

Abstract
The significant variation among solved structures of the λ Cro dimer suggests its flexibility. However, contacts in the crystal lattice could have stabilized a conformation which is unrepresentative of its dominant solution form. Here we report on the conformational space of the Cro dimer in solution using replica exchange molecular dynamics in explicit solvent. The simulated ensemble shows remarkable correlation with available x-ray structures. Network analysis and a free energy surface reveal the predominance of closed and semi-open dimers, with a modest barrier separating these two states. The fully open conformation lies higher in free energy, indicating that it requires stabilization by DNA or crystal contacts. Most NMR models are found to be unstable conformations in solution. Intersubunit salt bridging between Arg4 and Glu53 during simulation stabilizes closed conformations. Because a semi-open state is among the low-energy conformations sampled in simulation, we propose that Cro-DNA binding may not entail a large conformational change relative to the dominant dimer forms in solution.
Introduction
The Cro transcription factor found in bacteriophage λ was the first specific DNA repressor protein to have its structure solved (1). Ever since, Cro has served as a prototypical system for studies ranging from gene regulation (2, 3) to protein fold evolution (4, 5, 6), protein dimerization (7, 8), and DNA-protein interactions (9, 10, 11). Cro and another repressor protein, cI, exhibit variable affinity for multiple binding sites within two operator regions of bacteriophage λ DNA to control switching between the lysogenic and lytic cycles during host infection (2, 12).
Cro establishes its functional protein-DNA complex as a dimer. Each monomer consists of 66 amino acids and an α+β fold with three α-helices and three β-strands. Helices α2 and α3 participate in a helix-turn-helix (HTH) motif responsible for DNA recognition. In its dimeric state, the β-strands form a pliable network connecting the two α-helical domains (Fig. 1 A). The most C-terminal β-strand (β3) of one monomer inserts into the hydrophobic core of an adjacent monomer and, together with hydrogen bonding between residues 54–56, forms the homodimer interface. The intrinsic mobility of the β2β3-hairpin of each subunit, along with a ball-and-socket joint formed about F58 at the dimer interface (Fig. 1 B), permit a hinge motion and a relative rotation between the two domains, respectively (13). Indeed, significant conformational rearrangement appears to be crucial for Cro-DNA binding (14, 15, 16), indicating that the dynamics of the β-hairpin region may result in varying the distance between the DNA recognition helices (RHs; α3 and α3′).
Figure 1.
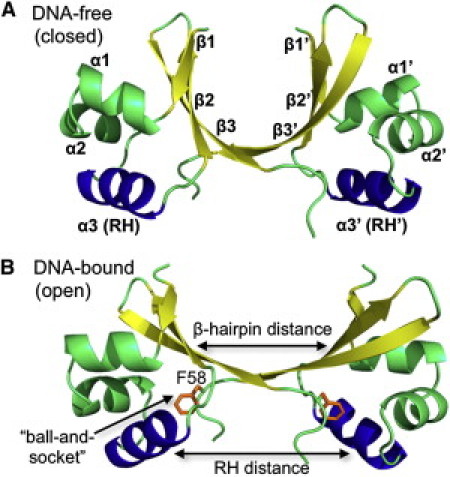
WT (A) apo (closed) and (B) DNA-bound (open) λ Cro dimer structures were the starting coordinates for REMD. Secondary structure motifs are labeled on the apo form, with β-strands shown in yellow and the DNA RHs (α3 and α3′) shown in blue. For clarity, termini added for simulation are not shown. The reaction coordinates defined as the distance between the β2β3-hairpin and RH of each subunit are labeled on the bound crystal structure in B and, along with RMSD measurements, were used to distinguish open/closed conformational transitions of the dimer throughout simulation. F58 of both subunits is shown in orange to highlight the location of the ball-and-socket joint in B.
The variation in global conformation observed in the solved Cro dimer crystal structures supports the notion of flexibility. Although the individual subunits of the apo (17) and bound (14) structures are relatively similar, the apo dimer displays a noticeably bent β-hairpin motif (representing a closed dimer structure; Fig. 1 A) in comparison with the DNA-complexed conformation, which has a relatively flat appearance in this region (i.e., an open structure; Fig. 1 B). This appears to be a textbook example of induced conformational transition. Recently, however, two crystal structures were solved for the same Cro dimer mutant (Q27P, A29S and K32Q; PSQ). They were crystallized unbound to DNA, although cognate DNA was present in the crystallization mixture (13). One crystal structure of the mutant (PSQ1) resembles the fully open DNA-bound form, whereas the other (PSQ2) displays a semi-open conformation intermediate between the apo and bound crystal structures. The mutations were located in the DNA RHs, far from the β-hairpin region, and are unlikely to be the cause of conformational differences. Instead, they may have resulted in a structural change by forming new crystal contacts. These x-ray images capture just a snapshot of the Cro dimer conformational ensemble in solution, and, from the standpoint of dimer flexibility, crystal packing could have selected a structure unrepresentative of its dominant solution conformation.
As suggested by the crystal structures, the NMR ensemble also emphasizes the notion of dimer flexibility through significant structural variation (18). The models display a 0.7–5.3 Å Cα root mean-square deviation (RMSD) with respect to one another. Compared with the x-ray images, the NMR models are more similar to open Cro than to closed Cro (2.7–4.8 Å and 2.0–6.7 Å RMSD to the open and closed dimers, respectively). However, the NMR dimer conformation is not well defined and must be interpreted with caution (18). Thus, further studies are needed to elucidate the true nature of the solution structure.
Here, we add dynamical information to the static x-ray images. We used replica exchange molecular dynamics (REMD) in an attempt to uncover the dominant forms and extent of accessible conformations of the dimer in solution. Simulations were performed starting from the wild-type (WT) apo (closed) and DNA-bound (open) x-ray structures. The x-ray images show remarkable consistency with the conformational space sampled by REMD, whereas the NMR structures are inconsistent with the simulated ensemble. Network analysis and a free energy profile show that two major conformational states exist in solution. One is a closed conformation similar to the apo Cro crystal structure. The other is a stable form that is similar to the DNA-bound structure, but is not fully open. Instead, this semi-open conformation closely resembles the recent PSQ2 mutant x-ray structure, for which no biological context was previously identified. The closed and semi-open states reside relatively close in energy, such that crystal contacts could easily stabilize one conformation over the other. Intersubunit salt bridging between Arg4 and Glu53 contributes to the stability of closed conformations. The fully open DNA-bound state lies higher in free energy, indicating that it requires the presence of DNA to be stable in solution. This also suggests that the PSQ1 mutant closely resembling the DNA-bound form was likely stabilized by crystal contacts. These results support an extension of the conformational selection model (19, 20) for Cro-DNA recognition in which the partially populated semi-open conformation initiates DNA contact before undergoing a residual degree of conformational adjustment to form the fully open state in the functional complex.
Materials and Methods
Setup and general MD
We performed REMD simulations starting from two x-ray structures: DNA-free (closed) and DNA-bound (open) dimeric λ Cro (Protein Data Bank (PDB) ID: 5CRO (17) and 6CRO (14), respectively; Fig. 1). These simulations were used for subsequent analyses. The term “combined trajectory” refers to a set of coordinates from both of these simulations. The asymmetric unit of 5CRO consists of a tetramer of two dimers (chains A-C and chains B-O). Both dimers are relatively similar (0.4 Å Cα RMSD for residues 4–56 of each subunit), and chains A-C were chosen. The missing terminal residues of the x-ray structures were added by alignment with an NMR model (PDB ID: 1COP) (18). Based on the observations from the trajectories started from the x-ray coordinates, a third REMD simulation was run from an NMR model as a control. The seventh reported model was chosen as the starting coordinates for this simulation because it corresponds to the centroid of the largest population from average linkage clustering of the NMR ensemble. Thus, we ran three independent simulations from three sets of initial coordinates (two x-ray structures and one NMR model) to ensure that our conclusions would not depend on the initial conditions.
Simulations were run using the Amber10 package (21, 22). Each simulated dimer had one neutral histidine (protonated at the epsilon position of the imidazole ring) per subunit and an overall +6 charge. The structures were aligned at the origin in VMD (23) and solvated in a cube of length 77 Å with TIP3P water molecules such that the longest direction of the dimer would always have at least ∼14 Å minimum padding. The systems were neutralized and protonated using tleap (24). The systems with x-ray coordinates contained 14,715 waters and 46,259 total atoms. The system with the NMR model had 14,702 waters and 46,220 total atoms. The FF99SB force field parameter set (25) was used. Energy minimization was performed as previously described (26). An additional minimization step relaxed backbone torsion angles of the added termini, with 500/500 steepest descent/conjugate gradient cycles with harmonic restraints applied to the rest of the protein backbone (500 kcal/(mol · Å2) force constant). The systems were then heated in the NPT ensemble to 300K over 20 ps with nonterminal main chain atoms still fixed. An initial equilibration with no positional restraints was run in the NPT ensemble at 300 K for 200 ps for each system.
REMD
REMD embodies the principles of MD as well as Monte Carlo methods to efficiently sample the conformational space of biomolecules (27, 28). The method combines frequent barrier crossing at higher temperatures with proper Boltzmann statistics at each temperature state. Thus, REMD significantly increases conformational sampling and has shown exceptional capability in studying protein dynamics in implicit (29, 30) and explicit (31, 32, 33) solvent. The algorithm independently runs several identical copies of a system, each of which exists in a unique temperature state. After a short interval of MD, a Monte Carlo step is performed to determine whether neighboring replicas may exchange their temperature states based on a Boltzmann-weighted probability.
Here, we applied REMD to a relatively large system, a dimer consisting of 66 amino acids per subunit solvated with >14,000 waters. Because both dimer dissociation and backbone denaturation of the λ Cro dimer are observed experimentally at 318 K (34), we were limited in choosing an adequate temperature range for REMD. Placement of a covalent bond at the dimer interface diminished the dynamics between the two subunits (16), so no restraints were placed at the interface during simulation. The presence of explicit water dramatically increases the energy of the system in comparison with dielectric continuum models, necessitating sufficiently small gaps between replica temperatures to achieve acceptance ratios that do not vanish to zero. Furthermore, the number of replicas required for REMD increases as O(f1/2), where f represents the total number of degrees of freedom in the system (35), which is large for an explicit solvent simulation. Thus, we chose an exchange ratio of 0.15 to ensure proper potential energy distributions while covering a sufficient temperature range with 24 replicas between 287.9 and 338.7 K. Because we chose a moderate temperature range and explicit solvent, we did not use chirality restraints typically employed during REMD. Other than SHAKE constraints (36) applied to bonds involving hydrogen atoms, no other constraints were placed on the system. The velocity Verlet algorithm was used. An 8 Å nonbonded cutoff was employed in direct calculations of electrostatic and van der Waals interactions. Periodic boundary conditions were used, and the particle mesh Ewald method with a grid spacing of 1 Å was used to calculate long-range electrostatics. A Langevin thermostat with a collision frequency parameter of 1 ps−1 maintained the temperature at each state. The minimized and NPT-equilibrated dimers served as the input coordinates for heating and equilibration at each of the 24 temperatures. The total equilibration time at each temperature was 500 ps. For all three simulations, each replica was run in the NVT ensemble for a total of 30 ns (720 ns when considering all 24 replicas). Exchanges were attempted every 1 ps. This corresponds to 500 integration steps (2 fs each), which has been estimated to be a suitable time between exchanges to properly construct a canonical ensemble at each temperature state (27). Each replica traversed from the lowest to the highest temperatures (see Fig. S1 in the Supporting Material). Coordinates were saved every 2 ps. The trajectories were analyzed at 300 K and processed with the use of ptraj (24), VMD (23), and PyMOL (37).
Network analysis and clustering
We constructed a network representation of the combined trajectory based on pairwise Cα RMSD measurements of trajectory frames to visualize the conformational ensemble of the Cro dimer (26, 38, 39). The unweighted force-directed layout algorithm that is part of Cytoscape (40) was used. This method considers a conformation from simulation as a node and connects two nodes by an edge if the RMSD calculated between them in the pairwise matrix falls below a defined cutoff (26) (see Fig. S2 for details). The algorithm treats the resulting graph as a heuristic physical system in which the edges act as springs, and all nodes feel a repulsive Coulomb-type interaction with respect to each other. The final layout is generated by minimization of the potential energy of the system. An RMSD cutoff of 1.7 Å was used to create the layout, which comprises 500 frames from 15–30 ns of the simulations started from the x-ray coordinates (250 frames from both trajectories, corresponding to every 60 ps). Networks comprising 500, 1000, and 1500 frames were nearly identical (data not shown). We used 500 frames for computational efficiency. After constructing the network, we performed clustering using the average linkage algorithm that is part of ptraj (41). We calculated five representative structures for 3750 frames from the combined and individual trajectories, and subsequently included the ones from the combined trajectory (as well as the x-ray and NMR structures) in the pairwise RMSD calculation to embed them in the network layout. The relationship between the network layout and clustering by the average linkage algorithm is shown in Fig. S3.
Reaction coordinate measurements
To report on Cro conformational transitions, we used the open and closed crystal structures as reference points in conformational space. RMSD values were calculated for Cα atoms of residues 4–56 and 4′–56′ of the dimer and used to define a ΔRMSD (42) as
where qtraj is the trajectory coordinates, and qopen and qclosed are the bound and apo crystal structure coordinates, respectively. We also defined backbone center-of-mass distances between the DNA RHs (residues 27–36 (α3) to 27′–36′ (α3′)) and between the β-hairpins of each subunit (residues 41–43 (β2), 51–53 (β3) to 41′–43′ (β2′), 51′–53′ (β3′); Fig. 1 B) to characterize open/closed transitions. The free-energy surface was constructed in the plane of these intersubunit distances. The probability distributions of the β-hairpin and RH distances sampled in the combined 300 K REMD trajectory (3750 frames from 15 to 30 ns) were measured and used to calculate the free energy. The reaction coordinate values for the Cro dimer structures are listed in Table 1.
Table 1.
Reaction coordinates for experimentally solved and representative REMD Cro dimers
| R1 | R2 | R3 | WT-apo | WT-bound | PSQ1 | PSQ2 | NMR | |
|---|---|---|---|---|---|---|---|---|
| ΔRMSD | 3.0 | −2.5 | 0.7 | 4.1 | −4.1 | −3.0 | −1.3 | −2.8–1.0 |
| RH distance | 33.5 | 31.0 | 32.3 | 32.9/32.8∗ | 28.9 | 29.3 | 32.0 | 18.1–30.7 |
| β-hairpin distance | 16.3 | 20.3 | 18.3 | 16.5/16.4∗ | 20.4 | 20.0 | 19.3 | 18.1–21.1 |
All values are reported in Angstroms.
The asymmetric unit of the WT apo dimer is a tetramer with chains A, B, C, and O, where A-C and B-O are the biological dimers. The RH distances for chains A-C and B-O are 32.9 Å and 32.8 Å, respectively. Similarly, the β-hairpin distances for chains A-C and B-O are 16.5 Å and 16.4 Å, respectively.
Results
Trajectories show conformational transitions
The enhanced sampling of the REMD trajectories started from the x-ray structures yielded a large number of conformational transitions, and the simulations sampled the same magnitude of ΔRMSD values and distances encompassing the coordinates corresponding to the open and closed dimer crystal structures (Fig. 2). (Reaction coordinate values for experimentally solved Cro dimers are listed in Table 1.) Transitions along the ΔRMSD coordinate show that conformations similar to the fully open dimer crystal structure (∼ −4 Å) are rarely sampled by either trajectory (Fig. 2 A). Rather, semi-open dimer structures (between −1 Å and −2.5 Å) are frequently reached (discussed in further detail below). The trajectory from closed Cro shows several transitions from closed-like conformations to semi-open conformations throughout the simulation (Fig. 2, A–C). The trajectory from open Cro primarily samples intermediate and semi-open conformations during the first half of the simulation before showing transitions to closed dimer coordinates after 15 ns (Fig. 2, A–C), which indicates that the first half of the simulation (0–15 ns) is not fully equilibrated. Thus, we subsequently analyzed the last half of both simulations (15–30 ns) to describe the Cro dimer conformational space. We projected this portion of each trajectory onto the plane of RMSD-to-open and RMSD-to-closed Cro to further examine the conformational sampling (Fig. S4). Both simulations explored a comparable region of conformational space and visited closed and semi-open dimer structures despite starting from notably different sets of initial coordinates.
Figure 2.
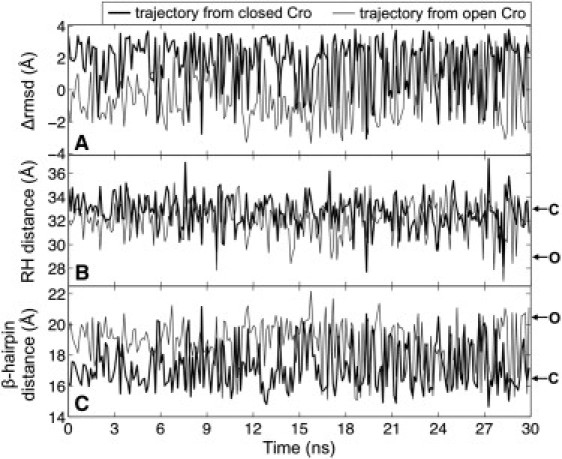
The (A) ΔRMSD, (B) RH distance, and (C) β-hairpin distance were the reaction coordinates of interest. In panel A, the maximum and minimum ΔRMSD values (+4.1 Å and −4.1 Å) correspond to the closed and open x-ray structure coordinates, respectively. Similarly, in B and C, the letters C and O respectively correspond to the coordinates for the closed and open dimer crystal structures. 15–30 ns of both trajectories was used for network analysis and constructing the free energy surface.
Network analysis illustrates the conformational ensemble
Network analysis permits visualization of the region of phase space sampled by the REMD trajectories (Fig. 3). The resulting interaction network defines similarity between the conformational substates visited during the simulation and shows how the experimentally solved Cro dimer structures relate to this space. The network layout comprises frames from both trajectories started from the crystal structures and was constructed with a 1.7 Å pairwise Cα RMSD cutoff. Four main clusters are evident, one of which belongs solely to NMR ensemble members. Both trajectories sample within the two largest populations, whereas only the trajectory from the closed dimer visits the smallest collection of nodes from simulation. Overall, the conformational space sampled by the REMD trajectories agrees exceptionally well with the x-ray images since all four of the crystal structures fall within the two major clusters of the layout. This is not the case for the NMR models: only two of the nodes of the NMR ensemble are connected to the nodes from the simulation, whereas the others only show connections among themselves. This is reasonable when considering that none of the NMR models yield an RMSD < 2 Å when aligned to either the closed or open x-ray structures (Table 2). The difference between the NMR ensemble and the conformational space sampled by REMD is further highlighted by the free energy surface as well as an additional REMD simulation begun from an NMR model (both discussed below).
Figure 3.
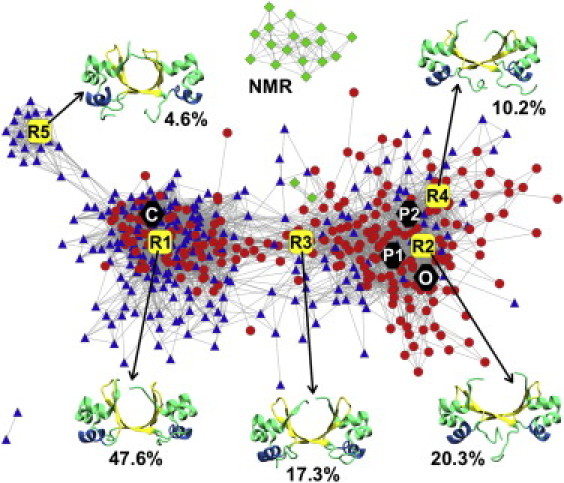
Network analysis highlights the conformational space of the Cro dimer. The force-directed layout algorithm and a 1.7 Å Cα pairwise RMSD cutoff for 500 frames from the combined REMD trajectory were used. Structures from the simulations started from the closed and open WT Cro dimer crystal structures are represented by blue triangles and red circles, respectively. The location of the closed (C), open (O), PSQ1 mutant (P1), and PSQ2 mutant (P2) dimer crystal structures within the layout are shown by black hexagons. The NMR ensemble is represented by green diamonds. The five representative structures calculated by clustering with the average linkage algorithm are shown in cartoon representation and depicted as large yellow squares in the network (R1 (47.6%), R2 (20.3%), R3 (17.3%), R4 (10.2%), and R5 (4.6%), where the percentages refer to the proportion of the combined REMD ensemble in each cluster.
Table 2.
Pairwise RMSD comparison of Cro dimer structures
| R1 | R2 | R3 | WT-apo | WT-bound | PSQ1 | PSQ2 | NMR | |
|---|---|---|---|---|---|---|---|---|
| R1 | N.A. | 4.2 | 2.4 | 1.7 | 4.6 | 4.3 | 3.9 | 3.0–7.4 |
| R2 | N.A. | 2.5 | 4.0 | 1.5 | 1.2 | 1.9 | 3.0–5.6 | |
| R3 | N.A. | 2.1 | 2.8 | 2.6 | 2.3 | 2.1–6.2 | ||
| WT-apo | (0.4)∗ | 4.1 | 4.0 | 3.7 | 2.0–6.7 | |||
| WT-bound | N.A. | 1.0 | 2.4 | 2.7–4.8 | ||||
| PSQ1 | N.A. | 1.9 | 2.9–5.3 | |||||
| PSQ2 | N.A. | 3.2–6.6 | ||||||
| NMR | (0.7–5.3) |
All RMSD values are reported in Angstroms and were calculated for residues 4–56 and 4′–56′ for the dimer. R1, R2, and R3 are the top three ranked representative structures as calculated by the average linkage algorithm for the combined REMD trajectory. WT-apo, WT-bound, PSQ1, and PSQ2 are the four Cro dimer crystal structures, and NMR is the experimentally solved solution structure.
A 0.4 Å difference exists between dimers A-C and B-O for the WT-apo x-ray structure. Dimer A-C was used for the pairwise calculations.
Representative conformations were calculated for the combined REMD trajectory. These structures show a high degree of similarity to those calculated for the individual REMD trajectories (Fig. S5 and Table S1), indicating that sufficient REMD sampling was achieved for our discussion. The highest-ranking representative structure from the average linkage algorithm (R1) denotes the centroid of the population that includes the closed (free) Cro crystal structure and represents 47.6% of the ensemble (Fig. 4 A and Table 2). The most notable differences between these two structures occur in the displacement of the N-terminal β1-strands and the increased rotation of one β-hairpin of R1. In the crystal of the DNA-free dimer, β1-β1′ strand interaction forms a closed ring between dimer structures, resulting in a tetrameric arrangement of the asymmetric unit that could have distorted the dimer in the lattice. For R1, the RH distance is 0.6 Å greater and the termini of these helices show a less tight helical geometry than the closed crystal structure. The β-hairpin distance is similar between the two structures. Appended to the largest population on the layout is the smallest cluster, corresponding to the lowest-ranked centroid (R5, 4.6%; Fig. 3). This structure shows an overall closed conformation, but differs from R1 in the noticeable asymmetry about the β-hairpin region.
Figure 4.
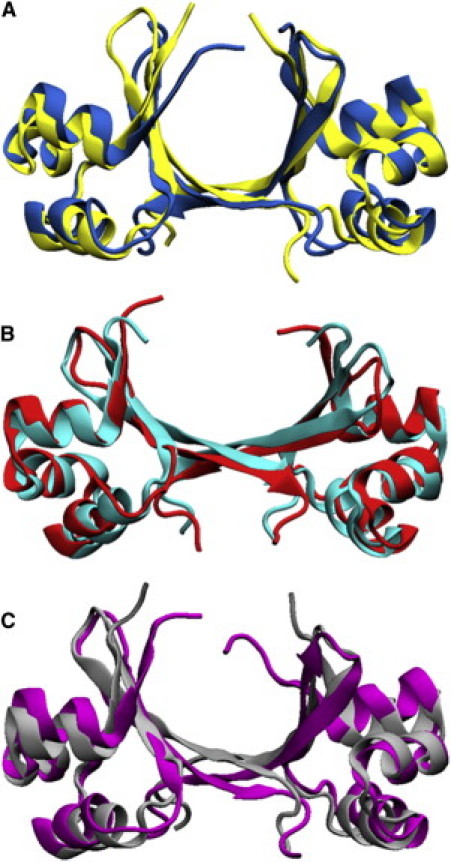
(A) The most representative closed dimer conformation from REMD (R1, blue) aligned with the closed Cro x-ray image (yellow; 1.7 Å RMSD). (B) The most representative open dimer from simulation (R2, red) overlaid with the DNA-bound open Cro crystal structure (cyan; 1.5 Å RMSD). (C) The representative intermediate structure from REMD (R3, purple) aligned with the ninth reported NMR model (gray; 2.1 Å RMSD).
The second-ranked representative conformation, R2 (20.3%), falls in the next-largest collection of nodes, and is an open-like dimer conformation (Fig. 4 B and Table 2). R2 displays roughly the same β-hairpin distance as the open crystal structure, and there is no notable difference in the rotation of either of the β-hairpins when the two structures are aligned. The RH distance of the open Cro x-ray image is 2.1 Å greater than R2, although the orientation of the helices is similar. R2 yields comparable RMSD values when aligned with either the fully open or semi-open x-ray structures (Table 2). The population comprising open-like dimer conformations is more disperse than the collection of nodes containing the closed crystal structure. As a result, it was split into two clusters (centroids R2 and R4) before the centroid for the smallest population (R5) could be determined. R2 and R4 display comparable overall conformations (1.9 Å RMSD) and together represent 30.5% of the ensemble. R4 is a semi-open dimer, displaying a 2.6 Å and 1.2 Å RMSD to the fully open DNA-bound and the semi-open PSQ2 mutant x-ray structures, respectively.
Between the two largest populations are structures that are intermediate of the open and closed dimer crystal structures. The third-ranked representative structure (R3, 17.3%) is located in this region (Fig. 3 and Table 2). In close proximity to R3 are the two NMR ensemble members that fall within the body of the network (the ninth and 18th reported structures). Alignment of R3 with the ninth and 18th reported NMR models yields a 2.1 Å and 2.3 Å RMSD, respectively. Fig. 4 C compares R3 and the ninth reported NMR model. The β3-β3′ portion of the dimer interface appears flatter in the NMR structure than in R3, and the β1 strands differ in conformation. The NMR model also displays a shorter RH distance than R3 by 2.2 Å. No significant difference due to β-hairpin rotation is present.
Free energy surface of Cro dimer conformations in solution
We calculated a two-dimensional free energy surface spanning the plane of the β-hairpin and RH distances for a set of coordinates from both REMD trajectories started from the x-ray structures after equilibration (Fig. 5). The landscape displays a topology similar to that of the network layout, and shows a moderate free energy range of 0–2.25 kcal/mol and two minima separated by a ∼1 kcal/mol barrier. An overall trend for decreasing RH distances with increasing β-hairpin distances (similar to the plotted crystal structures and NMR models) is evident as well. The most prominent basin falls within a 32–34.5 Å RH and 15–17.5 Å β-hairpin distance range. The coordinates of the closed crystal structure and R1 are located in this minimum. The other basin occurs at shorter RH (30.5–32.5 Å) and longer β-hairpin distances (19–20 Å), and most closely coincides with the PSQ2 mutant x-ray structure and R2, indicating that these structures represent semi-open conformations that are stable in solution. Such agreement is noteworthy because both of the two partially stable conformations observed in MD simulations are indeed observed as x-ray crystallographic structures. Small differences in the crystal packing interfaces would explain why different conformations were selected during the crystallization process. The DNA-bound and PSQ1 mutant crystal structures (the fully open forms) are located ∼1.25 and 1.0 kcal/mol from this minimum, respectively, and thus lie higher in free energy than the closed crystal structure. This indicates that the WT bound conformation was due to DNA binding, and the similar open form of the PSQ1 mutant was most likely stabilized by crystal contacts. The R3 structure falls at ∼0.75 kcal/mol and corresponds to coordinates intermediate of the open and closed basins.
Figure 5.
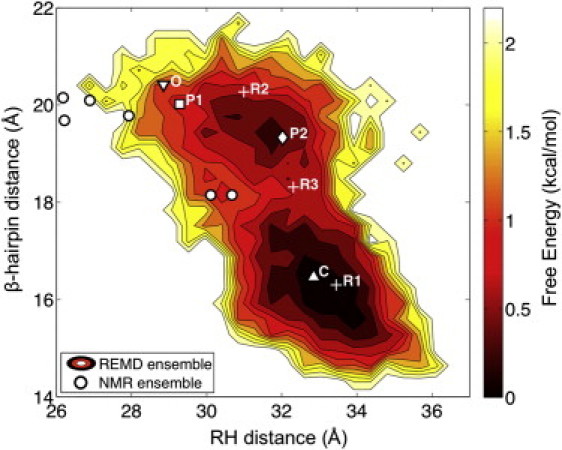
Free-energy surface in the plane of the β-hairpin and RH distances. The landscape was calculated for the combined REMD trajectory. Experimentally solved Cro dimer structures are plotted on the surface for comparison with the simulated ensemble. The representative and x-ray structures are labeled as in Fig. 3. The NMR structures that fall within the histogram bin windows are shown as open circles.
Perhaps the most striking feature of the landscape, given its functional relevance, is that the RH coordinate corresponding to the DNA-bound Cro x-ray structure (29 Å) is accessible only when the β-hairpins are in an open conformation (>19 Å). The RH distance for the DNA-free crystal structure (33 Å) is roughly <1 kcal/mol from the minimum over almost the entire range of β-hairpin coordinates. Despite the large population differences of the RH distances corresponding to the closed and open crystal structures, the β-hairpin coordinates corresponding to both of these structures are significantly sampled by the trajectories. This implies that the dynamics of the β-hairpins govern the transition to open dimer states with shorter RH distances (<30 Å) as displayed by the DNA-bound x-ray structure. Most of the open-like dimer conformations observed during the simulation display RH distances that are more in agreement with the PSQ2 mutant dimer.
The NMR ensemble was also plotted onto the free-energy surface. The NMR structures are notably biased toward large β-hairpin and small RH distances (Fig. 5 and Table 1)—just two of the 20 reported structures (the ninth and 18th) fall within ∼1 kcal/mol of the minimum. These are the only two NMR models that display an RH distance > 28 Å and a β-hairpin distance < 19.5 Å, and most display coordinates beyond the range of the conformational space sampled by the REMD simulation (RH and β-hairpin distances ranging from 18 Å to 26 Å and 19.5 Å to 21 Å, respectively). Consequently, all but four NMR models lie >2.25 kcal/mol.
To ensure that this observation was not the result of insufficient sampling, we started an additional REMD simulation from the NMR model that is the centroid of the separate NMR population shown in the network layout. This is the seventh reported model, which displays RH and β-hairpin distances of 24.0 Å and 20.4 Å, respectively. This trajectory immediately drifts from its initial coordinates within 10 ns, and over the course of 30 ns samples a range of conformational space similar to that sampled by the REMD simulations started from the x-ray structures (Fig. S6). This indicates no free-energy barrier separates the x-ray and NMR models, and that most of the conformations depicted by the NMR ensemble are not stable dimer states in solution.
Inter- and intrasubunit salt bridging
Given the predominance of both closed and semi-open dimer conformations, we searched for specific structural underpinnings that might stabilize these two states in solution. The REMD trajectories show significant inter- and intrasubunit interactions between Arg4 and Glu53 in the form of salt bridges. Due to the symmetric nature of the dimer, either intersubunit salt bridge may form (Arg4-Glu53′ or Arg4′-Glu53), and both appear to stabilize closed dimer conformations (Fig. 6, A–C). This salt bridge is shown to be a characteristic of the conformations comprising the largest population (closed-like dimers) of the network layout (Fig. 6 G). It is present throughout the entire simulation from the closed dimer and begins to form after approximately the first half of the simulation from open Cro, after which significant sampling of positive ΔRMSD values in this trajectory is observed (data not shown). At least one intrasubunit salt bridge (Arg4-Glu53 or Arg4′-Glu53′) is present throughout both simulations. The dimer does not show a preference for open or closed conformations upon formation of this interaction (Fig. 6, D–F). Salt bridging between Arg4 and Glu53 would contribute to the stability of both open and closed dimers in simulation without DNA, which is not obvious from the crystal structures, and the breaking of the intersubunit interaction may be a prerequisite for the transition from closed to open states.
Figure 6.
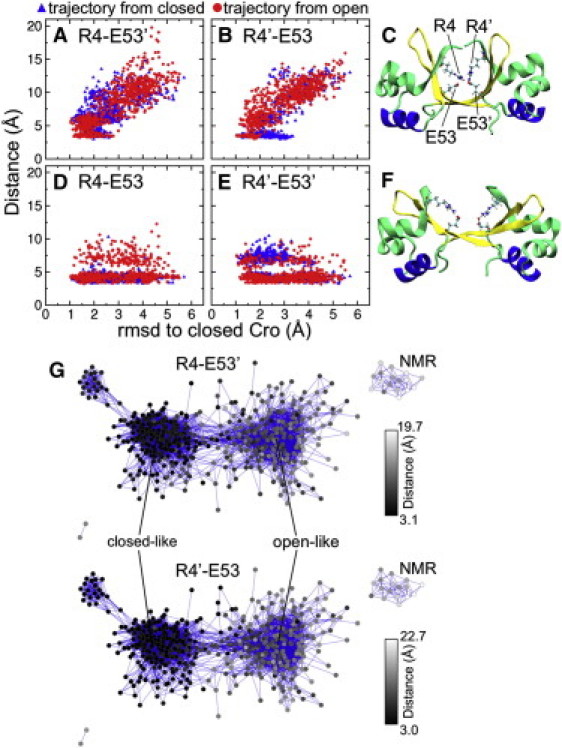
(A and B) Correlation between the intersubunit salt bridge distance (Arg4-Glu53′ and Arg4′-Glu53) and the RMSD to the closed crystal structure. (C) The structure from the combined REMD trajectory closest to the closed Cro x-ray image (1.0 Å RMSD) displays the intersubunit salt bridge between R4 and E53′. (D and E) Correlation between the intrasubunit salt bridge distance (Arg4-Glu53 and Arg4′-Glu53′) and the RMSD to closed Cro. (F) The conformation most like open Cro (1.0 Å RMSD) shows the intrasubunit salt bridges between these residues. (G) Intersubunit salt bridging as highlighted by the network layout. The Arg4-Glu53′ and Arg4′-Glu53 distances are represented as a color gradient in the network; black corresponds to shorter distances, and gray-white indicates larger distances. All data shown are for the last half of simulation (15–30 ns). Distances were measured between the centers-of-mass of Nη1 and Nη2 of arginine, and Oε1 and Oε2 of glutamate.
Discussion
Comparison of the REMD trajectories with x-ray and NMR
Despite a wealth of information from x-ray structures and NMR, the nature of the Cro dimer conformational equilibrium in solution has remained unknown. Our REMD-derived ensemble comprises both closed and open dimer structures separated by a modest energy barrier, such that different contacts in the crystal lattice could select either of these energetically close conformations. This would explain why one recently solved DNA-free mutant x-ray structure (PSQ1) displays remarkable resemblance to its DNA-bound conformation (13). The mutations, each within the DNA RHs, play a role in crystal packing and the new contacts may have stabilized a fully open conformation. This mutant was also solved in another crystal form (PSQ2). In addition to mutation, the extended β-sheet structure in this lattice may have shifted the conformational equilibrium of the dimer toward a semi-open state. Yet both mutant structures are accessible in solution and appear within the population of open-like dimers in the network layout. The free-energy surface clearly shows that semi-open conformations similar to that of PSQ2 are more stable than fully open ones.
A key structural feature revealed by the REMD simulations is intersubunit salt bridging, which is also a characteristic of the WT free Cro asymmetric unit. Crystal contacts between β1 strands in this lattice result in a dimer-dimer interface that satisfies salt bridging similarly to what is observed in simulation. Specifically, Arg4 of each monomer salt bridges with Glu2 of a neighboring chain (in contrast to the Arg4 and Glu53 interaction seen in simulation), likely stabilizing a closed dimer conformation similar to the most representative structure from the REMD trajectories. In fact, the WT DNA-free crystal form is the only Cro dimer x-ray structure that displays an intersubunit salt bridge. The presence of DNA or new crystal contacts due to mutation could have prevented the formation of this interaction in the bound and mutant Cro lattices. This appears to be a case in which x-ray crystallography revealed a crucial structural feature, but where the dynamical insight unique to simulation was needed to uncover the nature of this interaction in the context of the solution ensemble.
Unlike the x-ray structures, the Cro dimer NMR models differ markedly from the REMD-derived ensemble. The β-hairpin and RH distances are significantly over- and underestimated, respectively, in comparison with our simulations. Furthermore, no NMR ensemble member displays a conformation similar to the WT free crystal structure, whereas our simulations clearly show the predominance of such conformations. Matsuo et al. (18) stated, “In general, it is difficult to determine a dimer conformation precisely by means of NMR, since rather small numbers of inter-subunit constraints are obtained.” Because of their short-range nature, very few intersubunit nuclear Overhauser enhancement restraints (just 40 out of 1576 total) near the hinge region were available for constructing the models. Without sufficient distance information between subunits, the resulting model would strongly depend on the structure determination protocol, and inaccuracy in the energy function (which did not contain an electrostatic term) may be a source of the disagreement in conformational space. Additional information from residual dipolar couplings may help to refine the relative subunit orientations, perhaps yielding NMR models in better agreement with x-ray crystallography and simulation. In addition to the disparity in global conformation, none of the NMR models include the critical intersubunit salt bridge observed in simulation.
Biological implication
The REMD trajectories not only permit comparison with experimentally solved structures, but also provide new insight into Cro-DNA binding. Previous work suggested an induced-fit mechanism in which Cro may first bind nonspecifically to DNA in a form resembling its apo conformation (43). A structural transition may then take place as the protein diffuses to its cognate operator site, resulting in an open dimer structure that maximizes favorable contacts with DNA basepairs. In contrast to this view, Hall et al. (13), after solving the PSQ mutant structures, proposed that the dimer may instead undergo a significant conformational change upon initial DNA binding before transitioning to a structure similar to its dominant solution form in the specific complex.
A tentative model for Cro-DNA binding may be accrued from the free-energy surface obtained from REMD. Although conformations similar to the bound dimer are accessible during simulation, the RH distance of this structure is infrequently sampled. Instead, the stable open-like conformations from simulation, which closely resemble the PSQ2 mutant, display RH distances >2 Å longer than that of the bound crystal structure, indicating that the transition from the semi-open to the fully open form requires additional energetic contribution. Moreover, the intersubunit salt bridge observed during simulation appears to stabilize closed conformations. On the basis of these observations, we propose that closed conformations represent the main dimeric form of Cro in solution. Upon breakage of the intersubunit salt bridge, the dimer may adopt semi-open conformations similar to the PSQ2 mutant structure. Such conformations may bind initially to DNA before transitioning to the fully open state as the protein diffuses to its operator site. Thus, sequence-specific Cro-DNA binding may not necessitate large dimer distortions relative to the protein's dominant solution conformations (13), supporting an extension of the original conformational selection model in which initial binding is followed by a degree of residual conformational adjustment (19, 20). Because only a modest free-energy barrier separates dimer states in simulation, a significant portion of the energetic payoff from complex formation could be directed toward driving local conformational change in DNA. This would contribute to the bending of DNA as seen in the crystal structure of the specific complex between Cro and its operator sequence (14).
Conclusion
The REMD-derived ensemble describes the Cro dimer conformational equilibrium in solution. The x-ray images show remarkable consistency with the conformational space sampled by REMD. Closed and semi-open dimer states dominate the solution ensemble and are only separated by a modest free energy barrier such that crystal contacts can select either form. Closed conformations may be slightly more stable than semi-open forms in solution due to intersubunit salt bridging. The fully open conformation appears to be stabilized by DNA-binding or contacts in the crystal lattice since it lies higher in free energy in solution. On the other hand, the NMR structures are inconsistent with the global conformations sampled during REMD, probably due to the conditions under which the structure calculation was performed. The prevalence of semi-open states in simulation supports an extension of the conformational selection model in which such conformations similar to the PSQ2 mutant crystal structure may initially recognize DNA, and subsequent dimer distortions needed to achieve the specific complex may be relatively minor.
Acknowledgments
We thank Christian Roessler and Matthew H. J. Cordes for helpful discussions. The calculations were performed at the University of Arizona High Performance Computing Center.
This research was supported by the National Institutes of Health (training grant GM084905 to L.S.A.) and Achievement Rewards for College Scientists (Phoenix chapter, to L.S.A.).
Editor: Nathan Andrew Baker.
Footnotes
Six figures are available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(11)01206-9.
Supporting Material
References
- 1.Anderson W.F., Ohlendorf D.H., et al. Matthews B.W. Structure of the Cro repressor from bacteriophage λ and its interaction with DNA. Nature. 1981;290:754–758. doi: 10.1038/290754a0. [DOI] [PubMed] [Google Scholar]
- 2.Ptashne M. Cell Press; Cambridge: 1986. A Genetic Switch: Gene Control and Phage λ. [Google Scholar]
- 3.Svenningsen S.L., Costantino N., et al. Adhya S. On the role of Cro in λ prophage induction. Proc. Natl. Acad. Sci. USA. 2005;102:4465–4469. doi: 10.1073/pnas.0409839102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Newlove T., Konieczka J.H., Cordes M.H.J. Secondary structure switching in Cro protein evolution. Structure. 2004;12:569–581. doi: 10.1016/j.str.2004.02.024. [DOI] [PubMed] [Google Scholar]
- 5.Roessler C.G., Hall B.M., et al. Cordes M.H. Transitive homology-guided structural studies lead to discovery of Cro proteins with 40% sequence identity but different folds. Proc. Natl. Acad. Sci. USA. 2008;105:2343–2348. doi: 10.1073/pnas.0711589105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Van Dorn L.O., Newlove T., et al. Cordes M.H. Relationship between sequence determinants of stability for two natural homologous proteins with different folds. Biochemistry. 2006;45:10542–10553. doi: 10.1021/bi060853p. [DOI] [PubMed] [Google Scholar]
- 7.Darling P.J., Holt J.M., Ackers G.K. Coupled energetics of λ Cro repressor self-assembly and site-specific DNA operator binding I: analysis of Cro dimerization from nanomolar to micromolar concentrations. Biochemistry. 2000;39:11500–11507. doi: 10.1021/bi000935s. [DOI] [PubMed] [Google Scholar]
- 8.Jia H.F., Satumba W.J., et al. Mossing M.C. Slow assembly and disassembly of λ Cro repressor dimers. J. Mol. Biol. 2005;350:919–929. doi: 10.1016/j.jmb.2005.05.054. [DOI] [PubMed] [Google Scholar]
- 9.Jana R., Hazbun T.R., et al. Mossing M.C. A folded monomeric intermediate in the formation of λ Cro dimer-DNA complexes. J. Mol. Biol. 1997;273:402–416. doi: 10.1006/jmbi.1997.1256. [DOI] [PubMed] [Google Scholar]
- 10.Takeda Y., Kim J.G., et al. Matthews B.W. Different interactions used by Cro repressor in specific and nonspecific DNA binding. J. Biol. Chem. 1986;261:8608–8616. [PubMed] [Google Scholar]
- 11.Takeda Y., Sarai A., Rivera V.M. Analysis of the sequence-specific interactions between Cro repressor and operator DNA by systematic base substitution experiments. Proc. Natl. Acad. Sci. USA. 1989;86:439–443. doi: 10.1073/pnas.86.2.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Anderson W.F., Takeda Y., et al. Matthews B.W. The structure of a repressor: crystallographic data for the Cro regulatory protein of bacteriophage λ. J. Mol. Biol. 1979;130:507–510. doi: 10.1016/0022-2836(79)90437-6. [DOI] [PubMed] [Google Scholar]
- 13.Hall B.M., Roberts S.A., et al. Cordes M.H. Two structures of a λ Cro variant highlight dimer flexibility but disfavor major dimer distortions upon specific binding of cognate DNA. J. Mol. Biol. 2008;375:802–811. doi: 10.1016/j.jmb.2007.10.082. [DOI] [PubMed] [Google Scholar]
- 14.Albright R.A., Matthews B.W. Crystal structure of λ Cro bound to a consensus operator at 3.0 Å resolution. J. Mol. Biol. 1998;280:137–151. doi: 10.1006/jmbi.1998.1848. [DOI] [PubMed] [Google Scholar]
- 15.Kyogoku Y., Kojima C., et al. Shirakawa M. Induced structural changes in protein-DNA complexes. Methods Enzymol. 1995;261:524–541. doi: 10.1016/s0076-6879(95)61023-5. [DOI] [PubMed] [Google Scholar]
- 16.Shirakawa M., Matsuo H., Kyogoku Y. Intersubunit disulfide-bonded λ Cro protein. Protein Eng. 1991;4:545–552. doi: 10.1093/protein/4.5.545. [DOI] [PubMed] [Google Scholar]
- 17.Ohlendorf D.H., Tronrud D.E., Matthews B.W. Refined structure of Cro repressor protein from bacteriophage λ suggests both flexibility and plasticity. J. Mol. Biol. 1998;280:129–136. doi: 10.1006/jmbi.1998.1849. [DOI] [PubMed] [Google Scholar]
- 18.Matsuo H., Shirakawa M., Kyogoku Y. Three-dimensional dimer structure of the λ Cro repressor in solution as determined by heteronuclear multidimensional NMR. J. Mol. Biol. 1995;254:668–680. doi: 10.1006/jmbi.1995.0646. [DOI] [PubMed] [Google Scholar]
- 19.Csermely P., Palotai R., Nussinov R. Induced fit, conformational selection and independent dynamic segments: an extended view of binding events. Trends Biochem. Sci. 2010;35:539–546. doi: 10.1016/j.tibs.2010.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wlodarski T., Zagrovic B. Conformational selection and induced fit mechanism underlie specificity in noncovalent interactions with ubiquitin. Proc. Natl. Acad. Sci. USA. 2009;106:19346–19351. doi: 10.1073/pnas.0906966106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Case D.A., Cheatham T.E., 3rd, et al. Woods R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005;26:1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Case D.A., Darden T.A., et al. Kollman P.A. University of California; San Francisco: 2008. AMBER 10. [Google Scholar]
- 23.Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. 27–28. [DOI] [PubMed] [Google Scholar]
- 24.Case, D. A., editor. 2008. AmberTools Users' Manual. Available from: http://ambermd.org/.
- 25.Duan Y., Wu C., et al. Kollman P. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem. 2003;24:1999–2012. doi: 10.1002/jcc.10349. [DOI] [PubMed] [Google Scholar]
- 26.Vorontsov I.I., Miyashita O. Solution and crystal molecular dynamics simulation study of m4-cyanovirin-N mutants complexed with di-mannose. Biophys. J. 2009;97:2532–2540. doi: 10.1016/j.bpj.2009.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nymeyer H., Gnanakaran S., García A.E. Atomic simulations of protein folding, using the replica exchange algorithm. Methods Enzymol. 2004;383:119–149. doi: 10.1016/S0076-6879(04)83006-4. [DOI] [PubMed] [Google Scholar]
- 28.Sugita Y., Okamoto Y. Replica-exchange molecular dynamics method for protein folding. Chem. Phys. Lett. 1999;314:141–151. [Google Scholar]
- 29.Ganguly D., Chen J.H. Atomistic details of the disordered states of KID and pKID. Implications in coupled binding and folding. J. Am. Chem. Soc. 2009;131:5214–5223. doi: 10.1021/ja808999m. [DOI] [PubMed] [Google Scholar]
- 30.Pitera J.W., Swope W. Understanding folding and design: replica-exchange simulations of “Trp-cage” miniproteins. Proc. Natl. Acad. Sci. USA. 2003;100:7587–7592. doi: 10.1073/pnas.1330954100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.García A.E. In: Methods in Molecular Biology. 2nd ed. Zaki M., Bystroff C., editors. Humana Press; Totowa, NJ: 2008. Molecular dynamics simulations of protein folding; pp. 315–330. [DOI] [PubMed] [Google Scholar]
- 32.Paschek D., Nymeyer H., García A.E. Replica exchange simulation of reversible folding/unfolding of the Trp-cage miniprotein in explicit solvent: on the structure and possible role of internal water. J. Struct. Biol. 2007;157:524–533. doi: 10.1016/j.jsb.2006.10.031. [DOI] [PubMed] [Google Scholar]
- 33.Sanbonmatsu K.Y., García A.E. Structure of Met-enkephalin in explicit aqueous solution using replica exchange molecular dynamics. Proteins. 2002;46:225–234. doi: 10.1002/prot.1167. [DOI] [PubMed] [Google Scholar]
- 34.Shirakawa M., Lee S.J., et al. Kyogoku Y. In: Structure and Expression. Sarma R.H., Sarma H.H., editors. Adenine Press; New York: 1987. Interaction of λ-Cro repressor with operator DNA and induced conformational change; pp. 167–179. [Google Scholar]
- 35.Fukunishi H., Watanabe O., Takada S. On the Hamiltonian replica exchange method for efficient sampling of biomolecular systems: application to protein structure prediction. J. Chem. Phys. 2002;116:9058–9067. [Google Scholar]
- 36.Ryckaert J.P., Ciccotti G., Berendsen H.J.C. Numerical integration of Cartesian equations of motion of a system with constraints—molecular dynamics of N-alkanes. J. Comput. Phys. 1977;23:327–341. [Google Scholar]
- 37.DeLano W.L. DeLano Scientific; Palo Alto, CA: 2008. The PyMOL Molecular Graphics System. [Google Scholar]
- 38.Campbell Z.T., Baldwin T.O., Miyashita O. Analysis of the bacterial luciferase mobile loop by replica-exchange molecular dynamics. Biophys. J. 2010;99:4012–4019. doi: 10.1016/j.bpj.2010.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rao F., Caflisch A. The protein folding network. J. Mol. Biol. 2004;342:299–306. doi: 10.1016/j.jmb.2004.06.063. [DOI] [PubMed] [Google Scholar]
- 40.Shannon P., Markiel A., et al. Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shao J.Y., Tanner S.W., et al. Cheatham T.E., 3rd Clustering molecular dynamics trajectories: 1. Characterizing the performance of different clustering algorithms. J. Chem. Theory Comput. 2007;3:2312–2334. doi: 10.1021/ct700119m. [DOI] [PubMed] [Google Scholar]
- 42.Arora K., Brooks C.L., III Large-scale allosteric conformational transitions of adenylate kinase appear to involve a population-shift mechanism. Proc. Natl. Acad. Sci. USA. 2007;104:18496–18501. doi: 10.1073/pnas.0706443104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Albright R.A., Mossing M.C., Matthews B.W. Crystal structure of an engineered Cro monomer bound nonspecifically to DNA: possible implications for nonspecific binding by the wild-type protein. Protein Sci. 1998;7:1485–1494. doi: 10.1002/pro.5560070701. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.