

Abstract
Aims
Rapid eye movement (REM) sleep behaviour disorder (RBD) is characterized by loss of muscle atonia during REM sleep and is associated with dream enactment behaviour. RBD is often associated with α-synuclein pathology, and we examined if there is a relationship of RBD with cholinergic neuronal loss in the pedunculopontine/laterodorsal tegmental nucleus (PPN/LDT), compared to catecholaminergic neurons in a neighbouring nucleus, the locus coeruleus (LC).
Methods
This retrospective study, utilized human brain banked tissues of 11 Lewy body disease (LBD) cases with RBD, 10 LBD without RBD, 19 AD and 10 neurologically normal controls. Tissues were stained with choline acetyl transferase immunohistochemistry to label neurons of PPN/LDT and tyrosine hydroxylase for the LC. The burden of tau and α-synuclein pathology was measured in the same regions with immunohistochemistry.
Results
Both the LC and PPN/LDT were vulnerable to α-synuclein pathology in LBD and tau pathology in AD, but significant neuronal loss was only detected in these nuclei in LBD. Greater cholinergic depletion was found in both LBD groups, regardless of RBD status, when compared with normals and AD. There were no differences in either degree of neuronal loss or burden of α-synuclein pathology in LBD with and without RBD.
Conclusions
Whether decreases in brainstem cholinergic neurons in LBD contribute to RBD is uncertain, but our findings indicate these neurons are highly vulnerable to α-synuclein pathology in LBD and tau pathology in AD. The mechanism of selective α-synuclein-mediated neuronal loss in these nuclei remains to be determined.
Keywords: α-synuclein, cholinergic, Lewy body, laterodorsal tegmentum, locus coeruleus, pedunculopontine nucleus, REM behaviour disorder, tau
Introduction
Sleep disturbances are among the most common of the non-motor manifestations of Lewy body disease (LBD) [1]. Rapid eye movement (REM) sleep behaviour disorder (RBD), which is characterized by loss of muscle atonia during REM sleep and associated dream enactment behaviour, represents one such manifestation. RBD has been found to be an early clinical feature of Parkinson disease or dementia with Lewy bodies, often predating the onset of parkinsonism or dementia by many years[2]. In contrast, RBD is uncommon in Alzheimer’s disease (AD) [3]. LBD is characterized pathologically by cytoplasmic deposits of α-synuclein within Lewy bodies (LBs) and Lewy neurites [4], and anatomically by LBs in subcortical and cortical regions [5, 6]. The microtubule associated protein tau, the major molecular constituent of neurofibrillary tangles (NFTs) [7] in AD, is associated with a subset of LBs [8]. In addition, Alzheimer type pathology is frequent in LBD [9].
The pedunculopontine/laterodorsal tegmental nucleus (PPN/LDT) contains cholinergic neurons that are active during REM sleep [10–12]. Pharmacological and lesion studies of cats have shown that damage to the mesopontine region, including the PPN/LDT, is associated with RBD-like features [10, 13, 14]. Furthermore, human lesion studies suggest impaired function of neurons in the dorsal pontine tegmentum may provide the basis for loss of muscle atonia during REM sleep; the distribution of these lesions overlap considerably with vulnerable neurons in LBD [15, 16]. It is not clear, however, whether the PPN/LDT itself is partially or fully responsible for REM sleep muscle atonia and the behavioural manifestations of RBD. In LBD, cholinergic neurons in the PPN/LDT are depleted [17], in both LBD with and without RBD [18]. These studies, however, did not take into account possible interaction of tau and α-synuclein pathology and the role of concurrent Alzheimer type pathology in LBD. The catecholaminergic neurons of the locus coeruleus (LC) are in the vicinity of the PPN/LDT, and are known to degenerate in both LBD and AD, although degeneration is age- and location-dependent [19–21]. The LC is a key component of the ascending reticular activating system, and it also heavily innervates the PPN/LDT. The roles of the PPN/LDT and LC in humans are not entirely clear, but have been thought to be involved in the control of sleep transitions into and out of REM sleep [15, 22, 23].
To gain a better understanding of the molecular underpinnings of RBD, we examined whether proportion of cholinergic neurons in the PPN/LDT and found that catecholaminergic neurons in the LC were different in LBD with and without RBD. We also examined whether the burden of tau and α-synuclein pathology differed between the two groups. Neurologically normal individuals and AD without RBD served as controls.
Materials and methods
Cases
Autopsy material was obtained from 51 subjects, most of whom were enrolled in the Mayo Clinic Alzheimer’s Disease Research Center. A summary of the demographics is provided in Table 1. Allpatients or their proxies had signed informed consent for autopsy according to the Institutional Review Board guidelines. Cases were excluded if they had any significant degree of other pathological processes that might have contributed to neuronal loss or tau pathology (e.g., cerebrovascular disease and argyrophilic grain disease).
Table 1.
Summary of clinical and pathological characteristics.
| Normal | AD | LBD No RBD | LBD RBD | |
|---|---|---|---|---|
| N | 11 | 19 | 10 | 11 |
| Male (%) | 72% | 42% | 70% | 72% |
| PSG (%) | 27% | 31% | 30% | 45% |
| PMI (mean hr ± SDev) | 17.6 ± 9 | 11.1 ± 9 | 12.4 ± 10 | 12.3 ± 10 |
| Time in formalin (mean wk ± SDev) | 7.2 ± 5 | 5.1 ± 4 | 6.0 ± 4 | 3.7 ± 5 |
| Age (mean yr ± SDev) | 75.4 ± 5 | 79.8 ± 11 | 81.1 ± 4 | 79.2 ± 10 |
| Illness duration (mean yr ± SDev) | n/a | 7.9 ± 2 | 8.4 ± 4 | 8.4 ± 4 |
| Braak NFT stage (median, range) | II (I–III) | VI (IV–VI)* | III (II–VI) | III (II–VI) |
| LBD type | ||||
| Brainstem (%) | n/a | n/a | 10% | 0 |
| Limbic (Transitional) (%) | n/a | n/a | 60% | 64% |
| Neocortical (Diffuse) (%) | n/a | n/a | 30% | 36% |
No RBD- without RBD, RBD- with RBD, NFT- neurofibrillary tangle, PSG-Polysomnography conducted, LBD- Lewy body disease, PMI- post mortem interval
AD cases have significantly higher Braak NFT staging when compared to normals and LBD groups
Definitions/diagnoses
Pathological methods proposed by Kosaka and colleagues were used and LBD were classified as brainstem predominant, limbic (transitional) or neocortical (diffuse), accordingly [5]. AD was diagnosed according to NIA-Reagan guidelines [24]. Information regarding the presence or absence of clinically probable RBD was obtained on each patient based on the clinical documentation of presence or absence of dream enactment behaviour during sleep, from the Mayo Sleep Questionnaire (MSQ), or both. The core question on the MSQ regarding recurrent dream enactment behaviour during sleep has 98% sensitivity and 74% specificity for definite RBD based on a polysomnography (PSG) validation study [25]. Polysomnography was carried out when possible to confirm RBD, using established criteria by board certified sleep specialists [26]. Cases without RBD had no historical, MSQ or PSG evidence of RBD. There were 11 LBD with RBD (8 males) and 10 LBD without RBD (7 males). In the RBD group, 5 had polysomnography-confirmed RBD. In LBD without RBD group, 3 had overnight PSG, which confirmed the presence of normal atonia during REM sleep. The AD group included 19 cases (8 males) with a documented history of an absence of dream enactment behaviour during sleep and confirmation of normal REM sleep atonia in 6 individuals. The neurologically normal control group included 11 cases (8 men) with documentation of an absence of dream enactment behaviour during sleep and confirmation of normal REM sleep atonia in 3 cases. There were no differences in age, sex, time in formalin fixation till processing, or post mortem interval between all cohorts; and no differences with disease duration between AD and LBD groups (see Table 1). As expected, AD cases had higher Braak NFT stages compared to normal and LBD groups [24]. There were no significant differences in distribution of LBD type; however, LBD with RBD did not include any brainstem predominant cases [5].
Immunohistochemistry
Based on the retrospective nature of the study, whole brainstems were not available for stereological analysis. To compensate for possible rostral/caudal differences and to understand the nuclei as a whole, three 5-μm thick paraffin sections at least 20-μm a part were analyzed for each stain and in each nucleus. Nuclei were identified by anatomical landmarks as determined by an atlas of the human brainstem [27]. Paraffin blocks were obtained from the pons at the level of the inferior colliculus and the decussation of the superior cerebellar peduncle for the PPN/LDT, and at the level of the isthmus for the locus coeruleus (LC). After removal of paraffin, sections were rehydrated using a graded series of alcohols. Sections were steamed in distilled water for 30 minutes then subjected to uniform processing for immunoreactivity via a DAKO Autostainer (DAKO, Carpinteria CA) for either choline acetyl transferase (ChAT, goat polyclonal, 1:500, Chemicon Billerica, MA) or tyrosine hydroxylase (TH, rabbit polyclonal, 1:600, Affinity Bioreagents, Golden, CO) to identify the PPN/LDT and LC, respectively. For both nuclei, α-synuclein (LB509, mouse monoclonal, 1:100, Zymed, San Francisco, CA) and tau (CP13, mouse monoclonal, 1:1000 provided by Peter Davies, Albert Einstein college of Medicine) were used to assess the burden of α-synuclein and tau pathology. The chromogen was 3, 3 diaminobenzidine (DAB, DAKO) for all antibodies. Immunoreactive neuronswere identified under bright-field illumination by a characteristic brown reaction product that densely fills theperikarya and their processes. Omission of the primary antibodyor incubation with normal sera resulted in lack of immunostaining. All sections were counterstained with haematoxylin to identify surroundingstructures, and to determine whether loss of immunoreactivity reflected neuronal loss or lack of expression of the antigen.
Image analysis and quantitation
All slides were digitally scanned using the brightfield scanning console, ScanScope XT (Aperio, Vista, CA). ImageScope software (Aperio, Vista, CA) was used to view and analyze all stained sections. Borders for the PPN/LDT, due to its diffuse nature, were standardize across all cohorts using anatomical landmarks to avoid bias due to cell loss. The PPN/LDT landmarks were the ventromedial border of the inferior colliculus and dorsolateral border of the decussation of the superior cerebellar peduncle. Presence of ChAT staining aided in the confirmation of identification of the PPN/LDT. For the LC, the fourth ventricle and medial border of the superior cerebellar peduncle served as boundaries along with the presence of neuromelanin pigment in LC neurons. All objects (neurons, LBs, or NFTs) were counted that lay within the traced border of the region (LC or PPN/LDT). Neurons were counted based upon their distinct polarity and presence of a nucleolus within the plane of section and were sub-classified as to whether they were positive or negative for ChAT (in the PPN/LDT) or TH (in the LC). Presence of a NFT or a LB within the neuron was also recorded [28]. Once counts were complete, all raw data was then divided by the total number of neurons in that area to generate a proportion, which was expressed as percentage.
For quantitation of total α-synuclein and tau immunoreactivity (neuronal and extra-somal), a modified positive pixel count algorithm was utilized on the same traced areas used for counts (Aperio Technologies, Inc, Vista, CA). To parcellate DAB positive staining from neuromelanin or other similar hues (e.g., lipofuscin), the algorithm was adjusted to consider DAB positive staining as a strong positive and other hues as medium positive. These data were then divided by total pixels (representing the entire traced area) then multiplied by 100 to yield percent burden. Similar methods have been published elsewhere [29]. The same investigator, blinded tothe clinical and neuropathological diagnoses, performedall counts/analyzes.
Statistical analysis
Statistical analyses and graphs were performed with Sigma Plot 11.0 (Systat Software, Inc., San Jose, CA). Group differences in LC and PPN/LDT counts were not normally distributed and did not show equal variance; therefore, analyses were preformed using non-parametric methods. The relationship of counts (neurons, LBs or NFT) and burden (tau or α-synuclein) to diagnostic group (normal, AD, LBD with and without RBD) was analyzed using Kruskal-Wallis One Way Analysis of Variance on Ranks followed by Dunn’s post hoc method to detect significant differences. Spearman correlations were used to explore relationships between neuronal phenotypes (TH, ChAT, LB or NFT) and burdens (α-synuclein and tau); with no statistical significant correlations noted. Logistic regression of neuron counts and tau/α-synuclein measures in relations to RBD status were also done, again no statistical significant correlations noted. Kruskal-Wallis one-way analysis of variance on ranks was used to determineif age of death, gender, disease duration, postmortem delay, clinical procedure for RBD diagnosis (history, PSG or sleep questionnaire), or time in formalin fixation had any effect on counts. The criterion for significant difference is P <0.05.
Results
Neuronal phenotypes in the PPN/LDT and LC
In triplicate mesopontine sections stained for ChAT, we found a significant reduction in the percentage of cholinergic neurons in LBD compared to normal controls and AD (p < 0.004). The PPN/LDT cholinergic neuronal densities did not differentiate LBD groups with and without RBD (Figure 1). There were no differences in cholinergic neuronal densities in the PPN/LDT between AD and normal controls. There were no group differences for non-ChAT neurons in the PPN/LDT.
Figure 1.
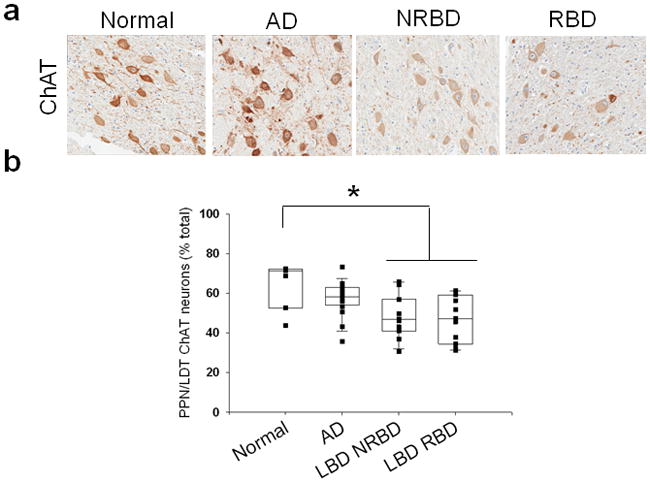
a. ChAT staining in the PPN/LDT of normal, AD, and LBD cases without RBD (LBD NRBD) and with RBD (LBD RBD). Photos taken at 20x. b. quantification of percentages of ChAT positive neurons out of total neurons in the PPN/LDT. * LBD RBD contained significantly less ChAT positive neurons than normals and AD.
In triplicate rostral pontine sections stained for TH, we found significantly lower percentages of catecholaminergic neurons in the LC in LBD compared to AD and normal controls (Figure 2, p < 0.003). Further examination revealed no LBD subgroup differences when compared to AD and normal controls. There was no difference in catecholaminergic neurons between AD and normal controls, and no difference in LC neurons in the LBD groups with and without RBD for catecholaminergic and non-catecholaminergic neurons.
Figure 2.
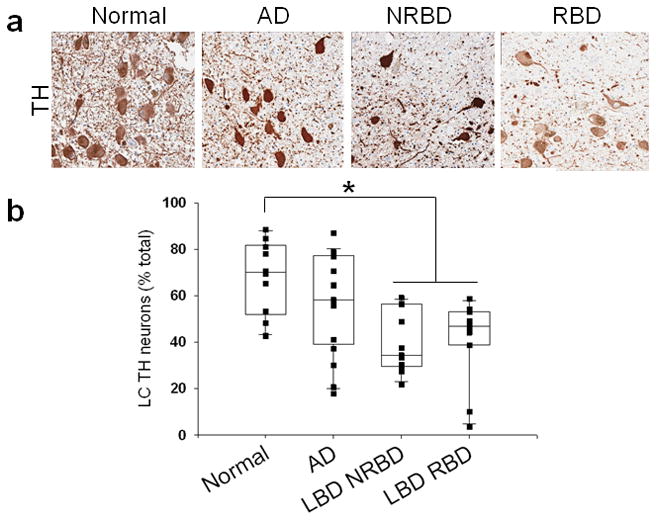
a. TH staining in the LC of normal, AD, and LBD cases without RBD (LBD NRBD) and with RBD (LBD RBD). Photos taken at 20x. b. quantification of percentages of TH positive neurons out of total neurons in the LC. * LBD cohorts contained significantly less TH positive neurons than normals and AD.
Tau and α-synuclein pathology in PPN/LDT and LC
The PPN/LDT and LC were vulnerable to α-synuclein pathology in LBD and tau pathology in AD. There was no difference in tau or α-synuclein measures (counts or burden) in either the PPN/LDT or LC in LBD with and without RBD. In the PPN/LDT, there was a higher percentage of neurons with LBs and with α-synuclein burden in both LBD groups compared to the AD and normal controls (Figure 3, p < 0.001). As expected, AD had greater NFTs and tau burden in the PPN/LDT compared to LBD and normal controls (Figure 3, p <0.001). LBD groups also had higher tau burden in the PPN/LDT, but did not differ in the percentage of neurons with NFTs when compared to normal controls; suggesting pathology differences may only be neuritic in origin (Figure 3, p <0.001). LBD groups had a greater percentage of neurons with LBs and with α-synuclein burden in the LC compared to the AD and normal controls (Figure 4, p < 0.01). There was also greater percentages of neurons with NFTs and greater tau burden in the LC in AD compared to LBD and normal controls (Figure 4, p< 0.01).
Figure 3.
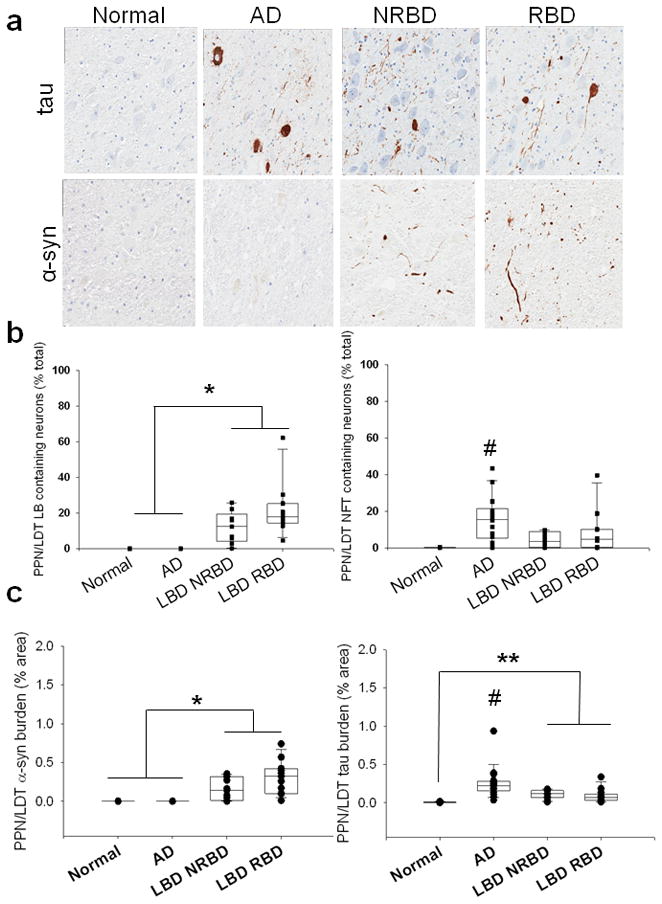
a. Tau and α-synuclein staining in the PPN/LDT of normal, AD, and LBD cases without RBD (LBD NRBD) and with RBD (LBD RBD). Photos taken at 20x. b. Quantification of α-syn (LBs-left) and tau (NFTs- right) intra-neuronal inclusions in the PPN/LDT. c. Quantification of IHC staining (burden) of α-syn (left) and tau (right) in the PPN/LDT. * LBD cohorts had significantly greater LBs/α-syn than normals and AD; # AD had significantly greater NFTs/tau compared to normal and LBD groups. ** LBD cohorts had significantly greater tau compared to normals.
Figure 4.
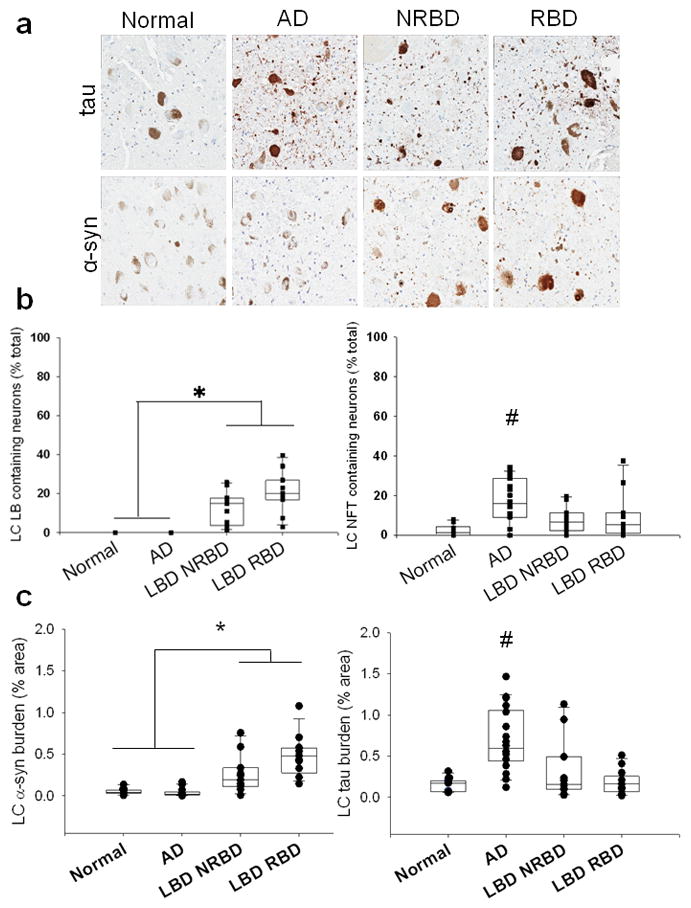
a. Tau and α-synuclein staining in the LC of normal, AD, and LBD cases without RBD (LBD NRBD) and with RBD (LBD RBD). Photos taken at 20x. b. Quantification of α-syn (LBs-left) and tau (NFTs- right) intra-neuronal inclusions in the LC. c. Quantification of IHC staining (burden) of α-syn (left) and tau (right) in the LC. * LBD cohorts had significantly greater LBs/α-syn than normals and AD; # AD had significantly greater NFTs/tau compared to normal and LBD groups.
Discussion
This study used image analysis and neuronal counting methods to assess tau and α-synuclein pathology as well as the percentage of neurons with NFT and LB in anatomically defined nuclei with distinct neurotransmitter profiles (i.e. catecholaminergic for LC and cholinergic for PPN/LDT) in a series of patients evaluated antemortem for clinically probable RBD using clinical history, a validated sleep questionnaire and in a subset of cases, PSG. We found the LC and PPN/LDT were vulnerable to α-synuclein pathology in LBD and tau pathology in AD, but significant neuronal loss was only detected in these nuclei in LBD. The mechanism of selective α-synuclein mediated neuronal loss in these nuclei remains to be determined.
This study supports previous literature demonstrating LBD is associated with significantly fewer cholinergic neurons in the PPN/LDT compared to normal controls and AD[17, 30]. The failure to detect a difference in cholinergic neuronal density between LBD cases with and without RBD in this study and a previous report [18] raises the possibility of heterogeneity of LBD with respect to cholinergic neuronal loss in the PPN/LDT in LBD. Both studies had large variance in PPN/LDT neuronal counts in LBD, and we also found large variance in a neighboring nucleus, the locus coeruleus (Figure 1, personal communication with Schmeichel et al. [18]). It is also possible RBD is a product of pathology at multiple sites in the integrated sleep circuitry, with variable involvement in the different nodes of the network affected differentially in each individual, but producing the same clinical phenotype.
The PPN/LDT and the LC are located within the group of lower brainstem nuclei where lesions that produce an RBD phenotype overlap [15]. Research in cats by Hendricks and colleagues determined that bilateral pontine tegmental lesions resulted in loss of atonia during sleep [10]. These data suggests a particular behavioural syndrome could be produced by a specific lesion. In spite of this insight into behavioural mechanisms, these lesions are acute in nature and do not mimic the chronic insult of a neurodegenerative disease. Further quantitative neuropathological studies in a larger number of patients with prospective clinical assessments are needed to address this hypothesis. There is clearly cholinergic deficiency in LBD as a whole, and demonstrating an additional greater loss in those with RBD will require larger numbers of cases and controls with as many as possible having PSG evaluations or at least information from a validated sleep questionnaire.
Patients with LBD tend to exhibit other clinical features, including daytime somnolence and parkinsonism. Other potential roles of the PPN/LDT should be considered, including arousal, postural control and locomotion [31–33]. We found no correlations within the LBD group on severity of extrapyramidal motor signs using the Unified Parkinson’s Disease Rating Scale [34] or daytime somnolence, utilizing scores from the Epworth Sleepiness Scale-collateral version [35, 36] (data not shown). Previous studies have examined the relation of the PPN/LDT to gait and postural control, with loss of PPN/LDT cholinergic neurons correlating with these disorders [32].
This is the first neuropathological report that measures α-synuclein and tau burden in these nuclei in LBD, as well as AD and neurologically normal controls, in relation to presence or absence of antemortem RBD. The present study is based on stringent cohort selection with respect to clinical assessment of presence or absence of RBD. In addition, cases were matched for severity of concomitant Alzheimer type pathology [37] and for LBD subtype [5], and evaluated with state-of-the-art digital imaging analysis that permits tracing and analysis of the entire region of interest based upon standardized anatomical landmarks. Several limitations should, however, be considered. Being a retrospective anatomical study, formal stereological analysis of the entire region of interest was not possible. It is possible that differences in disease-specific neuronal vulnerability across the span of the LC [21] or PPN/LDT could have contributed to variance in data, limiting our ability to find significant group differences. We attempted to compensate for this by measuring three 5-μm thick paraffin sections at least 20-μm apart for each stain in each nucleus. In support of the validity of limited sampling of brainstem nuclei, a study comparing single section counts of dopaminergic neurons in the substantia nigra to counts derived from the stereological dissector method [38], showed no differences between methods. On the other hand, this may not be applicable for more diffuse nuclei such as the PPN/LDT. Despite this, our findings are in agreement with a previous study, which averaged counts throughout the entire extent of the PPN/LDT [18]. Furthermore, unpublished results from our lab, based upon a larger cohort without a priori matching for severity of Alzheimer type pathology, demonstrated higher concomitant Alzheimer’s pathology in LBD cases without RBD compared to those with RBD (Dugger unpublished observations). Use of stringent cohort selection criteria may have masked true differences on these parameters with respect to RBD status. While this is one of the largest studies to date of LBD, AD and neurologically normal controls with ante-mortem sleep status evaluations by PSG and/or sleep questionnaire, in addition to historical annotations, the sample size is still small, especially for patients with PSG. The PSG is the gold standard for diagnosis of RBD, but PSG is usually used when a patient has a probable diagnosis of a sleep disturbance. This expensive and demanding procedure is seldom performed on neurologically normal controls or patients with AD.
The PPN/LDT is damaged in LBD with and without RBD due to α-synuclein accumulation and cholinergic neuronal loss. Recent lesion studies in animal models and humans suggest involvement of other nuclei, as well as possible afferent and efferent connections of these nuclei in RBD pathogenesis. Other nuclei that need to be studied in the future include the sub-coeruleus region and magnocellular reticular formation [9, 33]. Future research analyzing nuclei implicated in both upstream and downstream connectivity of the PPN/LDT in humans will be crucial for a full understanding of the clinical correlates of PPN/LDT pathology in LBD.
Acknowledgments
The authors thank Tae-Beom Ahn, Monica Castanedes Casey, Linda Rousseau and Virginia Phillips their histopathological expertise and technical support. The authors would also like to thank Ann Schmeichel for reviewing her previously published PPN/LDT data. This research would not be possible without brain donations from patients and their families. This study was supported by grants from the National Institutes of Health (P50 AG16574, P50 NS72187 & R01 AG15866.) and Mayo Foundation for Education & Research.
References
- 1.Comella CL. Sleep disorders in Parkinson’s disease: An overview. Mov Disord. 2007;22:S367–S73. doi: 10.1002/mds.21682. [DOI] [PubMed] [Google Scholar]
- 2.Boeve BF, Silber MH, Ferman TJ, Lucas JA, Parisi JE. Association of REM sleep behavior disorder and neurodegenerative disease may reflect an underlying synucleinopathy. Mov Disord. 2001;16:622–30. doi: 10.1002/mds.1120. [DOI] [PubMed] [Google Scholar]
- 3.Schenck CH, Garcia-Rill E, Skinner RD, Anderson ML, Mahowald MW. A case of REM sleep behavior disorder with autopsy-confirmed Alzheimer’s disease: postmortem brain stem histochemical analyses. Biol Psychiatry. 1996;40:422–5. doi: 10.1016/0006-3223(96)00070-4. [DOI] [PubMed] [Google Scholar]
- 4.Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388:839–40. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- 5.Kosaka K, Yoshimura M, Ikeda K, Budka H. Diffuse type of Lewy body disease: progressive dementia with abundant cortical Lewy bodies and senile changes of varying degree--a new disease? Clin Neuropathol. 1984;3:185–92. [PubMed] [Google Scholar]
- 6.McKeith IG, Dickson DW, Lowe J, Emre M, O’Brien JT, Feldman H, Cummings J, Duda JE, Lippa C, Perry EK, Aarsland D, Arai H, Ballard CG, Boeve B, Burn DJ, Costa D, Del Ser T, Dubois B, Galasko D, Gauthier S, Goetz CG, Gomez-Tortosa E, Halliday G, Hansen LA, Hardy J, Iwatsubo T, Kalaria RN, Kaufer D, Kenny RA, Korczyn A, Kosaka K, Lee VM, Lees A, Litvan I, Londos E, Lopez OL, Minoshima S, Mizuno Y, Molina JA, Mukaetova-Ladinska EB, Pasquier F, Perry RH, Schulz JB, Trojanowski JQ, Yamada M. Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology. 2005;65:1863–72. doi: 10.1212/01.wnl.0000187889.17253.b1. [DOI] [PubMed] [Google Scholar]
- 7.Grundke-Iqbal I, Iqbal K, Quinlan M, Tung YC, Zaidi MS, Wisniewski HM. Microtubule-associated protein tau. A component of Alzheimer paired helical filaments. J Bio Chem. 1986;261:6084–9. [PubMed] [Google Scholar]
- 8.Ishizawa T, Mattila P, Davies P, Wang D, Dickson DW. Colocalization of tau and alpha-synuclein epitopes in Lewy bodies. J Neuropathol Exp Neurol. 2003;62:389–97. doi: 10.1093/jnen/62.4.389. [DOI] [PubMed] [Google Scholar]
- 9.Fujishiro H, Ferman TJ, Boeve BF, Smith GE, Graff-Radford NR, Uitti RJ, Wszolek ZK, Knopman DS, Petersen RC, Parisi JE, Dickson DW. Validation of the neuropathologic criteria of the third consortium for dementia with Lewy bodies for prospectively diagnosed cases. J Neuropathol Exp Neurol. 2008;67:649–56. doi: 10.1097/NEN.0b013e31817d7a1d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hendricks JC, Morrison AR, Mann GL. Different behaviors during paradoxical sleep without atonia depend on pontine lesion site. Brain Res. 1982;239:81–105. doi: 10.1016/0006-8993(82)90835-6. [DOI] [PubMed] [Google Scholar]
- 11.Lai YY, Siegel JM. Muscle tone suppression and stepping produced by stimulation of midbrain and rostral pontine reticular formation. J Neurosci. 1990;10:2727–34. doi: 10.1523/JNEUROSCI.10-08-02727.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shouse MN, Siegel JM. Pontine regulation of REM sleep components in cats: integrity of the pedunculopontine tegmentum (PPT) is important for phasic events but unnecessary for atonia during REM sleep. Brain Res. 1992;571:50–63. doi: 10.1016/0006-8993(92)90508-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baghdoyan HA, Spotts JL, Snyder SG. Simultaneous pontine and basal forebrain microinjections of carbachol suppress REM sleep. Journal of Neuroscience. 1993;13:229–42. doi: 10.1523/JNEUROSCI.13-01-00229.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lai YY, Hsieh KC, Nguyen D, Peever J, Siegel JM. Neurotoxic lesions at the ventral mesopontine junction change sleep time and muscle activity during sleep: an animal model of motor disorders in sleep. Neuroscience. 2008;154:431–43. doi: 10.1016/j.neuroscience.2008.03.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Boeve BF, Silber MH, Saper CB, Ferman TJ, Dickson DW, Parisi JE, Benarroch EE, Ahlskog JE, Smith GE, Caselli RC, Tippman-Peikert M, Olson EJ, Lin SC, Young T, Wszolek Z, Schenck CH, Mahowald MW, Castillo PR, Del Tredici K, Braak H. Pathophysiology of REM sleep behaviour disorder and relevance to neurodegenerative disease. Brain. 2007;130:2770–88. doi: 10.1093/brain/awm056. [DOI] [PubMed] [Google Scholar]
- 16.Xi Z, Luning W. REM sleep behavior disorder in a patient with pontine stroke. Sleep Med. 2008 doi: 10.1016/j.sleep.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 17.Jellinger K. The pedunculopontine nucleus in Parkinson’s disease, progressive supranuclear palsy and Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 1988;51:540–3. doi: 10.1136/jnnp.51.4.540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schmeichel AM, Buchhalter LC, Low PA, Parisi JE, Boeve BW, Sandroni P, Benarroch EE. Mesopontine cholinergic neuron involvement in Lewy body dementia and multiple system atrophy. Neurology. 2008;70:368–73. doi: 10.1212/01.wnl.0000298691.71637.96. [DOI] [PubMed] [Google Scholar]
- 19.Grinberg LT, Rueb U, Alho AT, Heinsen H. Brainstem pathology and non-motor symptoms in PD. J Neurol Sci. 2010;289:81–8. doi: 10.1016/j.jns.2009.08.021. [DOI] [PubMed] [Google Scholar]
- 20.Haglund M, Sjobeck M, Englund E. Locus ceruleus degeneration is ubiquitous in Alzheimer’s disease: possible implications for diagnosis and treatment. Neuropathology. 2006;26:528–32. doi: 10.1111/j.1440-1789.2006.00725.x. [DOI] [PubMed] [Google Scholar]
- 21.Marcyniuk B, Mann DM, Yates PO. The topography of nerve cell loss from the locus caeruleus in elderly persons. Neurobiol Aging. 1989;10:5–9. doi: 10.1016/s0197-4580(89)80004-1. [DOI] [PubMed] [Google Scholar]
- 22.Lu J, Sherman D, Devor M, Saper CB. A putative flip-flop switch for control of REM sleep. Nature. 2006;441:589–94. doi: 10.1038/nature04767. [DOI] [PubMed] [Google Scholar]
- 23.Samuels ER, Szabadi E. Functional neuroanatomy of the noradrenergic locus coeruleus: its roles in the regulation of arousal and autonomic function part I: principles of functional organisation. Curr Neuropharmacol. 2008;6:235–53. doi: 10.2174/157015908785777229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hyman BT, Trojanowski JQ. Consensus recommendations for the postmortem diagnosis of Alzheimer disease from the National Institute on Aging and the Reagan Institute Working Group on diagnostic criteria for the neuropathological assessment of Alzheimer disease. J Neuropathol Exp Neurol. 1997;56:1095–7. doi: 10.1097/00005072-199710000-00002. [DOI] [PubMed] [Google Scholar]
- 25.Boeve BF, Molano JR, Ferman TJ, Smith GE, Lin SC, Bieniek K, Haidar W, Tippmann-Peikert M, Knopman DS, Graff-Radford NR, Lucas JA, Petersen RC, Silber MH. Validation of the Mayo Sleep Questionnaire to screen for REM sleep behavior disorder in an aging and dementia cohort. Sleep Med. 2011 doi: 10.1016/j.sleep.2010.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Medicine AAoS. International Classification of Sleep Disorders: Diagnostic and Coding Manual. 2. Westchester, IL: American Academy of Sleep Medicine; 2005. [Google Scholar]
- 27.Paxinos G, Huang X-F. Atlas of the human brainstem. San Diego, CA: Academic Press, Inc; 1995. [Google Scholar]
- 28.Dugger BN, Dickson DW. Cell type specific sequestration of choline acetyltransferase and tyrosine hydroxylase within Lewy bodies. Acta Neuropathol. 2010;120:633–9. doi: 10.1007/s00401-010-0739-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dugger BN, Tu M, Murray ME, Dickson DW. Disease specificity and pathologic progression of tau pathology in brainstem nuclei of Alzheimer’s disease and progressive supranuclear palsy. Neurosci Lett. 2011 doi: 10.1016/j.neulet.2011.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Manaye KF, Zweig R, Wu D, Hersh LB, De Lacalle S, Saper CB, German DC. Quantification of cholinergic and select non-cholinergic mesopontine neuronal populations in the human brain. Neuroscience. 1999;89:759–70. doi: 10.1016/s0306-4522(98)00380-7. [DOI] [PubMed] [Google Scholar]
- 31.Garcia-Rill E. The pedunculopontine nucleus. Prog Neurobiol. 1991;36:363–89. doi: 10.1016/0301-0082(91)90016-t. [DOI] [PubMed] [Google Scholar]
- 32.Karachi C, Grabli D, Bernard FA, Tande D, Wattiez N, Belaid H, Bardinet E, Prigent A, Nothacker HP, Hunot S, Hartmann A, Lehericy S, Hirsch EC, Francois C. Cholinergic mesencephalic neurons are involved in gait and postural disorders in Parkinson disease. J Clin Invest. 2010;120:2745–54. doi: 10.1172/JCI42642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reese NB, Garcia-Rill E, Skinner RD. The pedunculopontine nucleus--auditory input, arousal and pathophysiology. Prog Neurobiol. 1995;47:105–33. doi: 10.1016/0301-0082(95)00023-o. [DOI] [PubMed] [Google Scholar]
- 34.van Hilten JJ, van der Zwan AD, Zwinderman AH, Roos RA. Rating impairment and disability in Parkinson’s disease: evaluation of the Unified Parkinson’s Disease Rating Scale. Mov Disord. 1994;9:84–8. doi: 10.1002/mds.870090113. [DOI] [PubMed] [Google Scholar]
- 35.Johns MW. A new method for measuring daytime sleepiness: the Epworth sleepiness scale. Sleep. 1991;14:540–5. doi: 10.1093/sleep/14.6.540. [DOI] [PubMed] [Google Scholar]
- 36.Ferman TJ, Smith GE, Boeve BF, Ivnik RJ, Petersen RC, Knopman D, Graff-Radford N, Parisi J, Dickson DW. DLB fluctuations: specific features that reliably differentiate DLB from AD and normal aging. Neurology. 2004;62:181–7. doi: 10.1212/wnl.62.2.181. [DOI] [PubMed] [Google Scholar]
- 37.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–59. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 38.Ma SY, Roytta M, Rinne JO, Collan Y, Rinne UK. Single section and disector counts in evaluating neuronal loss from the substantia nigra in patients with Parkinson’s disease. Neuropathol Appl Neurobiol. 1995;21:341–3. doi: 10.1111/j.1365-2990.1995.tb01068.x. [DOI] [PubMed] [Google Scholar]