± 0.7 fold, P<0.05 and 2.0 ±0.2 fold, P<0.01, respectively); an effect that was confined to the cytoplasmic p27 fraction (Fig. 2B).
Fig. 2. H. pylori induces p27 phosphorylation at T157 and T198.
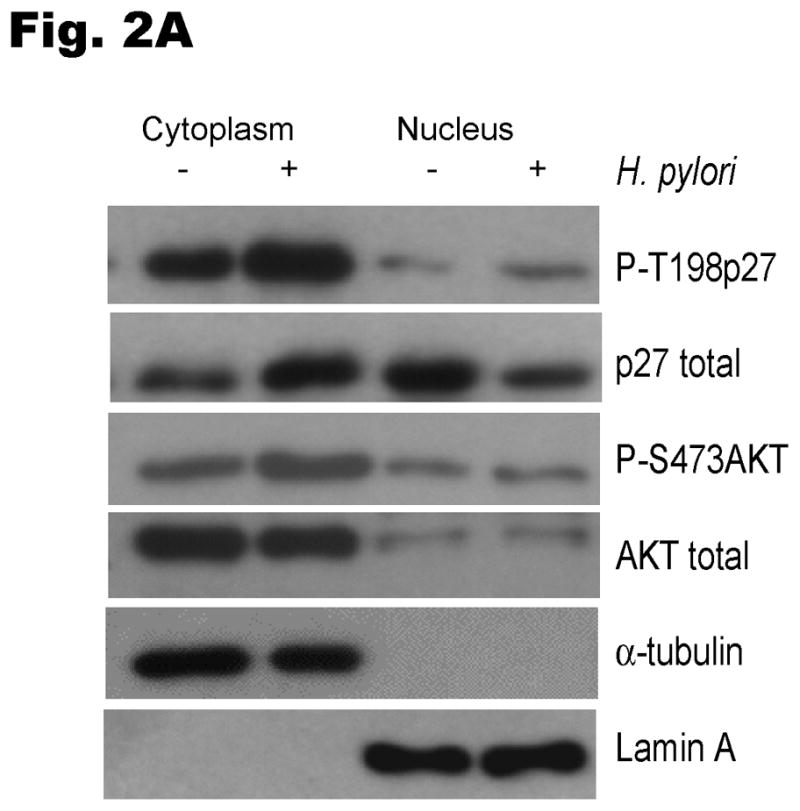
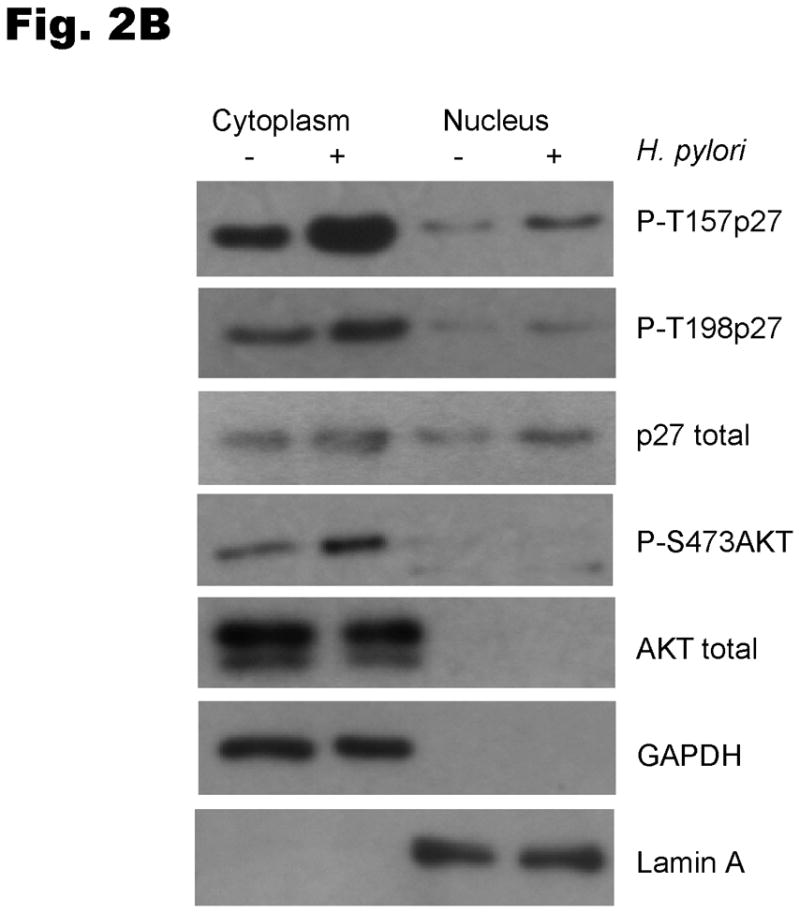
The levels of phosphorylated T157p27 and T198p27 in cell lysates were analyzed by immunoblot using p27 phospho-site specific antibodies. (A) To examine the regulation of endogenous p27, AGS cells were infected with H. pylori and nuclear and cytoplasmic extracts immunoblotted with phospho-site specific p27 and AKT antibodies. (B) AGS cells were transiently transfected with a Myc-tagged wild type p27 plasmid for 24 hours, deprived of serum for 24 hours and then infected with H. pylori for 6 hours.
Dependence of H. pylori-induced phosphorylation of p27 at T198 and cytoplasmic localization of p27 upon the PI3K pathway
To investigate the involvement of PI3K in H. pylori-mediated up-regulation of p27 T157 and T198 phosphorylation, AGS cells were pretreated with either AKT siRNA targeting all three isoforms of Akt1 as well as Akt2 (or a scrambled siRNA as control), or the PI3Kinhibitor LY294002 at 20μm, and were then co-cultured with H. pylori. Inhibiting AKT activation decreased the upregulation of H. pylori’s induction of p27T198 phosphorylation, whether AKT activation was inhibited through the use of AKT siRNA (1.7 ± 0.6 fold decrease in p27T198 phosphorylation, P<0.05) (Fig. 3A) or by PI3K inhibition (2.9 ± 0.1 fold decrease in p27T198 phosphorylation, P<0.05) (Fig. 3B). To determine if activation of the PI3K pathway is required for the H. pylori-mediated cytoplasmic localization of p27, AGS cells transfected with a wild type p27-YFP fusion plasmid were treated with LY294002 at 20μm for 1 hour and then infected with H. pylori for another 6 hours. In uninfected controls, 19±4% of AGS cells exhibited cytoplasmic p27 staining. This increased to 48 ± 6% after H. pylori infection. However, treatment with the PI3K inhibitor LY294002 inhibited cytoplasmic expression of p27, with only 15 ± 2% of cells showing cytoplasmic p27 staining before and 18 ± 3% after H. pylori infection (Fig. 3C), (P <0.01). Similar effects of LY294002 were observed on endogenous p27 expression, or with AKT inhibition by AKT siRNA (data not shown). These results suggest that the cytoplasmic localization of p27 induced by H. pylori is PI3K dependent.
Fig. 3. H. pylori-induced p27T198 phosphorylation is decreased by AKT siRNA (A) or PI3K inhibition (B).
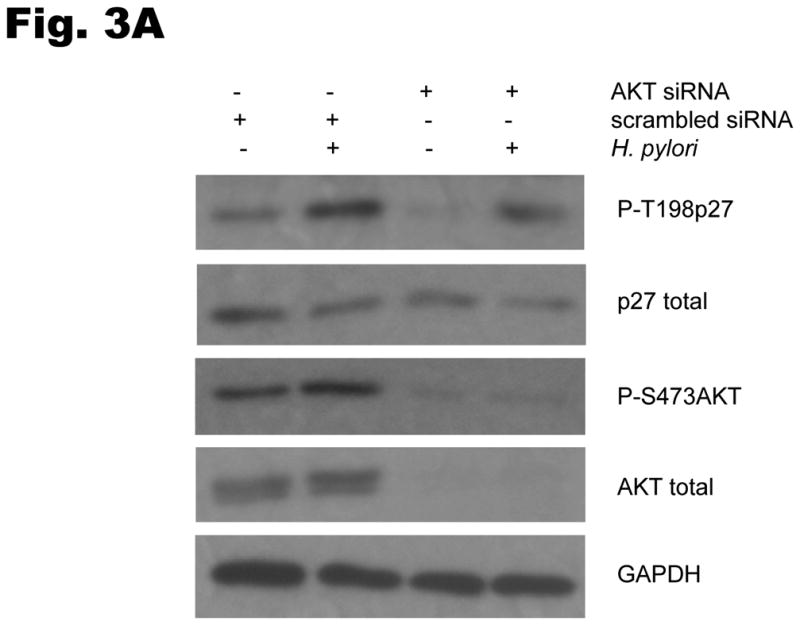
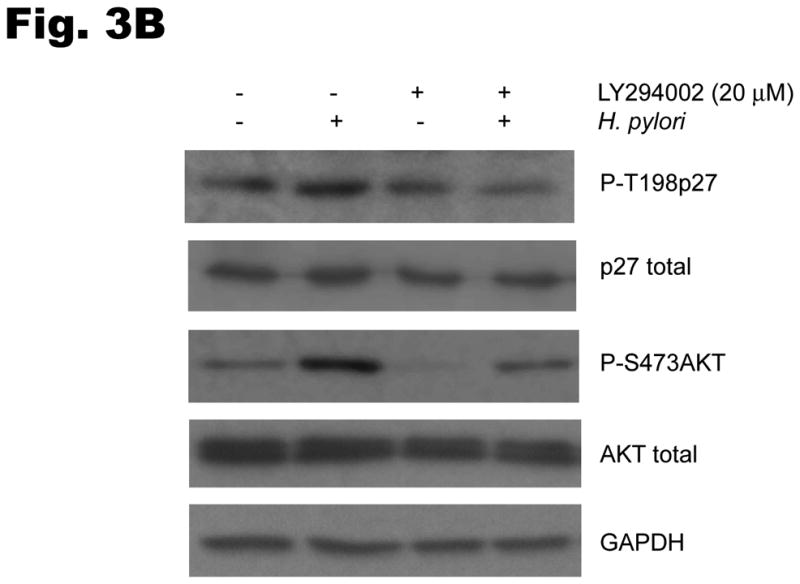

AGS cells were pretreated with either AKT siRNA (A) for 24 hours or LY294002 (B) at 20 μm for 1 hour and then infected with H. pylori for 6 hrs. Phosphorylation of p27 and AKT was measured by immunoblot using phospho-site specific antibodies. PI3K inhibition decreases the cytoplasmic localization of p27 induced by H. pylori (C). AGS cells were transfected with a wild type p27-YFP fusion construct, treated with LY294002 (LY) at 20μm for 1 hour and then infected with H. pylori for 6 hours. The percentages of cells exhibiting p27 staining in the cytoplasm or nucleus were quantified in a minimum of 100 cells in at least three fields. Data shown as percentage of cells expressing p27 in cytoplasm (mean ± standard deviation), representative of 3 separate experiments. *: P < 0.01.
Requirement of phosphorylation of p27 at T157 and T198 for the altered subcellular distribution of p27 induced by H. pylori
To determine if phosphorylation of p27 at T157 and T198 is required for H. pylori-mediated changes in the subcellular localization of p27, we transfected AGS cells with either a wild type p27-YFP fusion construct or a series of YFP-p27 mutants generated by site-directed mutagenesis. In these mutants T157 of p27 was substituted by either non-phosphorylated alanine (p27T157A) or the phosphomimetic aspartic acid (p27T157D), T198 of p27 was replaced with either alanine (p27T198A) or aspartic acid (p27T198D), and T157 and T198 of p27 were both mutated to alanine in a double mutant (p27T157AT198A). Following transfection, p27 localization in AGS cells after H. pylori infection was investigated by confocal immunofluorescent microscopy. Following H. pylori infection, wild type p27 was predominantly expressed in the cytoplasm, as it was with the phosphomimetic mutants YFPp27T157D and YFPp27T198D (even in the absence of H. pylori). In contrast, the p27 mutants lacking threonine phosphorylation sites at 157 and 198 (YFPp27T157A, YFPp27T198A and YFPp27T157AT198A) localized exclusively to the cytoplasm (Fig. 4) thus indicating that phosphorylation of p27 at T157 and T198 are necessary for the cytoplasmic accumulation of p27 following H. pylori infection.
Fig. 4. Localization of wild type YFPp27 and specific p27 mutants after H. pylori infection in AGS cells.

AGS cells were transfected with either a wild type p27-YFP fusion protein or with specific YFP-p27 mutants as indicated. The distribution of the p27-YFP fusion proteins in AGS cells was investigated by confocal immunofluorescent microscopy after H. pylori infection (Hp+) and in uninfected (Hp−) cells as controls. Original magnification x 600.
Dependence of specific H. pylori genes on the induction of p27 phosphorylation and AKT activation
To evaluate the role of specific H. pylori factors in the induction of p27 and AKT phosphorylation, wild type H. pylori strain 60190 or its isogenic mutants lacking the cagA cagE cagL or VacA genes were cocultured with AGS cells. The wild type strain and the VacA knockout strain strongly induced the phosphorylation of endogenous of AKT at Ser 437 (2.3 ± 0.8 fold, P<0.05 and 2.8 ± 1.4 fold P<0.05 respectively) and of p27 at T198 (3.4 ± 0.7 fold, P<0.01 and 4.7 ± 1.7 fold, P<0.01 respectively). In comparison, mutants lacking the cagA cagE or cagL gene had much less effect on p27T198 phosphorylation, that were not significantly different to uninfected control cells. Similar results were obtained with wild type H. pylori strain 11637 and its isogenic mutants. These results suggest that structural integrity of H. pylori’s type 4 secretory system is necessary for the induction of p27 phosphorylation and activation of AKT (Fig. 5)
Fig. 5. Effects of H. pylori factors on phosphorylation of p27 and activation of AKT.

Wild type (WT) or isogenic mutant strains of H. pylori lacking specific genes as shown (cagA, cagE, cagL or VacA) were co-cultured with AGS cells for 6 hours, and specific proteins and phosphoproteins assayed by immunoblot.
Nuclear and cytoplasmic localization of p27 in human gastric cancer and association with clinical outcome
p27 expression and subcellular localization were investigated by immunohistochemical staining of human gastric tissue. The normal (non-neoplastic) stomach mucosa and the intestinal metaplastic epithelium showed uniformly strong nuclear and very weak cytoplasmic p27 expression limited to the epithelial cells lining the lower third of glands. In contrast, 81 of 164 cases (49%) of gastric cancer had low nuclear p27 expression and 67 of 164 cases (41%) of cases had moderate to strong cytoplasmic p27 expression. Examples of the patterns and range of p27 expression in normal gastric mucosa and gastric cancer are depicted in Fig. 6A–C. No significant differences in cytoplasmic or nuclear p27 expression were observed between gastric cancers of the intestinal and diffuse histological subtypes.
Fig. 6. Expression of p27 in gastric cancer.
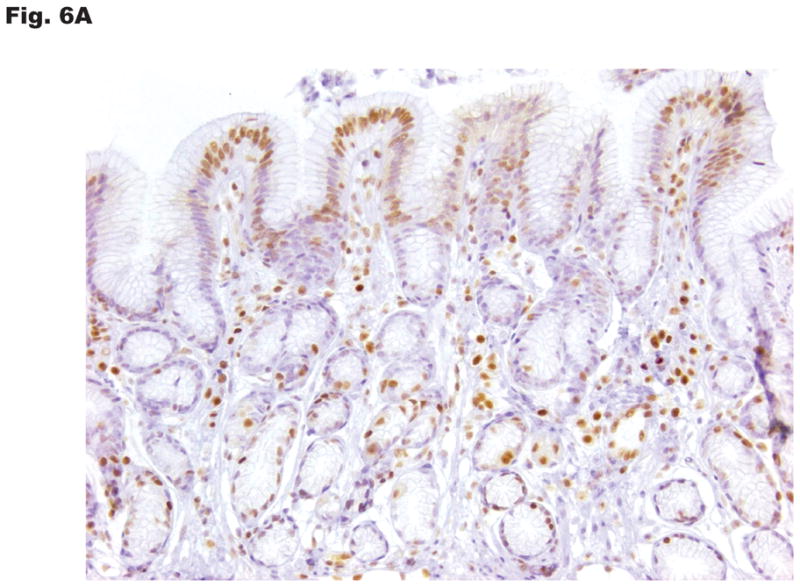
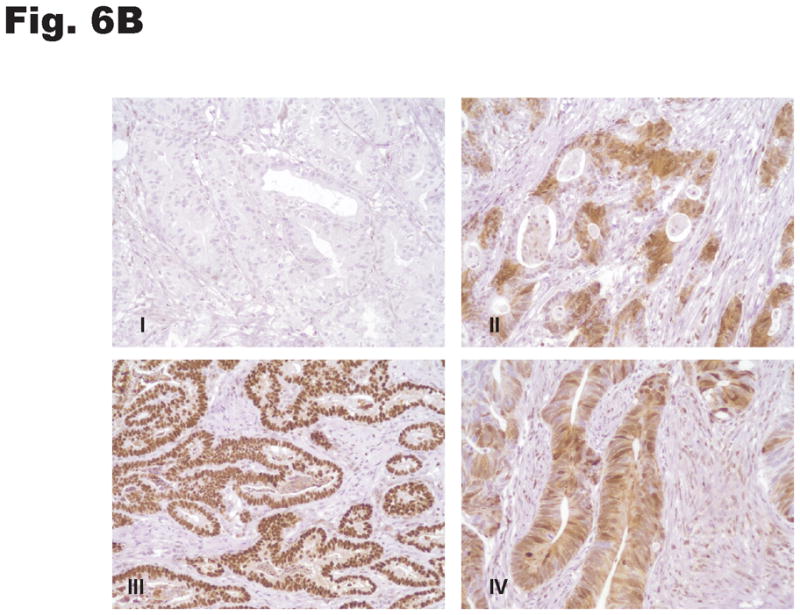
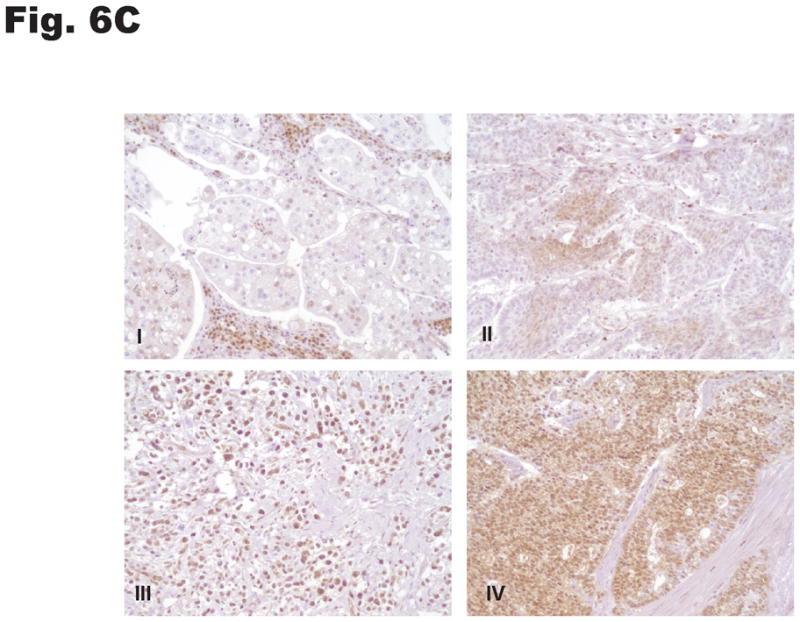
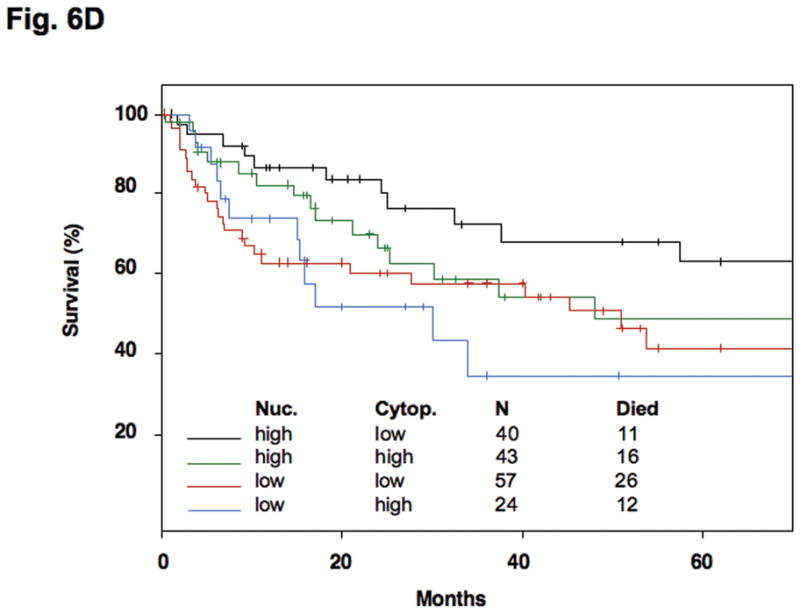
Subcellular distribution of p27 was evaluated by immunohistochemistry in formalin-fixed, paraffin-embedded specimens. Photomicrographs depict the range of findings from representative cases. (A) Normal gastric antral mucosa with nuclear p27 expression. (B) Intestinal-type gastric cancers showing I: No nuclear or cytoplasmic expression, II: Patchy nuclear and cytoplasmic expression. III: Strong p27 staining, nucleus only, IV: Strong nuclear and cytoplasmic p27 expression. (C) Diffuse-type gastric carcinomas. I: Rare nuclear and low cytoplasmic expression, II: Low nuclear expression and patchy cytoplasmic p27 expression, III: Strong nuclear p27 staining only, IV: Diffuse high nuclear and strong cytoplasmic p27 expression. Original magnification x 200. (D) Effect of cytoplasmic mislocalization of p27 on prognosis in gastric cancer. Immunostaining of p27 in 164 cases of gastric cancer in tissue microarrays was evaluated and 4 groups of cases defined based upon high and low nuclear and cytoplasmic expression, with cutoffs selected by regression tree analysis. Kaplan-Meier plots of cancer-specific mortality after surgery show that cytoplasmic p27 expression is associated with lower survival in gastric cancer. (P= 0.017 comparing high nuclear, low cytoplasmic with low nuclear, high cytoplasmic group, P= 0.03 comparing high nuclear, low cytoplasmic with low nuclear, low cytoplasmic group). Nuc, nuclear p27 expression; Cytop, cytoplasmic p27 expression. Cut-off values for low and high expression were selected by regression tree analysis, as described in Methods.
Regression tree analysis defined nuclear and cytoplasmic cut-off values with which to separate the 4 patterns of p27 expression queried in survival modeling: high nuclear, low cytoplasmic expression (40 cases); high nuclear, high cytoplasmic (43 cases); low nuclear, low cytoplasmic (57 cases) and low nuclear, high cytoplasmic (24 cases). The Kaplan-Meier curves show the influence of p27 localization on patient survival from gastric cancer (Fig. 6D). Overall, those gastric cancers with high cytoplasmic p27 expression are associated with a poorer prognosis than those cases with low cytoplasmic p27 expression. The very best survival was seen in cases with high nuclear but low cytoplasmic p27 expression (similar to the subcellular expression of p27 of non-neoplastic gastric tissues) and the group with the worst survival had low nuclear with high cytoplasmic p27 expression. For each nuclear subclassification group (high nuclear or low nuclear expression), the presence of cytoplasmic mislocalization was associated with poorer prognosis.
In the multivariate Cox proportional hazards model, p27 expression pattern and tumor stage were statistically significant independent factors associated with survival. The estimated hazard ratios and their 95% confidence intervals are listed in Table I. In particular, the estimated hazard ratio for cancer-associated mortality from the Cox model is 2.8 (P = 0.030) for low nuclear, high cytoplasmic expression versus the “normal” high nuclear low cytoplasmic group, and 2.6 (P = 0.016) for low nuclear, low cytoplasmic expression versus the “normal” high nuclear low cytoplasmic expression tumors. The hazard ratio is 3.8 (P < 0.0001) between stage 3–4 versus stage 1–2 tumors. Patient age, gender, tumor subtype (intestinal versus diffuse) and whether or not the patient received adjunctive chemotherapy or radiation therapy were not independent prognostic variables. Taken together, these data indicate that both low nuclear p27 and high cytoplasmic p27 expression are associated with poor survival in gastric carcinoma.
Table 1.
Cytoplasmic p27 localization and low nuclear p27 expression are associated with high cancer-specific mortality
| Factor | Hazard Ratio | CI: lower 95% | CI: upper 95% | p-value |
|---|---|---|---|---|
| p27 expression | ||||
| (nucleus, cytoplasm) | ||||
| (high, low) | reference | NA | NA | NA |
| (high, high) vs (high, low) | 1.214 | 0.526 | 2.800 | 0.650 |
| (low, low) vs (high, low) | 2.618 | 1.195 | 5.740 | 0.016 |
| (low, high) vs (high, low) | 2.799 | 1.106 | 7.080 | 0.030 |
| Stage(3 or 4 vs 1 or 2) | 3.785 | 2.018 | 7.100 | 0.000 |
| Tumor Type (Intestinal vs Diffuse) | 1.284 | 0.680 | 2.430 | 0.440 |
| Patient age | 1.019 | 0.995 | 1.040 | 0.120 |
| Patient gender | 1.725 | 0.944 | 3.150 | 0.076 |
| Adjunctive chemotherapy | 0.524 | 0.244 | 1.120 | 0.097 |
| Adjunctive radiation therapy | 0.988 | 0.427 | 2.290 | 0.980 |
Multivariate analysis. Cytoplasmic p27 localization and low nuclear p27 expression are associated with high cancer-specific mortality in gastric cancer. Advanced tumor stage is also significantly associated with poor prognosis. CI, confidence interval. NA, not applicable
DISCUSSION
Chronic gastritis induced by H. pylori infection is well established as an etiologic factor underlying gastric cancer, (Forman and Burley 2006, Parkin 2006, Wroblewski et al 2010) but the molecular mechanisms whereby this bacterium facilitates neoplastic transformation have not been not fully elucidated. Our results demonstrate that infection of gastric cancer cells with H. pylori leads to phosphorylation of p27 at T157 and T198, resulting in the mislocalization of p27 into the cytoplasm. Cytoplasm p27 accumulation was also observed in a majority of primary human gastric cancer specimens assayed. Our findings suggest that these post-translational effects of H. pylori on p27 may be of pathogenic significance in gastric carcinogenesis.
Cytoplasmic p27 localization has been reported in several tumor types, in association with a particularly poor prognosis (Duncan et al, Hidaka et al 2009, Li et al 2006, Liang et al 2002, Shin et al 2002, Viglietto et al 2002) and reviewed in (Chu et al 2008). While the canonical role of p27 is as a tumor suppressor, interacting with nuclear cyclin-dependent kinases (especially CDK2) to inhibit cell cycle progression and tumorigenesis (Chu et al 2008), recent work indicates that this tumor suppressor can play a pro-oncogenic role when mislocalized to the cytoplasm. However, an oncogenic function for cytoplasmic p27 has been suggested based on the demonstration that cytoplasmic p27 interacts with RhoA to alter cytoskeletal function and increase cell migration (Besson et al 2004). Moreover, a genetic model in which p27 deficient in cyclin CDK-binding (p27CK-) was knocked in to a p27 null strain, indicates that cytoplasmic p27 may be pro-oncogenic (Besson et al 2007). In this p27 CK knock-in (p27 CK-) mouse, 4 amino-acid substitutions were created in the CDKN1B gene, such that the resultant mutant p27 protein cannot bind to cyclins or CDKs but its other functions are preserved. The p27 CK- mouse developed tumors in multiple organs.
Certain prokaryotes have devised mechanisms to modulate and dysregulate the cell cycle of the host cells with which they are in contact. Examples include the cyclomodulin family of proteins that have acetyltransferase and protease activities (Jubelin et al 2009). These proteins are translocated inside eukaryotic host cells by diverse bacterial species to induce cell cycle arrest and cyclin-dependent kinase accumulation. In the case of H. pylori, Cag A, a putative oncoprotein, is translocated by the type IV secretion system inside gastric epithelial cells where it may be tyrosine phosphorylated by host cell kinases and then activate multiple signal transduction pathways, eventuating in cell cycle dysregulation and apoptosis (Mimuro et al 2007, Saito et al). Our results show cag-dependent effects on the PI3K/AKT pathway, thus confirming the results of others (Nagy et al 2009, Tabassam et al 2009). In addition we demonstrate that p27 is consequently phosphorylated and mislocalized, thus linking for the first time translocation of a specific bacterial virulence factor to post-translational regulation of a host cell cyclin-dependent kinase inhibitor.
Several groups have demonstrated that p27 is phosphorylated on T157 and T198 by different effectors of PI3K pathway activation including AKT, SKG and RSK1 and these lead to cytoplasmic mislocalization of the protein (Chu et al 2008). It has recently been established that the phosphorylation of p27 at T198 not only stabilizes p27 (Kossatz et al 2006, Liang et al 2007), but also promotes its binding to RhoA leading to loss of actin filament stability and a p27-dependent increase in cell motility (Larrea et al 2009). Whereas there have been no prior reports of cytoplasmic mislocalization of p27 and phosphorylation of p27 at T157 and T198 following experimental exposure of cells to chemical carcinogens in vitro, the present study shows that an etiologic agent, H. pylori, linked to chronic gastritis and ultimately to gastric carcinoma, activates the PI3K/AKT pathway, leading to increased phosphorylations at T157 and T198 and mislocalization of p27 to the cytoplasm. AKT-mediated phosphorylation of T157, which is located within p27’s nuclear localization sequence, has previously been shown to impair p27 nuclear import (Liang et al 2002); an effect that has been ascribed to interference by T157 phosphorylation with the physical interaction between p27 and the importin alpha carrier protein (Shin et al 2005). These events have been shown to mediate increased cell motility and invasion, to promote cyclin D-CDk4 assembly and, by reducing p27 access to nuclear CDks, activate the Cyclin-Cdk2 complexes that drive G1-to-S phase progression.
The functional consequences of H. pylori-induced p27 phosphorylation at T157 and T198 remain to be fully elucidated. In some studies the phosphorylation of T198 has been linked to the promotion of cell migration (Larrea et al 2009, Schiappacassi et al), and invasion (Wu et al 2006) and indeed tumor metastasis (Denicourt et al 2007) and cytoplasmic p27 following serine 10 phosphorylation has been implicated in the regulation of apoptosis (Kajihara et al). Evaluating the expression of p27 in a large series of gastric cancer resections indicates that cytoplasmic p27 mislocalization is common in gastric cancer and that cytoplasmic p27 expression is an independent marker of poor prognosis in patients following resective gastric surgery, consistent with observations in some other tumor types (Duncan et al, Hidaka et al 2009, Li et al 2006, Liang et al 2002, Shin et al 2002, Viglietto et al 2002). Interestingly, the association of cytoplasmic p27 expression with adverse outcome was evident for tumors expressing “normal” (high) nuclear expression and for tumors that had lost nuclear p27 expression. These results suggest that cytoplasmic p27 expression promotes a more aggressive gastric cancer phenotype in vivo, and imply that consideration of cytoplasmic p27 status might be of clinical utility, for example in choosing to select adjuvant therapy following potentially curative surgery in this disease.
In conclusion, cag-dependent activation of the PI3K/AKT pathway is implicated in phosphorylation of p27 at T157 and T198 by H. pylori, causing cytoplasmic p27 mislocalization in gastric cancer cells. This is of pathophysiological importance and clinical relevance given the important role of H. pylori in the development of gastric cancer, the frequent occurrence of cytoplasmic p27 expression in gastric cancer and the association of cytoplasmic p27 expression with adverse clinical outcome. Further studies are needed to define how the mislocalization of p27 into the cytoplasm by H. pylori promotes oncogenic cellular behavior in gastric cancer.
MATERIALS AND METHODS
Cell lines and reagents
Human gastric epithelial lines used were: AGS (CRL-1739) from American Type Culture Collection (Manassas, VA), and MKN-28 (JCRB0253) and MKN-45 (JCRB0254); both from Japan Health Sciences Foundation, Tokyo, Japan. Cells were maintained in Ham’s F12 medium (AGS) or RPMI 1640 (MKN-28 and MKN-45) from Lonza, Biologics Inc., Hopkinton, MA) with 10% fetal bovine serum (Sigma, St. Louis, MO) in 5% CO2 at 37°C. LY294002 (Cell Signaling Technology, Beverly, MA) was used at 20μM.
H. pylori strains and culture conditions
Wild type H. pylori strain 60190 (ATCC #49503) was from American Type Culture Collection and its isogenic cagA cagE, cagL and VacA negative mutants were provided by Richard Peek (Vanderbilt University, Nashville, TN). Strain NCTC11639 and its isogenic cagE, PAI and OipA negative mutants were provided by Yoshio Yamaoka (Baylor College of Medicine, Houston, TX). H. pylori was grown on trypticase soy agar with 5% sheep blood cells (BD Biosciences, San Jose, CA) at 37°C in 5% CO2, as described previously (Shirin et al 1999). H. pylori were added to gastric cells that had been serum-starved for 16 hours and co-cultured at a ratio of 100:1 unless otherwise stated.
Protein extraction and Western blot analysis
Cells were harvested, washed twice with cold PBS, and centrifuged. The cell pellet was lysed in NP40 cell lysis buffer (Invitrogen) for 30 minutes on ice, vortexing every 10 minutes. After centrifugation at 13,000 rpm for 10 minutes the supernatant was saved for immunoblotting. For subcellular fractionation, nuclear and cytoplasmic extracts were separated using the NE-PER Nuclear and Cytoplasmic Extraction Kit (Thermo Scientific Waltham, MA) according to manufacturer’s instructions. All buffers included 1x protease and phosphatase inhibitor cocktail (Thermo Fisher Scientific). Protein concentrations were determined using the BCA Protein Assay (Thermo Scientific).
For western blotting, 20 μg protein samples were separated on SDS-PAGE and transferred to a polyscreen PVDF membrane (PerkinElmer Life Sciences Inc., Boston, MA). The antibodies used were: p27kip1 antibody (clone 57) (BD Bioscience) at 1:2500, phospho-p27 T157 and T198 (R&D Systems, Minneapolis, MN), each at 1:10,000, total AKT (used at 1:2500) or phospho-AKT (S473) (1:2500) from Cell Signaling Technology. Lamin A (1:2500) and α-tubulin (1:2500) antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA). GAPDH (1:10,000) antibody from Millipore (Billerica, MA) and β-actin antibody (1:20,000) from Sigma. Horseradish peroxidase conjugated secondary antibodies were from Santa Cruz. Immune detection was performed by enhanced chemiluminescence (Perkin-Elmer Life Sciences Inc., Boston, MA). Amersham ECL Advance Western Blotting Detection Kit was used for detection of phospho-p27 T157 and T198 (GE Healthcare, Piscataway, NJ). Numerical data were derived by densitometry, using ImageJ 1.42q software (NIH, Bethesda, Maryland).
Immunofluorescence assays
AGS cells were seeded in eight-well chamber slides (Thermo Scientific, Rochester, NY) and co-cultured with H. pylori for 10 hours. Epithelial cells were then fixed with 4% paraformaldehyde for 10 minutes and permeabilized with 0.1% triton X-100 for 15 minutes. Endogenous p27 was identified by primary mouse anti-p27 antibody (Cat. # 18-2370, Invitrogen, Carlsbad, CA), with Alexa Fluor 594 rabbit anti-mouse secondary antibody (Invitrogen #A-11062). Nuclei were labeled using Vectashield mounting medium with DAPI (Vector Laboratories, Burlingame, CA). Confocal images were acquired with a Nikon C1si confocal microscope (Nikon Inc., Mellville, NY) using diode lasers 402 and 561. Serial optical sections were performed with EZ-C1 computer software (Nikon Inc.) and Z-series sections collected at 0.2μm with a 60x PlanApo lens and scan zoom of 2. Images were processed in Elements computer software (Nikon Inc.).
Plasmids and small interfering RNA (siRNA)
A series of plasmids encoding wild type and mutant p27 YFP fusion proteins were utilized (Larrea et al 2008). The p27 mutants included a threonine to alanine substitution at position 157 (p27T157A), an aspartic acid substitution of the same residue (p27T157D), a threonine to alanine substitution at position 198 (p27T198A), an aspartic acid substitution of the same residue (p27T198D), and a double threonine to alanine substitution (p27T157AT198A). YFP-p27 constructs were subcloned into the pCMV-Myc-tag plasmid (Agilent Technologies, Marlborough, MA) using PCR Cloning, In-Fusion Advantage Kits (Clontech, Mountain View, CA). SKP2 (Cat. # AM51331) and AKT (Cat. # 6511) siRNAs were purchased from Ambion (Austin, TX) and Cell Signaling respectively
Transient transfection of plasmid and siRNA
AGS cells were transiently transfected with p27 plasmids and siRNA using GenJet Plus DNA In Vitro Tranfection reagent and GenMute siRNA & DNA Transfection Reagents (both from SignaGen Laboratories, Gaithersburg, MD).
Gastric cancer cases and p27 immunohistochemistry
Human gastric cancer tissue microarrays were constructed with four tumor tissue cores per case from surgically resected gastric cancers (Resnick et al 2005). Five histologically normal gastric biopsies and 5 cases of intestinal metaplasia were also included. Four-micron sections were immunostained using mouse anti-p27 antibody (clone 57, 1:100 dilution, Invitrogen) and the DAB Map Kit on an automated immunohistochemistry platform (Ventana Medical Systems Inc, Tuscon, AZ). The extent (0–100%) and intensity (scale 0 –3) of p27 immunostaining was evaluated separately for nucleus and cytoplasm of tumor cells by KS and MBR. Scores from all 4 cores per case were averaged for overall extent and intensity. Because there is no standard scoring system for p27 immunostaining (Wander et al) cytoplasmic p27 was scored as high (intensity scale >1) or low (intensity scale ≤1), and levels of nuclear p27 were scored as high (≥52%) or low (<52%). These cut-offs were based on regression tree analysis, and in accordance with previous studies(Liang et al 2002). Patient demographics, tumor histology, stage, adjuvant therapy, and recurrence and survival data were ascertained through the Rhode Island Tumor Registry and hospital chart review. This study was approved by the Institutional Review Board of Rhode Island Hospital.
Statistical methods
All cell culture experiments were repeated at least 3 times and paired t-tests were used to determine statistical significance (GraphPad Prism, Graphpad Software Inc., La Jolla, CA). Regression tree analysis was used to identify subsets and conditions by which p27 expression could be used to predict survival. We estimated the survival distributions using the Kaplan-Meier method and tested for equivalence of distributions using the log rank test. A multivariate Cox proportional hazards regression model was used to examine the association of nuclear and cytoplasmic p27 expression with survival, adjusting for demographic, tumor specific and adjuvant therapy variables; namely age, gender, tumor stage, tumor histological subtype, and whether or not the patient had undergone adjuvant chemotherapy or radiation therapy.
Acknowledgments
Grant Support: NIH R01CA111533 (SFM); NIH P20RR17695 (MBR)
We appreciate the help of Virginia Hovanesian for technical assistance with confocal microscopy, and Leila Noble and Rose Tavares for immunohistochemistry. The study was funded by U S Public Health Service Grants NIH R01CA111533 (to SFM) and NIH P20RR17695 (to MBR).
Footnotes
Conflict of interest: No competing financial interests
References
- Besson A, Gurian-West M, Schmidt A, Hall A, Roberts JM. p27Kip1 modulates cell migration through the regulation of RhoA activation. Genes Dev. 2004;18:862–876. doi: 10.1101/gad.1185504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besson A, Hwang HC, Cicero S, Donovan SL, Gurian-West M, Johnson D, et al. Discovery of an oncogenic activity in p27Kip1 that causes stem cell expansion and a multiple tumor phenotype. Genes Dev. 2007;21:1731–1746. doi: 10.1101/gad.1556607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu IM, Hengst L, Slingerland JM. The Cdk inhibitor p27 in human cancer: prognostic potential and relevance to anticancer therapy. Nat Rev Cancer. 2008;8:253–267. doi: 10.1038/nrc2347. [DOI] [PubMed] [Google Scholar]
- Denicourt C, Saenz CC, Datnow B, Cui XS, Dowdy SF. Relocalized p27Kip1 tumor suppressor functions as a cytoplasmic metastatic oncogene in melanoma. Cancer Res. 2007;67:9238–9243. doi: 10.1158/0008-5472.CAN-07-1375. [DOI] [PubMed] [Google Scholar]
- Duncan TJ, Al-Attar A, Rolland P, Harper S, Spendlove I, Durrant LG. Cytoplasmic p27 expression is an independent prognostic factor in ovarian cancer. Int J Gynecol Pathol. 29:8–18. doi: 10.1097/PGP.0b013e3181b64ec3. [DOI] [PubMed] [Google Scholar]
- Eguchi H, Carpentier S, Kim SS, Moss SF. P27Kip1 regulates the apoptotic response of gastric epithelial cells to Helicobacter pylori. Gut. 2004;53:797–804. doi: 10.1136/gut.2003.032144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feakins RM, Mulcahy HE, Quaglia A, Jawhari A, Zhang Z, Patchett SE. p27(Kip1) loss does not predict survival in patients with advanced gastric carcinoma. Cancer. 2000;89:1684–1691. doi: 10.1002/1097-0142(20001015)89:8<1684::aid-cncr6>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- Fero ML, Randel E, Gurley KE, Roberts JM, Kemp CJ. The murine gene p27Kip1 is haplo-insufficient for tumour suppression. Nature. 1998;396:177–180. doi: 10.1038/24179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forman D, Burley VJ. Gastric cancer: global pattern of the disease and an overview of environmental risk factors. Best Pract Res Clin Gastroenterol. 2006;20:633–649. doi: 10.1016/j.bpg.2006.04.008. [DOI] [PubMed] [Google Scholar]
- Fuccio L, Zagari RM, Eusebi LH, Laterza L, Cennamo V, Ceroni L, et al. Meta-analysis: can Helicobacter pylori eradication treatment reduce the risk for gastric cancer? Ann Intern Med. 2009;151:121–128. doi: 10.7326/0003-4819-151-2-200907210-00009. [DOI] [PubMed] [Google Scholar]
- Hartgrink HH, Jansen EP, van Grieken NC, van de Velde CJ. Gastric cancer. Lancet. 2009;374:477–490. doi: 10.1016/S0140-6736(09)60617-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hidaka T, Hama S, Shrestha P, Saito T, Kajiwara Y, Yamasaki F, et al. The combination of low cytoplasmic and high nuclear expression of p27 predicts a better prognosis in high-grade astrocytoma. Anticancer Res. 2009;29:597–603. [PubMed] [Google Scholar]
- Hong F, Larrea MD, Doughty C, Kwiatkowski DJ, Squillace R, Slingerland JM. mTOR-raptor binds and activates SGK1 to regulate p27 phosphorylation. Mol Cell. 2008;30:701–711. doi: 10.1016/j.molcel.2008.04.027. [DOI] [PubMed] [Google Scholar]
- Jubelin G, Chavez CV, Taieb F, Banfield MJ, Samba-Louaka A, Nobe R, et al. Cycle inhibiting factors (CIFs) are a growing family of functional cyclomodulins present in invertebrate and mammal bacterial pathogens. PLoS One. 2009;4:e4855. doi: 10.1371/journal.pone.0004855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajihara R, Fukushige S, Shioda N, Tanabe K, Fukunaga K, Inui S. CaMKII phosphorylates serine 10 of p27 and confers apoptosis resistance to HeLa cells. Biochem Biophys Res Commun. 401:350–355. doi: 10.1016/j.bbrc.2010.09.051. [DOI] [PubMed] [Google Scholar]
- Kim SS, Meitner P, Konkin TA, Cho YS, Resnick MB, Moss SF. Altered expression of Skp2, c-Myc and p27 proteins but not mRNA after H. pylori eradication in chronic gastritis. Mod Pathol. 2006;19:49–58. doi: 10.1038/modpathol.3800476. [DOI] [PubMed] [Google Scholar]
- Kossatz U, Vervoorts J, Nickeleit I, Sundberg HA, Arthur JS, Manns MP, et al. C-terminal phosphorylation controls the stability and function of p27Kip1. EMBO J. 2006;25:5159–5170. doi: 10.1038/sj.emboj.7601388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzushita N, Rogers AB, Monti NA, Whary MT, Park MJ, Aswad BI, et al. p27Kip1 deficiency confers susceptibility to gastric carcinogenesis in Helicobacter pylori-infected mice. Gastroenterology. 2005;129:1544–1556. doi: 10.1053/j.gastro.2005.07.056. [DOI] [PubMed] [Google Scholar]
- Larrea MD, Liang J, Da Silva T, Hong F, Shao SH, Han K, et al. Phosphorylation of p27Kip1 regulates assembly and activation of cyclin D1-Cdk4. Mol Cell Biol. 2008;28:6462–6472. doi: 10.1128/MCB.02300-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larrea MD, Hong F, Wander SA, da Silva TG, Helfman D, Lannigan D, et al. RSK1 drives p27Kip1 phosphorylation at T198 to promote RhoA inhibition and increase cell motility. Proc Natl Acad Sci U S A. 2009;106:9268–9273. doi: 10.1073/pnas.0805057106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Wheeler TM, Dai H, Sayeeduddin M, Scardino PT, Frolov A, et al. Biological correlates of p27 compartmental expression in prostate cancer. J Urol. 2006;175:528–532. doi: 10.1016/S0022-5347(05)00151-5. [DOI] [PubMed] [Google Scholar]
- Liang J, Zubovitz J, Petrocelli T, Kotchetkov R, Connor MK, Han K, et al. PKB/Akt phosphorylates p27, impairs nuclear import of p27 and opposes p27-mediated G1 arrest. Nat Med. 2002;8:1153–1160. doi: 10.1038/nm761. [DOI] [PubMed] [Google Scholar]
- Liang J, Shao SH, Xu ZX, Hennessy B, Ding Z, Larrea M, et al. The energy sensing LKB1-AMPK pathway regulates p27(kip1) phosphorylation mediating the decision to enter autophagy or apoptosis. Nat Cell Biol. 2007;9:218–224. doi: 10.1038/ncb1537. [DOI] [PubMed] [Google Scholar]
- Mimuro H, Suzuki T, Nagai S, Rieder G, Suzuki M, Nagai T, et al. Helicobacter pylori dampens gut epithelial self-renewal by inhibiting apoptosis, a bacterial strategy to enhance colonization of the stomach. Cell Host Microbe. 2007;2:250–263. doi: 10.1016/j.chom.2007.09.005. [DOI] [PubMed] [Google Scholar]
- Mori M, Mimori K, Shiraishi T, Tanaka S, Ueo H, Sugimachi K, et al. p27 expression and gastric carcinoma. Nat Med. 1997;3:593. doi: 10.1038/nm0697-593. [DOI] [PubMed] [Google Scholar]
- Nagy TA, Frey MR, Yan F, Israel DA, Polk DB, Peek RM., Jr Helicobacter pylori regulates cellular migration and apoptosis by activation of phosphatidylinositol 3-kinase signaling. J Infect Dis. 2009;199:641–651. doi: 10.1086/596660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkin DM. The global health burden of infection-associated cancers in the year 2002. Int J Cancer. 2006;118:3030–3044. doi: 10.1002/ijc.21731. [DOI] [PubMed] [Google Scholar]
- Resnick MB, Gavilanez M, Newton E, Konkin T, Bhattacharya B, Britt DE, et al. Claudin expression in gastric adenocarcinomas: a tissue microarray study with prognostic correlation. Hum Pathol. 2005;36:886–892. doi: 10.1016/j.humpath.2005.05.019. [DOI] [PubMed] [Google Scholar]
- Saito Y, Murata-Kamiya N, Hirayama T, Ohba Y, Hatakeyama M. Conversion of Helicobacter pylori CagA from senescence inducer to oncogenic driver through polarity-dependent regulation of p21. J Exp Med. 207:2157–2174. doi: 10.1084/jem.20100602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiappacassi M, Lovisa S, Lovat F, Fabris L, Colombatti A, Belletti B, et al. Role of T198 Modification in the Regulation of p27 Protein Stability and Function. PLoS One. 6:e17673. doi: 10.1371/journal.pone.0017673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin I, Yakes FM, Rojo F, Shin NY, Bakin AV, Baselga J, et al. PKB/Akt mediates cell-cycle progression by phosphorylation of p27(Kip1) at threonine 157 and modulation of its cellular localization. Nat Med. 2002;8:1145–1152. doi: 10.1038/nm759. [DOI] [PubMed] [Google Scholar]
- Shin I, Rotty J, Wu FY, Arteaga CL. Phosphorylation of p27Kip1 at Thr-157 interferes with its association with importin alpha during G1 and prevents nuclear re-entry. J Biol Chem. 2005;280:6055–6063. doi: 10.1074/jbc.M412367200. [DOI] [PubMed] [Google Scholar]
- Shirin H, Sordillo EM, Oh SH, Yamamoto H, Delohery T, Weinstein IB, et al. Helicobacter pylori inhibits the G1 to S transition in AGS gastric epithelial cells. Cancer Res. 1999;59:2277–2281. [PubMed] [Google Scholar]
- Shirin H, Sordillo EM, Kolevska TK, Hibshoosh H, Kawabata Y, Oh SH, et al. Chronic Helicobacter pylori infection induces an apoptosis-resistant phenotype associated with decreased expression of p27(kip1) Infect Immun. 2000;68:5321–5328. doi: 10.1128/iai.68.9.5321-5328.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabassam FH, Graham DY, Yamaoka Y. Helicobacter pylori activate epidermal growth factor receptor- and phosphatidylinositol 3-OH kinase-dependent Akt and glycogen synthase kinase 3beta phosphorylation. Cell Microbiol. 2009;11:70–82. doi: 10.1111/j.1462-5822.2008.01237.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vervoorts J, Luscher B. Post-translational regulation of the tumor suppressor p27(KIP1) Cell Mol Life Sci. 2008;65:3255–3264. doi: 10.1007/s00018-008-8296-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viglietto G, Motti ML, Bruni P, Melillo RM, D’Alessio A, Califano D, et al. Cytoplasmic relocalization and inhibition of the cyclin-dependent kinase inhibitor p27(Kip1) by PKB/Akt-mediated phosphorylation in breast cancer. Nat Med. 2002;8:1136–1144. doi: 10.1038/nm762. [DOI] [PubMed] [Google Scholar]
- Wander SA, Zhao D, Slingerland JM. p27: a barometer of signaling deregulation and potential predictor of response to targeted therapies. Clin Cancer Res. doi: 10.1158/1078-0432.CCR-10-0752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wroblewski LE, Peek RM, Jr, Wilson KT. Helicobacter pylori and gastric cancer: factors that modulate disease risk. Clin Microbiol Rev. 2010;23:713–739. doi: 10.1128/CMR.00011-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu FY, Wang SE, Sanders ME, Shin I, Rojo F, Baselga J, et al. Reduction of cytosolic p27(Kip1) inhibits cancer cell motility, survival, and tumorigenicity. Cancer Res. 2006;66:2162–2172. doi: 10.1158/0008-5472.CAN-05-3304. [DOI] [PubMed] [Google Scholar]
- Yasui W, Kudo Y, Semba S, Yokozaki H, Tahara E. Reduced expression of cyclin-dependent kinase inhibitor p27Kip1 is associated with advanced stage and invasiveness of gastric carcinomas. Jpn J Cancer Res. 1997;88:625–629. doi: 10.1111/j.1349-7006.1997.tb00428.x. [DOI] [PMC free article] [PubMed] [Google Scholar]