

Abstract
Electron microscopy structural determinations suggest that the membrane-proximal external region (MPER) of glycoprotein 41 (gp41) may associate with the HIV-1 membrane interface. It is further proposed that MPER-induced disruption and/or deformation of the lipid bilayer ensue during viral fusion. However, it is predicted that the cholesterol content of this membrane (∼45 mol %) will act against MPER binding and restructuring activity, in agreement with alternative structural models proposing that the MPER constitutes a gp41 ectodomain component that does not insert into the viral membrane. Here, using MPER-based peptides, we test the hypothesis that cholesterol impedes the membrane association and destabilizing activities of this gp41 domain. To that end, partitioning and leakage assays carried out in lipid vesicles were combined with x-ray reflectivity and grazing-incidence diffraction studies of monolayers. CpreTM, a peptide combining the carboxyterminal MPER sequence with aminoterminal residues of the transmembrane domain, bound and destabilized effectively cholesterol-enriched membranes. Accordingly, virion incubation with this peptide inhibited cell infection potently but nonspecifically. Thus, CpreTM seems to mimic the envelope-perturbing function of the MPER domain and displays antiviral activity. As such, we infer that CpreTM bound to cholesterol-enriched membranes would represent a relevant target for anti-HIV-1 immunogen and inhibitor development.
Introduction
Cell infection by the human immunodeficiency virus type-1 (HIV-1) begins with the merger of viral and cellular membranes by the envelope glycoprotein transmembrane subunit (gp41) (1, 2). The membrane-proximal external region (MPER) of gp41 is enriched in conserved aromatic residues and is required for fusogenic activity (3, 4) (Fig. 1). Cryoelectron microscopy and 3D reconstructions of intact virions and detergent-solubilized glycoproteins display each trimer's stalk as composed of three legs that project obliquely from the trimer head, tripodlike, and insert independently through the MPER into the external monolayer of the viral membrane (5, 6) (Fig. 1 A, left). Supporting the relevance of this interfacial structure, hydrophobicity distribution and structural analyses suggest that the MPER favorably inserts into the water-bilayer interface as an α-helix that runs almost parallel to the membrane plane (7, 8, 9). Recent mutational studies further indicate that insertion of the MPER and the adjacent transmembrane domain (TMD) stretch may disrupt the membrane during gp41-induced fusion (10, 11).
Figure 1.
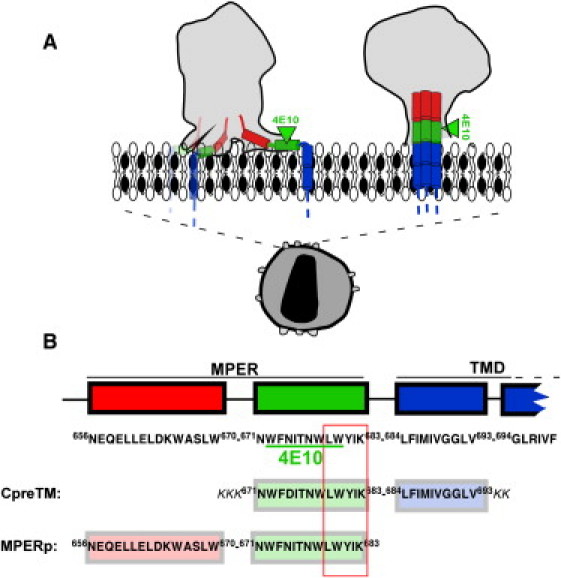
Models for gp41 MPER-TMD region organization at the HIV membrane and designation of the CpreTM and MPERp sequences used in this study. (A) Proposed models for the location of gp41 MPER-TMD at the Chol-enriched viral membrane. The cryoET density map contours of envelope spikes reported by Zhu et al. (5) and Zanetti et al. (12) are compared (left and right gray areas, respectively). Sequences depicted roughly parallel to the membrane plane (left) span amphipathic-at-interface and interfacial MPER subdomains (7). Cylinders inserted into the membrane represent the TMD anchors. (B) Sequences of the peptides used in this study. The diagram also shows positions and sequences for the potential cholesterol recognition/interaction amino acid consensus (outlined residue sequence) and 4E10 epitope (underlined residues). Sequence and numbering are according to the prototypic HXBc2 isolate.
In contrast to this view, a competing model postulates that MPER remains associated to the native gp41 ectodomain stalk region, i.e., without establishing contacts with the viral membrane (12, 13) (Fig. 1 A, right). In addition, interactions of this element with the viral membrane are not considered at any stage of the fusion mechanism (14). This model is supported by the presence of high cholesterol (Chol) concentration at the viral envelope (∼45 mol % (15)). This Chol concentration corresponds to that of a highly ordered membrane domain, and precludes coexistence with laterally segregated, more fluid domains (15). Lorizate et al. (16) actually determined for infectious virions lipid order levels at 37°C corresponding to those found in liquid-ordered (lo) domains. In general, high levels of Chol exert a phospholipid condensing effect that hinders water-membrane partitioning and subsequent assembly of lytic pores by peptides that bind tightly to the membrane interface (17, 18), as is predicted to be the case for MPER (7). Accordingly, Chol interferes with the membrane-interacting properties of antimicrobial, cytolytic, and cell-penetrating peptides such as magainin (19), gramicidin S (20), cathelicidin LL-37 (21, 22), melittin (17, 23), or transportan (24), although exceptions to this general rule can be found in the literature (25, 26). In addition, certain cytolytic peptides, such as δ-lysin, are mostly excluded from Chol-enriched ordered domains in systems that exhibit separation between liquid-disordered (ld) and lo domains (27, 28). Thus, despite MPER hydrophobicity, the Chol concentration existing at the viral membrane would likely preclude MPER-membrane binding and subsequent restructuring activity.
To test this hypothesis, in this work, we investigate the effects of biologically relevant Chol concentrations on MPER-membrane interactions. To that end, first, we compare the membrane-binding and lytic activities of two overlapping, MPER-based peptides as a function of the Chol content in lipid vesicles (Fig. 1 B): MPERp, representing the sequence characterized in previous structural studies (9) and currently targeted by anti-MPER vaccines (29); and CpreTM, a peptide designed to preserve the sequence environment of the MPER-TMD hinge, which embodies an important gp41 functional determinant (4, 11, 30). Our data suggest that the sequence represented by MPERp (so far considered the canonical MPER peptide) would be incapable of perturbing the Chol-enriched viral envelope. In contrast, CpreTM bound to and permeabilized effectively vesicles containing Chol levels comparable to those existing at the viral envelope. Consistent with its potential for perturbing the viral membrane, CpreTM inhibited cell-assembled enveloped virions potently but nonspecifically. X-ray diffraction analyses of lipid monolayers containing the peptide, together with previously reported biophysical data on the short LWYIK oligopeptide (31, 32), provided molecular mechanisms for the interactions of CpreTM with Chol-enriched membranes. Our results support the physiological relevance of membrane-bound states of the MPER sequence represented by CpreTM, and the capacity of this gp41 domain to destabilize the viral lipid-bilayer architecture during fusion. Hence, we propose that the functional MPER sequence should be redefined to include N-terminal TMD residues. Finally, we discuss the implications of our findings for the development of peptide-based vaccines and inhibitors targeting MPER.
Materials and Methods
See the Supporting Material for the materials and methods used in this work.
Results
Chol effects on the membrane-perturbing properties of MPER-derived peptides
Mutational studies have established a correlation between MPER lytic activity and gp41 fusogenic function (10). The vesicle leakage kinetics reflect the lytic activities of CpreTM and MPERp peptides (Fig. 2, A and B, left). In consonance with the role proposed for the MPER domain during fusion, both peptides permeabilized vesicles devoid of Chol. However, whereas CpreTM was capable of inducing even higher levels of leakage from vesicles containing 0.5 mole fraction of Chol (Fig. 2 A, left), MPERp activity was markedly reduced in these vesicles (Fig. 2 B, left). The high Chol levels correlate with increased bilayer elastic moduli (33, 34). The increase in the area compressibility modulus (KA) induced by this compound has been shown to reduce the capacity of peptides to transfer from water into membranes (18). Thus, we first sought to establish a correlation between leakage and peptide-membrane partitioning (Fig. 2, right, and Fig. S1 in the Supporting Material). The lipid titration curves rendered comparable apparent mole fraction partition coefficients for CpreTM interacting with POPC and POPC/Chol (1:1) vesicles (Kx(app) values of 6.5 × 106 and 5.5 × 106, respectively). In contrast, as compared to POPC vesicles, the partitioning coefficient was reduced by two orders of magnitude for MPERp interacting with POPC/Chol (1:1) vesicles (Kx(app) values of 1.4 × 107 and 1.7 × 105, respectively).
Figure 2.
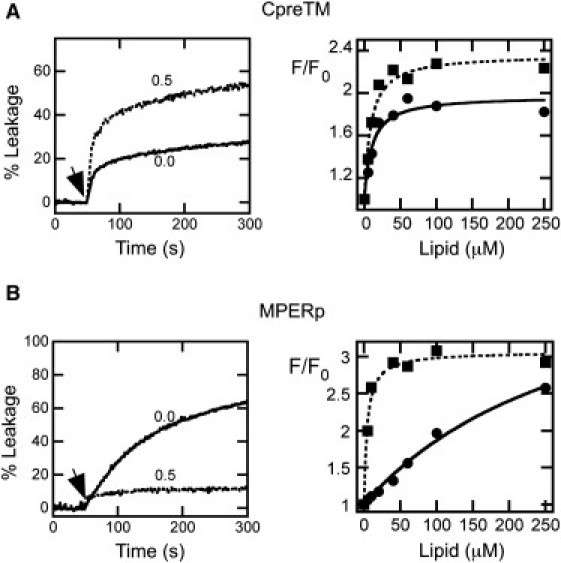
Effect of Chol on the lytic activities and binding capacities of MPER peptides. (A, left) Effect of Chol on CpreTM-induced leakage kinetics. The peptide was added at the time indicated by the arrow (t = 50 s) at a peptide/lipid ratio of 1:500. Lipid concentration was 100 μM. Chol mole fractions are indicated for each curve. (A, right) Partitioning curves estimated from the fractional change in Trp fluorescence with increasing POPC or POPC/Chol (1:1) concentrations (squares and circles, respectively). Peptide concentration was 0.5 μM. Lines correspond to the best fit of the experimental values to a hyperbolic function (see Materials and Methods in the Supporting Material). (B) MPERp peptide was used at a peptide/lipid ratio of 1:150 in the leakage assays (left). Otherwise, conditions are as in A.
To illustrate this observation further, data displayed in Fig. 3 compare KA increase, peptide association to vesicles, and resulting leakage as a function of the Chol content in lipid vesicles. Chol did not appreciably affect CpreTM partitioning into membranes in the range of the tested concentrations (Fig. 3, upper, open symbols). In contrast, the MPERp capacity for associating with the vesicles was impaired above the onset of the KA increase (∼0.2–0.3 mole fraction (33, 34)) (Fig. 3, lower, open symbols). The percentages of leakage induced by CpreTM and MPERp peptides, as a function of Chol content, were also comparatively different. Below the onset of the KA increase, CpreTM-induced vesicle permeabilization was enhanced (Fig. 3, upper, solid symbols). This effect reached a maximum and began to decrease slightly at the onset of the KA increase, but leakage was not inhibited. A comparable behavior was reported for the cytolytic peptide δ-lysin interacting with Chol-containing vesicles (27, 28). In contrast, low levels of Chol barely affected MPERp-induced permeabilization (Fig. 3, lower, solid symbols). Above the onset of the KA increase, leakage induced by this peptide was inhibited.
Figure 3.
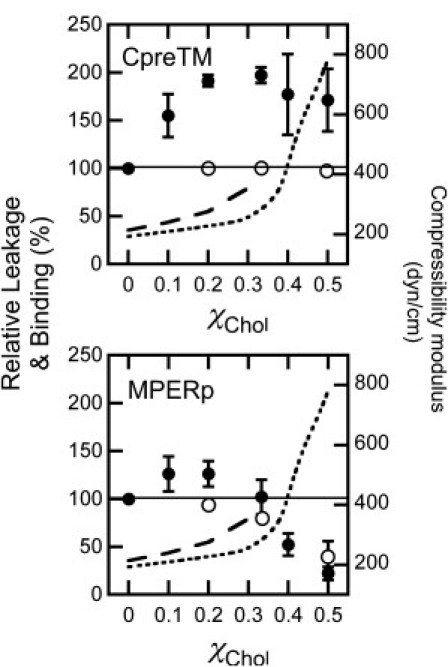
Binding to vesicles and ANTS leakage induced by CpreTM (upper) and MPERp (lower) as a function of the Chol mole fraction. Plotted values correspond to leakage (extents measured 60 min after peptide addition) and binding to membranes relative to those measured for vesicles devoid of Chol (solid and open circles, respectively). Dotted and dashed curves indicate values of the area compressibility moduli as measured by Needham and Nunn (33) and Henriksen et al. (34), respectively. Otherwise, experimental conditions are as in Fig. 2.
In conclusion, Chol-induced KA increase correlated with a reduction of MPERp binding to vesicles and the subsequent inhibition of its lytic activity. Since MPERp, and closely related peptides, are considered to embody the canonical MPER sequence (3, 7, 9), these results would be consistent with the absence of interactions between this gp41 domain and the viral membrane (Fig. 1 A, right). However, the CpreTM data, i.e., binding and permeabilization of vesicles containing Chol mole fractions >0.2–0.3, indicate that the envelope-perturbing activity of the MPER domain might be retained within this sequence. The following results support this notion.
Inhibition of viral infectivity by MPER-derived peptides
Recent works describe the discovery and development of antiviral compounds that inhibit cell infection by a broad range of enveloped viruses (35, 36). These compounds target the viral membrane and are believed to alter its structural and/or material properties. Therefore, to test the possibility that CpreTM could perturb the lipid bilayer surrounding the HIV particle, we evaluated its effect on HIV virion functionality (Fig. 4). We assumed that, given its limited capacity for partitioning into viral membranes, MPERp would comparatively exert a lower effect on virus infectivity. However, the MPERp peptide contains the NEQELLELDKWASLWNWF sequence representing the C-terminal portion of the fusion inhibitor T-20 and overall accounting for 50% of its residues. Thus, in principle, MPERp-induced infection inhibition, if any, was also expected to be more specific than that exerted by CpreTM.
Figure 4.
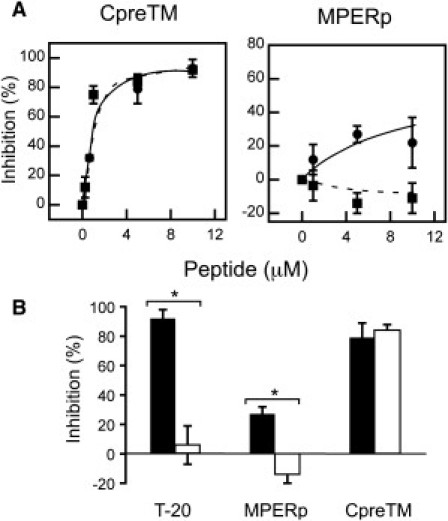
Inhibition of viral infectivity induced by CpreTM and MPERp. (A) Dose dependency of pseudovirus infection inhibition. Viruses were pseudotyped with HIV-1 Env (HXB2) or VSV-G (solid and dotted lines, respectively). The infection of TZM-bl target cells was monitored in both cases by flow cytometry as the green fluorescent protein signal in infected cells. (B) Specificity of the inhibitory effect. HIV-1 and VSV pseudovirions (solid and open columns, respectively) were incubated with 5 μM of CpreTM or MPERp. T-20 fusion inhibitor (0.1 μM) was included as a positive control for HIV-1 specificity. The asterisk denotes p < 0.005 by Student's test for unpaired data. Graphs in both panels display the mean ± SD of four measurements in two independent experiments.
Results displayed in Fig. 4 confirmed our predictions. Both peptides inhibited HIV infectivity as measured in the pseudovirus assay (Fig. 4 A, circles and solid lines). From the dose-dependency curves, IC50 values on the order of 0.5 and 50 μM were inferred for CpreTM and MPERp, respectively. For comparison, IC50 values in the range of 10 nM were measured for the HIV-1 specific fusion inhibitor T-20 in our assay (not shown). Peptide-induced inhibition was also tested by pseudotyping the HIV particles with the VSV-G-protein (Fig. 4 A, squares and dashed lines). VSV-G-protein-mediated infection could be inhibited by CpreTM to a comparable extent, whereas MPERp was totally ineffective. Fig. 4 B illustrates the specificity of the inhibitory activities. Significant differences between capacities for inhibiting HIV or VSV pseudoviruses could be observed for T-20 and MPERp, whereas CpreTM inhibited both types of virions with equal potency.
Mechanism of CpreTM-induced perturbation of Chol-containing membranes
The lack of specificity observed in the previous experiments supports the hypothesis that the virion membrane, rather than the gp41 protein, is the target for CpreTM-induced infection inhibition. The high content of aromatic residues (Fig. 1 B) suggests that CpreTM could tightly bind to the viral membrane interface and disrupt lipid packing according to an interfacial activity mechanism similar to that proposed for antimicrobial peptides (AMPs) (10, 37). Leakage according to such a mechanism is usually observed to occur in 100-nm large unilamellar vesicles (LUVs) at peptide/lipid mole ratios in the vicinity of 1:100 or higher. By comparison, in the case of true pore-forming peptides, a limited number of monomers may establish an aqueous channel in a single LUV, and the resulting leakage could be observed at peptide/lipid ratios as low as 1:10,000 (37, 38).
Thus, to test the CpreTM mode of action, we next determined the range of peptide/lipid ratios required to cause leakage of Chol-enriched vesicles. Fig. 5 A illustrates CpreTM partitioning into POPC, POPC/Chol (4:1), and POPC/Chol (2:1) vesicles. The water-membrane partitioning curves disclosed a similar capacity of the peptide for partitioning into these vesicles, under the experimental conditions selected for measuring leakage. In contrast, the effective CpreTM doses required for vesicle permeabilization were lower in the Chol-containing vesicles (Fig. 5 B and Table S1). Thus, for the same amount of membrane-bound peptide, pore formation was more efficient in the Chol-containing vesicles. Again, comparable Chol effects have been reported for leakage induced by the cytolytic peptide δ-lysin (27, 28).
Figure 5.
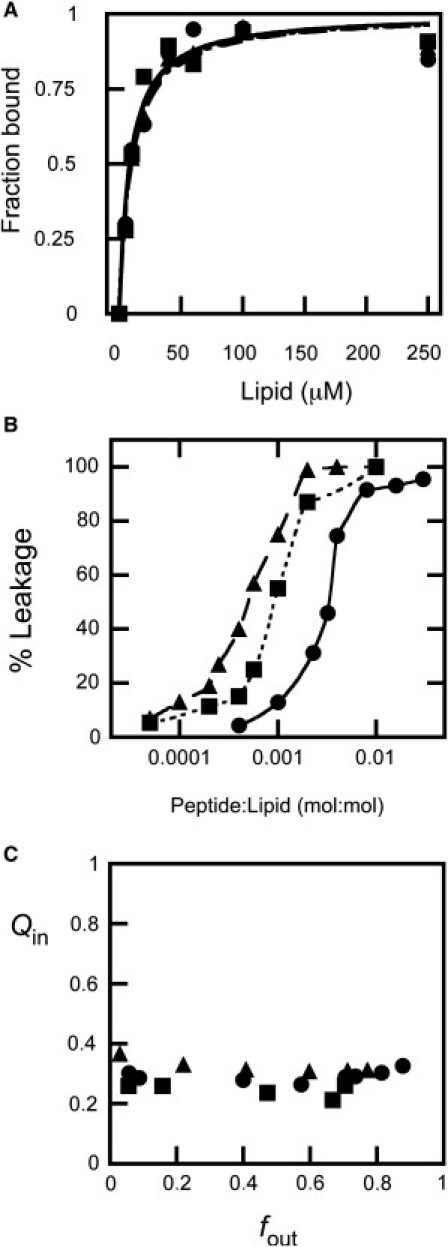
Dose dependency and mechanism of CpreTM-induced content leakage from POPC (circles and solid lines), POPC/Chol (4:1) (squares and dotted lines), and POPC/Chol (2:1) (triangles and dashed lines) LUVs. (A) Partitioning curves estimated from the fractional change in Trp fluorescence with increasing lipid concentration. The fitting curves are superimposed. (B) ANTS leakage (extent measured 60 min after peptide addition) measured as a function of the peptide/lipid mole ratio. The lines link the experimental values for better appreciation of the differences. (C) Fluorescence requenching analysis to establish the mechanism of membrane permeabilization. Internal quenching (Qin) was measured as a function of the ANTS released (fout) after incubation with peptide (see Supporting Material).
Finally, the requenching data shown in Fig. 5 C confirmed that CpreTM-induced leakage in POPC, POPC/Chol (4:1), and POPC/Chol (2:1) vesicles followed a similar all-or-none mechanism. The goal of the requenching assay (39) is to establish the dependence of the quenching inside vesicles (Qin) on the ANTS fraction outside vesicles (fout). Qin is defined as the ratio between the ANTS fluorescence inside vesicles at any time (Fi) and its maximum possible value in the absence of DPX (Fimax). Qin remains constant and low for any fout value when peptide-induced leakage follows an all-or-none mechanism, i.e., the population of vesicles consists of those that did not leak at all and those releasing all of their aqueous contents. Consistent with vesicle permeabilization according to a well-defined all-or-none mechanism, the CpreTM experimental Qin values were low and constant for all compositions.
In summary, the low membrane doses required for permeabilizing POPC/Chol 2:1 vesicles (in the peptide/lipid mole ratio range 1:10,000–1:1000) together with a defined all-or-none mechanism of leakage would qualify CpreTM as a true pore-forming peptide (37).
CpreTM and lipid organization in fluid phospholipid monolayers containing Chol
X-ray reflectivity (XR) and grazing-incidence x-ray diffraction (GIXD) studies of lipid monolayers containing CpreTM sustained a membrane-perturbation mechanism dissimilar to that of AMPs (Tables 1 and 2). Analysis of specular XR data yields information on the electron density distribution in a monolayer in a direction perpendicular to the interface, ρ(z), averaged over the beam footprint and thus over ordered and disordered regions of the film. Changes in ρ(z) after introduction of CpreTM can be related to the depth of membrane insertion, orientation within a film, and interfacial concentration (lipid/peptide ratio) of the peptide. On the other hand, the membrane-disruptive activity of the peptide can be assessed with GIXD, which provides direct structural information on a nanoscale lateral molecular order in Langmuir monolayers. Consequently, these techniques were chosen to investigate at the molecular level the Chol effects on CpreTM-lipid interactions and destabilizing activity, which are not derived from changes in the elastic properties of the bilayers. Thus, experiments were carried out on planar phospholipid monolayers with low and high levels of Chol with and without the peptide (Tables 1 and 2).
Table 1.
Results of x-ray reflectivity data analysis
| Slab | Li (Å) | ρi (e−/Å3)exp | ρi (e−/Å3)est | e−extra | e−extra total | σ (Å) | LT (Å) | A (Å2) | L:P100 | L:P50 | Apeptide (Å2) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| DPPC/cholesterol (87:13) | |||||||||||
| DPPCAC + CHOLAC | 7.9 | 0.311 | NA | NA | NA | 3.3 ± 0.2 | 24.3 | 47.7 | NA | NA | NA |
| DPPCAC + CHOLRB | 7.5 | 0.331 | |||||||||
| DPPCHG + CHOLHG | 8.9 | 0.424 | |||||||||
| DPPC/cholesterol (87:13) + CpreTM | |||||||||||
| DPPCAC + CHOLAC + CpreTM | 7.9 | 0.278 | 0.273 | 2.2 | 57.1 | 2.7 ± 0.2 | 25.2 | 54.4 | 43:1 | 38:1 | 255–288 |
| DPPCAC + CHOLRB + CpreTM | 7.4 | 0.326 | 0.294 | 12.9 | |||||||
| DPPCHG + CHOLHG + CpreTM | 9.9 | 0.412 | 0.334 | 42 | |||||||
| DPPC/cholesterol (54:46) | |||||||||||
| DPPCAC + CHOLAC | 7.5 | 0.281 | NA | NA | NA | 3.1 ± 0.2 | 24.5 | 40.9 | NA | ||
| DPPCAC + CHOLRB | 9.9 | 0.359 | |||||||||
| DPPCHG + CHOLHG | 7.1 | 0.417 | |||||||||
| DPPC/cholesterol (54:46) + CpreTM | |||||||||||
| DPPCAC + CHOLAC + CpreTM | 6.0 | 0.279 | 0.279 | 0 | 96.1 | 3.3 ± 0.7 | 24.9 | 51.5 | 25:1 | 22:1 | 233–265 |
| DPPCAC + CHOLRB + CpreTM | 9.6 | 0.347 | 0.294 | 26.2 | |||||||
| DPPCHG + CHOLHG + CpreTM | 9.3 | 0.399 | 0.253 | 69.9 | |||||||
AC, acyl chains; RB, ring body of cholesterol; HG, headgroups; L, slab thickness; ρexp, experimental average electron density of a slab; ρest, estimated average electron density of a slab; e−extra, number of extra electrons in a slab; e−extra total, total number of extra electrons in a film; LT, total thickness of the film; A, area/average lipid molecule; L:P100 and L:P50, lipid/peptide ratios assuming 100% and 50% hydration of peptide polar groups; Apeptide, estimated area/peptide molecule.
Table 2.
GIXD data
| Experiment | d-Spacing (Å) | Unit cell parameters a and b (Å), γ (°), and area A (Å2) | Coherence length (Å) |
|---|---|---|---|
| 13:87 | d(1,1),(1,−1) = 4.55 d(0,2) = 4.30 | a = 5.37, b = 8.6, γ = 90, A = 46.2 | L(1,1),(1,−1) = 52; L(0,2) = 301 |
| + CpreTM | d(1,1),(1,−1) = 4.59 d(0,2) = 4.28 | a = 5.44, b = 8.56, γ = 90, A = 46.6 | L(1,1),(1,−1) = 19; L(0,2) = 158 |
| 46:54 | 4.69 | a = b = 5.41, γ = 120, A = 25.3 | 58 |
| + CpreTM | 4.7 | a = b = 5.42, γ = 120, A = 25.5 | 22 |
Using constant-pressure insertion assays, we first tested the propensity of CpreTM to incorporate into DPPC/cholesterol (87:13 and 54:46) mixed monolayers at a constant surface pressure of 20 mN/m. The final relative increase in mean molecular area, ΔA/A, upon introduction of CpreTM comprised 14% in the 87:13 DPPC/cholesterol monolayer and 26% in the 54:46 DPPC/cholesterol film. Table 1 summarizes the results of our analysis of XR data from monolayers before and after introduction of CpreTM. Reflectivity curves giving rise to those data are shown in Fig. S2. In a recent x-ray study (40), it was shown that DPPC/cholesterol monolayers can be approximated by a three-slab system, where each of the slabs is characterized by an individual electron density (ρi) and thickness (Li) (Table 1). The top slab (closest to the air) contains the acyl chain (AC) of cholesterol molecule and a portion of DPPC acyl chains. The middle slab includes the rest of the DPPC acyl chains and the cholesterol rigid-ring body structure (RB), and the bottom slab (closest to the aqueous buffer) represents DPPC and cholesterol headgroups (HGs). The packing density of the hydrophobic core of 87:13 DPPC/cholesterol mixture is lower than that of the 54:46 mixture, as follows from the corresponding electron densities/unit area of 4.94 e−/Å2 and 5.66 e−/Å2, respectively. Although the electron density/unit area of the HGs of the 87:13 DPPC/cholesterol mixture, 3.77 e−/Å2, is 25% higher than that of the 54:46 mixture, 2.96 e−Å2, this is mainly due to the difference in hydration between the HGs in two monolayers (3.6 vs. 2.9 water molecules/lipid HG). Therefore, we anticipate that the peptide should incorporate with similar propensity into the HGs of the 54:46 and 87:13 mixtures, but the hydrophobic core of the 54:46 mixed monolayer should represent a more challenging barrier to peptide penetration.
However, the reflectivity data for the lipid monolayer/peptide systems revealed similar degrees of CpreTM penetration into both monolayers. The XR curves for the lipid/peptide films were also fitted using a three-slab model (Table 1). In both films, ∼30% of the CpreTM peptide's mass penetrates into the hydrophobic core of the lipid, and the rest localizes in the lipid HGs. Consistent with similar topologies, a 1.7-fold decrease in the lipid/peptide ratio in the CpreTM/(54:46) mixed monolayer system as compared to CpreTM/(87:13) mixed film system is matched by even 1.7-1.8 times increase in peptide's electrons in the lipid hydrophobic and HG regions. In addition, the observed increase in monolayer thickness upon peptide insertion, mainly localized at the HG slab, supports that CpreTM interactions preferentially occur within this monolayer region. By comparison, at similar monolayer peptide doses, the AMP LL-37 mainly localized within the hydrocarbon-core and provoked a significant reduction in the layer thickness (41).
Our conclusions about the depth of peptide penetration into the mixed monolayers are further indirectly supported by the GIXD data (Table 2). The diffraction of DPPC/cholesterol monolayers is due to the lipid hydrophobic region (DPPC acyl chains and cholesterol rigid fused ring body). Therefore, should the peptide insert mainly into the lipid HGs, it would create voids in the monolayer hydrophobic region, resulting in a less dense packing of the acyl chains and cholesterol molecules in the ordered domains. On the other hand, if a peptide penetrates deep into the film hydrophobic core, it may create additional stress on the lipid acyl chains and force them to pack more densely. Indeed, Table 2 indicates that CpreTM insertion leads to an increase in the unit cell area of the crystalline domains of both monolayers. It is noteworthy that CpreTM was not capable of disrupting the in-plane order of the mixed monolayers completely, as follows from the preserved diffraction peaks (Fig. S3). This again is opposite to the effect reported for the AMP LL-37 peptide (41). After LL-37 insertion, no Bragg peaks or rods could be observed, consistent with the total disruption by the peptide of the ordered structure of the monolayer (41).
In conclusion, CpreTM peptide interacted favorably with Chol-containing lipid monolayers, and its topology was not significantly altered upon increasing the concentration of this compound. In addition, it did not display monolayer-disrupting activities as those described for AMPs (41, 42).
Discussion
Early Wimley-White interfacial hydrophobicity computation, together with partitioning experiments (7), supported the model of MPER insertion into the viral membrane interface (Fig. 1 A, left). It was further proposed that the shallow insertion of MPER into the envelope external leaflet might poise the viral membrane for fusion (7, 43). However, those estimates did not take into consideration the high Chol concentration at the viral membrane (15). Phospholipid cohesion induced by this compound would act against the opening of cavities required for transferring MPER residues into the biologically relevant viral membrane (18). Viral Chol levels would in addition alter the bilayer mechanical properties to oppose MPER-induced restructuring (17, 33, 34, 44). In line with the phospholipid-condensing properties of the compound, Chol reduced partitioning into vesicles of MPERp, the peptide so far considered to be representative of the MPER domain (Figure 2, Figure 3). Also consistent with the lack of interaction with the Chol-enriched viral membrane, the infectivity experiments revealed a low, but HIV-specific, MPERp-induced inhibition, which points to the gp41 protein, rather than the lipid envelope, as the target for this peptide (Fig. 4). Impeded insertion into the viral membrane and restrained deformability would be consistent with an alternative model proposing that MPER remains associated to the gp41 globular ectodomain (Fig. 1 A, right), both in the native glycoprotein and during fusion (13).
However, the MPER-TMD LWYIK-LFIMIV hinge sequence represents an important gp41 functional determinant (4, 11, 30) not included in the MPERp peptide sequence. The CpreTM peptide used in this study was actually conceived to preserve the sequence environment of this determinant (Fig. 1 B). We report here Chol effects on peptide-membrane interactions that are comparatively different for MPERp and CpreTM. In fact, our data support an adaptation of the gp41 sequence represented by CpreTM for inserting into and destabilizing the rigid viral envelope.
Implications of CpreTM-lipid interactions for MPER insertion into the viral membrane
The increase in phospholipid cohesion that ensues with high concentrations of Chol did not inhibit CpreTM partitioning (Figure 2, Figure 3), which might reflect the capacity of this sequence for inserting into the viral envelope. In support of this assumption, infectivity of HIV virions was effectively inhibited by CpreTM (Fig. 4). Of note, VSV-G-mediated cell infection was inhibited by this peptide to the same extent. The lack of specificity observed in these experiments is highly suggestive of a CpreTM-induced inhibition mechanism involving insertion into the viral membrane, i.e., following a pattern similar to that of antiviral compounds that target a broad range of unrelated enveloped viruses (35, 36). Effective CpreTM insertion into Chol-enriched membranes was further confirmed using lipid monolayers. The use of this system allowed us to imitate the in vivo situation when MPER putatively comes into contact with the outer leaflet of the virion membrane. XR data for monolayers containing CpreTM and different Chol concentrations did not reveal significant changes in peptide topology, whereas the area occupied by the peptide at the interface was smaller in monolayers with high Chol content (Table 1). This reduction of the monolayer area occupied by the peptide implies smaller membrane cavities to accommodate peptide residues and, therefore, optimization of the energy cost for membrane insertion (18).
A recent study by Tristam-Nagle and co-workers (32) has provided an additional explanation for the CpreTM capacity for inserting into Chol-enriched membranes. Their x-ray synchrotron scattering data indicate that the LWYIK sequence may counteract the chain-ordering effect caused by Chol. Thus, we speculate that tethering to the gp41 TMD N-terminus enables LWYIK insertion into the Chol-enriched external monolayer of the viral membrane, which may then reduce phospholipid cohesion therein. This effect might also enable insertion of CpreTM, even with Chol concentrations in the range of 50 mol %. Altogether, the experimental evidence presented in this work and previous x-ray structural determinations suggest that the MPER-TMD gp41 region represented by CpreTM may favorably associate with the viral membrane (Fig. 1 A, left).
Implications of CpreTM-lipid interactions for viral membrane perturbation by MPER
Consistent with the CpreTM capacity for perturbing the rigid viral membrane upon insertion, leakage induced by the peptide was stimulated by Chol (Figure 3, Figure 5). It has been reported that membrane permeabilization after treatment of the virus with Chol-binding β-cyclodextrin actually abolishes HIV infectivity (45). Notably, this compound was effective in the millimolar range of concentrations (IC50 of ∼3 mM in our assay (not shown)). Thus, the fact that the IC50 values measured for CpreTM are three to four orders of magnitude lower underlines its higher potency and suggests a different mechanism of action.
The low peptide doses required for permeabilizing Chol-containing vesicles (Fig. 5 B and Table S1) rather suggest that CpreTM generates true pores in the viral membrane. Assembly of discrete aqueous pores is further supported by the all-or-none mechanism of lekage observed in LUVs (Fig. 5 C) and giant vesicles (46) that contain Chol. In the latter system, CpreTM was actually shown to establish permanently opened pores (46). All-or-none leakage induced by true pore-forming peptides has been subject to mathematical analysis based on a pore model that takes into account aggregation reversibility of peptide monomers at the membrane surface (38). A preliminary analysis of the CpreTM-induced leakage data (Fig. 5 B) displayed good fits to this model (Table S1). Finally, the results of GIXD experiments, which indicate that CpreTM insertion did not disrupt the in-plane order of the lipid monolayers (Table 2 and Fig. S3) would also be consistent with the assembly of CpreTM true pores in Chol-enriched membranes. In contrast, interfacially active AMPs (37), such as LL-37 or protegrin-1, destabilize lipid packing and produce disruption of monolayers upon insertion (41, 42).
In conclusion, CpreTM exhibits features of a true pore-forming peptide modulated by Chol. However, this phenomenon would be at odds with the lack of lipid selectivity postulated for the potent, ideal pore-forming peptides (37). Lipid modulation of the process is often ascribed to the formation of toroidal, lipidic pores (37, 47). To establish a lipidic pore, the bilayer must bend and deform so that lipid HGs interspersed with peptide units line the walls of a toroidal aqueous channel (17, 48, 49). Peptides forming lipidic pores induce monolayer area expansion and concomitant thinning (18, 47). Our XR data indicate opposite effects for CpreTM interacting with monolayers (Table 1). In both monolayers, peptide insertion resulted in an increase of the monolayer thickness. In addition, the area occupied by the peptide was reduced in the monolayer containing high levels of Chol, i.e., under conditions of optimal pore formation. Given the geometry of the toroid, permeabilization according to this mechanism is distinctively promoted by lipids that impart a positive spontaneous curvature (1/R0) to the bilayer (17, 49). As compared to Chol, including into the bilayer composition LPC or DG, two lipids with known positive and negative spontaneous curvatures, respectively, had little effect on CpreTM binding or vesicle permeabilization (Fig. S4). Thus, it appears that CpreTM did not promote lipidic pore formation in the Chol-enriched membranes.
Previous structural characterizations of the LWYIK sequence by Epand and colleagues provide a putative molecular mechanism to explain Chol effects on CpreTM-induced leakage (30, 31). These authors have proposed that LWYIK sequence may directly bind to and sequester Chol (31). Based on calculations of the most favorable conformations it was inferred that LWYIK interactions with Chol would occur at the membrane interface and involve H-bonding of its hydroxyl group (31). The observed increase in monolayer HG slab thickness upon peptide insertion would actually support such an interaction for CpreTM (Table 1). Satisfying polar head requirements set Chol rings rather free for intermolecular interactions. We surmise that this interfacial interaction may first prevent deep insertion of the CpreTM sequence into the acyl-chain region of the bilayer and then drive self-association of peptides and Chol. Thus, we infer that the local increase of the CpreTM concentration in the Chol-containing membranes might promote pore formation by this peptide.
Concluding Remarks: Implications For Anti-Hiv Vaccine And Inhibitor Development
Altogether, the experimental evidence reported in this work suggests that the MPER region represented by the CpreTM peptide is adapted for bilayer binding and destabilization under the conditions existing in the HIV envelope milieu, i.e., low spike density (10–20 trimers/virion) (5) and high Chol content (∼45 mol %) (15). In contrast, the MPERp peptide was unable to reproduce the binding and membrane-perturbing activities attributed to the functional MPER domain. In addition, we provide data indicating that CpreTM might perturb viral membrane integrity through the establishment of discrete aqueous pores. These observations might have important implications for development of anti-HIV inhibitors and immunogens. On one hand, the functional sequence of MPER should be redefined to include the TMD N-terminus. On the other hand, CpreTM pore structures assembled in Chol-enriched membranes seem to represent an unprecedented, biologically relevant MPER target.
Thus, the CpreTM sequence embodies a new scaffold, prospectively useful for expanding the spectrum of antiviral drugs targeting the membranes of enveloped viruses (35, 36). In particular, CpreTM-based compounds might be devised to target viral membranes enriched in Chol. Moreover, mimicking the epitope recognized by the broadly neutralizing 4E10 antibody by model systems (Fig. 1) constitutes a timely goal for the development of effective anti-HIV immunogens (29, 50). In that regard, our data suggest that the native structures adopted by this MPER sequence on the surface of the viral envelope might depend on the MPER-TMD hinge region and Chol.
Acknowledgments
This research was supported by Spanish MCINN, Basque Government and University of the Basque Country grants (BIO 2008-00772, GIU 06/42, and DIPE08/12 to J.L.N.). B.A. was the recipient of a predoctoral fellowship of the Spanish MEU. D.G. was supported by National Institutes of Health R01 AI073892 and DARPA W911NF-09-1-378 grants.
Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357. ChemMatCARS Sector 15 is principally supported by the National Science Foundation/Department of Energy under grant No. NSF/CHE-0822838.
Editor: Paulo F. Almeida.
Footnotes
Materials and methods, four figures, and a table are available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(11)01194-5.
Supporting Material
References
- 1.Gallo S.A., Finnegan C.M., et al. Blumenthal R. The HIV Env-mediated fusion reaction. Biochim. Biophys. Acta. 2003;1614:36–50. doi: 10.1016/s0005-2736(03)00161-5. [DOI] [PubMed] [Google Scholar]
- 2.Melikyan G.B. Common principles and intermediates of viral protein-mediated fusion: the HIV-1 paradigm. Retrovirology. 2008;5:111. doi: 10.1186/1742-4690-5-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Salzwedel K., West J.T., Hunter E. A conserved tryptophan-rich motif in the membrane-proximal region of the human immunodeficiency virus type 1 gp41 ectodomain is important for Env-mediated fusion and virus infectivity. J. Virol. 1999;73:2469–2480. doi: 10.1128/jvi.73.3.2469-2480.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen S.S., Yang P., et al. Huang S.C. Identification of the LWYIK motif located in the human immunodeficiency virus type 1 transmembrane gp41 protein as a distinct determinant for viral infection. J. Virol. 2009;83:870–883. doi: 10.1128/JVI.01088-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhu P., Liu J., et al. Roux K.H. Distribution and three-dimensional structure of AIDS virus envelope spikes. Nature. 2006;441:847–852. doi: 10.1038/nature04817. [DOI] [PubMed] [Google Scholar]
- 6.Wu S.R., Löving R., et al. Garoff H. Single-particle cryoelectron microscopy analysis reveals the HIV-1 spike as a tripod structure. Proc. Natl. Acad. Sci. USA. 2010;107:18844–18849. doi: 10.1073/pnas.1007227107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sáez-Cirión A., Arrondo J.L., et al. Nieva J.L. Structural and functional roles of HIV-1 gp41 pretransmembrane sequence segmentation. Biophys. J. 2003;85:3769–3780. doi: 10.1016/S0006-3495(03)74792-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schibli D.J., Montelaro R.C., Vogel H.J. The membrane-proximal tryptophan-rich region of the HIV glycoprotein, gp41, forms a well-defined helix in dodecylphosphocholine micelles. Biochemistry. 2001;40:9570–9578. doi: 10.1021/bi010640u. [DOI] [PubMed] [Google Scholar]
- 9.Sun Z.Y., Oh K.J., et al. Reinherz E.L. HIV-1 broadly neutralizing antibody extracts its epitope from a kinked gp41 ectodomain region on the viral membrane. Immunity. 2008;28:52–63. doi: 10.1016/j.immuni.2007.11.018. [DOI] [PubMed] [Google Scholar]
- 10.Vishwanathan S.A., Hunter E. Importance of the membrane-perturbing properties of the membrane-proximal external region of human immunodeficiency virus type 1 gp41 to viral fusion. J. Virol. 2008;82:5118–5126. doi: 10.1128/JVI.00305-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shang L., Yue L., Hunter E. Role of the membrane-spanning domain of human immunodeficiency virus type 1 envelope glycoprotein in cell-cell fusion and virus infection. J. Virol. 2008;82:5417–5428. doi: 10.1128/JVI.02666-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zanetti G., Briggs J.A., et al. Fuller S.D. Cryo-electron tomographic structure of an immunodeficiency virus envelope complex in situ. PLoS Pathog. 2006;2:e83. doi: 10.1371/journal.ppat.0020083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harris A., Borgnia M.J., et al. Subramaniam S. Trimeric HIV-1 glycoprotein gp140 immunogens and native HIV-1 envelope glycoproteins display the same closed and open quaternary molecular architectures. Proc. Natl. Acad. Sci. USA. 2011;108:11440–11445. doi: 10.1073/pnas.1101414108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Frey G., Peng H., et al. Chen B. A fusion-intermediate state of HIV-1 gp41 targeted by broadly neutralizing antibodies. Proc. Natl. Acad. Sci. USA. 2008;105:3739–3744. doi: 10.1073/pnas.0800255105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brügger B., Glass B., et al. Kräusslich H.G. The HIV lipidome: a raft with an unusual composition. Proc. Natl. Acad. Sci. USA. 2006;103:2641–2646. doi: 10.1073/pnas.0511136103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lorizate M., Brügger B., et al. Kräusslich H.G. Probing HIV-1 membrane liquid order by Laurdan staining reveals producer cell-dependent differences. J. Biol. Chem. 2009;284:22238–22247. doi: 10.1074/jbc.M109.029256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Allende D., Simon S.A., McIntosh T.J. Melittin-induced bilayer leakage depends on lipid material properties: evidence for toroidal pores. Biophys. J. 2005;88:1828–1837. doi: 10.1529/biophysj.104.049817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McIntosh T.J., Simon S.A. Roles of bilayer material properties in function and distribution of membrane proteins. Annu. Rev. Biophys. Biomol. Struct. 2006;35:177–198. doi: 10.1146/annurev.biophys.35.040405.102022. [DOI] [PubMed] [Google Scholar]
- 19.Matsuzaki K., Sugishita K., et al. Miyajima K. Molecular basis for membrane selectivity of an antimicrobial peptide, magainin 2. Biochemistry. 1995;34:3423–3429. doi: 10.1021/bi00010a034. [DOI] [PubMed] [Google Scholar]
- 20.Prenner E.J., Lewis R.N., et al. McElhaney R.N. Cholesterol attenuates the interaction of the antimicrobial peptide gramicidin S with phospholipid bilayer membranes. Biochim. Biophys. Acta. 2001;1510:83–92. doi: 10.1016/s0005-2736(00)00337-0. [DOI] [PubMed] [Google Scholar]
- 21.Thennarasu S., Tan A., et al. Ramamoorthy A. Antimicrobial and membrane disrupting activities of a peptide derived from the human cathelicidin antimicrobial peptide LL37. Biophys. J. 2010;98:248–257. doi: 10.1016/j.bpj.2009.09.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sood R., Kinnunen P.K. Cholesterol, lanosterol, and ergosterol attenuate the membrane association of LL-37(W27F) and temporin L. Biochim. Biophys. Acta. 2008;1778:1460–1466. doi: 10.1016/j.bbamem.2008.02.014. [DOI] [PubMed] [Google Scholar]
- 23.Raghuraman H., Chattopadhyay A. Interaction of melittin with membrane cholesterol: a fluorescence approach. Biophys. J. 2004;87:2419–2432. doi: 10.1529/biophysj.104.043596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Arsov Z., Nemec M., et al. Zorko M. Cholesterol prevents interaction of the cell-penetrating peptide transportan with model lipid membranes. J. Pept. Sci. 2008;14:1303–1308. doi: 10.1002/psc.1062. [DOI] [PubMed] [Google Scholar]
- 25.Rebolj K., Bakrac B., et al. Sepcić K. EPR and FTIR studies reveal the importance of highly ordered sterol-enriched membrane domains for ostreolysin activity. Biochim. Biophys. Acta. 2010;1798:891–902. doi: 10.1016/j.bbamem.2010.01.016. [DOI] [PubMed] [Google Scholar]
- 26.Bakás L., Chanturiya A., et al. Zimmerberg J. Paradoxical lipid dependence of pores formed by the Escherichia coliα-hemolysin in planar phospholipid bilayer membranes. Biophys. J. 2006;91:3748–3755. doi: 10.1529/biophysj.106.090019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pokorny A., Almeida P.F. Permeabilization of raft-containing lipid vesicles by δ-lysin: a mechanism for cell sensitivity to cytotoxic peptides. Biochemistry. 2005;44:9538–9544. doi: 10.1021/bi0506371. [DOI] [PubMed] [Google Scholar]
- 28.Pokorny A., Yandek L.E., et al. Almeida P.F. Temperature and composition dependence of the interaction of delta-lysin with ternary mixtures of sphingomyelin/cholesterol/POPC. Biophys. J. 2006;91:2184–2197. doi: 10.1529/biophysj.106.085027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Montero M., van Houten N.E., et al. Scott J.K. The membrane-proximal external region of the human immunodeficiency virus type 1 envelope: dominant site of antibody neutralization and target for vaccine design. Microbiol. Mol. Biol. Rev. 2008;72:54–84. doi: 10.1128/MMBR.00020-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vishwanathan S.A., Thomas A., et al. Epand R.M. Hydrophobic substitutions in the first residue of the CRAC segment of the gp41 protein of HIV. Biochemistry. 2008;47:124–130. doi: 10.1021/bi7018892. [DOI] [PubMed] [Google Scholar]
- 31.Epand R.F., Thomas A., et al. Epand R.M. Juxtamembrane protein segments that contribute to recruitment of cholesterol into domains. Biochemistry. 2006;45:6105–6114. doi: 10.1021/bi060245+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Greenwood A.I., Pan J., et al. Tristram-Nagle S. CRAC motif peptide of the HIV-1 gp41 protein thins SOPC membranes and interacts with cholesterol. Biochim. Biophys. Acta. 2008;1778:1120–1130. doi: 10.1016/j.bbamem.2008.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Needham D., Nunn R.S. Elastic deformation and failure of lipid bilayer membranes containing cholesterol. Biophys. J. 1990;58:997–1009. doi: 10.1016/S0006-3495(90)82444-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Henriksen J., Rowat A.C., et al. Ipsen J.H. Universal behavior of membranes with sterols. Biophys. J. 2006;90:1639–1649. doi: 10.1529/biophysj.105.067652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wolf M.C., Freiberg A.N., et al. Lee B. A broad-spectrum antiviral targeting entry of enveloped viruses. Proc. Natl. Acad. Sci. USA. 2010;107:3157–3162. doi: 10.1073/pnas.0909587107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.St Vincent M.R., Colpitts C.C., et al. Schang L.M. Rigid amphipathic fusion inhibitors, small molecule antiviral compounds against enveloped viruses. Proc. Natl. Acad. Sci. USA. 2010;107:17339–17344. doi: 10.1073/pnas.1010026107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wimley W.C. Describing the mechanism of antimicrobial peptide action with the interfacial activity model. ACS Chem. Biol. 2010;5:905–917. doi: 10.1021/cb1001558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nicol F., Nir S., Szoka F.C., Jr. Effect of cholesterol and charge on pore formation in bilayer vesicles by a pH-sensitive peptide. Biophys. J. 1996;71:3288–3301. doi: 10.1016/S0006-3495(96)79521-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ladokhin A.S., Wimley W.C., et al. White S.H. Mechanism of leakage of contents of membrane vesicles determined by fluorescence requenching. Methods Enzymol. 1997;278:474–486. doi: 10.1016/s0076-6879(97)78025-x. [DOI] [PubMed] [Google Scholar]
- 40.Ivankin A., Kuzmenko I., Gidalevitz D. Cholesterol-phospholipid interactions: new insights from surface x-ray scattering data. Phys. Rev. Lett. 2010;104:108101. doi: 10.1103/PhysRevLett.104.108101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Neville F., Cahuzac M., et al. Gidalevitz D. Lipid headgroup discrimination by antimicrobial peptide LL-37: insight into mechanism of action. Biophys. J. 2006;90:1275–1287. doi: 10.1529/biophysj.105.067595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Neville F., Ishitsuka Y., et al. Gidalevitz D. Protegrin interaction with lipid monolayers: Grazing incidence x-ray diffraction and x-ray reflectivity study. Soft Matter. 2008;4:1665–1674. doi: 10.1039/b718295c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Buzon V., Natrajan G., et al. Weissenhorn W. Crystal structure of HIV-1 gp41 including both fusion peptide and membrane proximal external regions. PLoS Pathog. 2010;6:e1000880. doi: 10.1371/journal.ppat.1000880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen Z., Rand R.P. The influence of cholesterol on phospholipid membrane curvature and bending elasticity. Biophys. J. 1997;73:267–276. doi: 10.1016/S0006-3495(97)78067-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Graham D.R., Chertova E., et al. Hildreth J.E. Cholesterol depletion of human immunodeficiency virus type 1 and simian immunodeficiency virus with beta-cyclodextrin inactivates and permeabilizes the virions: evidence for virion-associated lipid rafts. J. Virol. 2003;77:8237–8248. doi: 10.1128/JVI.77.15.8237-8248.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Apellániz B., Nieva J.L., et al. García-Sáez A.J. All-or-none versus graded: single-vesicle analysis reveals lipid composition effects on membrane permeabilization. Biophys. J. 2010;99:3619–3628. doi: 10.1016/j.bpj.2010.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Huang H.W., Chen F.Y., Lee M.T. Molecular mechanism of peptide-induced pores in membranes. Phys. Rev. Lett. 2004;92:198304. doi: 10.1103/PhysRevLett.92.198304. [DOI] [PubMed] [Google Scholar]
- 48.Qian S., Wang W., et al. Huang H.W. Structure of transmembrane pore induced by Bax-derived peptide: evidence for lipidic pores. Proc. Natl. Acad. Sci. USA. 2008;105:17379–17383. doi: 10.1073/pnas.0807764105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Matsuzaki K., Sugishita K., et al. Epand R.M. Relationship of membrane curvature to the formation of pores by magainin 2. Biochemistry. 1998;37:11856–11863. doi: 10.1021/bi980539y. [DOI] [PubMed] [Google Scholar]
- 50.Lorizate M., Huarte N., et al. Nieva J.L. Interfacial pre-transmembrane domains in viral proteins promoting membrane fusion and fission. Biochim. Biophys. Acta. 2008;1778:1624–1639. doi: 10.1016/j.bbamem.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.